

Abstract
Genome-wide association (GWA) studies have identified a large number of single-nucleotide polymorphisms (SNPs) associated with disease phenotypes. As most GWA studies have been performed primarily in populations of European descent, this review examines the issues involved in extending consideration of GWA studies to diverse worldwide populations. Although challenges exist with such issues as imputation, admixture, and replication, investigation of diverse populations in GWA studies has significant potential to advance the project of mapping the genetic determinants of complex diseases for the human population as a whole.
In the last few years, genome-wide association (GWA) studies have produced numerous successes in identifying genetic variants that contribute to complex human traits1,2. Several factors are recognized3,4 as having dramatically enlarged the number of genotype-phenotype associations documented for a wide range of phenotypes5,6. These include: increasingly dense sets of genetic markers, increasingly large sample sizes, improved resources on genomic variation, and new statistical techniques for genotype imputation 7,8 and meta-analysis9,10 that leverage these resources.
With few exceptions, however, GWA studies have been centered in populations of European descent (Box 1), and the degree to which knowledge gained from these studies is transferrable to other populations has not been extensively investigated. Recent reports such populations as Chinese11,12, Japanese13,14, Koreans15,16, and Pacific islanders from Kosrae17,18 represent some of the first in a new wave of GWA studies in non-European populations, as researchers seek to search additional groups for new findings on widely distributed phenotypes, to consider new phenotypes that are more prevalent in non-European populations, and to establish the generality of findings obtained initially in Europeans and European Americans.
Box 1. Populations in past GWA studies.
To assess the extent to which non-European populations have been incorporated into GWA studies, we examined the distribution of study populations across 492 GWA articles in the National Human Genome Research Institute catalog of GWA results6,130. This database provides a manually curated list of SNP-phenotype associations (P < 10−5) identified in studies with at least 100,000 SNPs. Article classifications were assessed independently by two raters, with discrepancies resolved by consensus in discussions with a third rater. The figure on the right tabulates classifications based on whether articles used individuals of European descent, individuals of non-European descent, or a combination of individuals of European and non-European descent. Eight articles that provided insufficient information about study subjects are omitted, so that each bar represents 80 or 81 articles, grouped by date. The later date ranges are narrower, indicating that in more recent time periods, more studies have been performed per unit time.
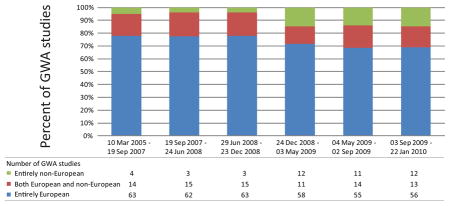
The figure illustrates that most studies, ~75%, utilize populations of European descent exclusively. It is likely that this value underestimates the true percentage of GWA effort devoted to populations of European descent, as the tabulation counts as “both European and non-European” studies in which non-Europeans comprise a small fraction of overall study subjects, or in which non-Europeans are part of replication samples examined only for a small number of SNPs. However, a slight trend over time suggests that studies with non-European populations have begun to constitute a larger proportion of the full collection of studies.
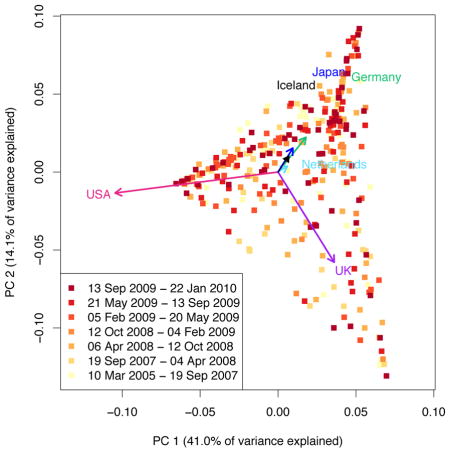
We further examined the representation of non-European populations by considering the diversity of the investigators performing the studies. For each article analyzed, we assigned weight nk/n to country k, where nk is the number of authors with affiliations in country k (splitting multiply affiliated authors evenly across affiliations), and n is the total number of authors of the article (excluding consortium authors). To examine temporal trends in country representation, the 473 articles (eleven articles with uncertain author affiliations or consortium-only authors were omitted) were divided into seven chronological groups of near-equal size, and for each country, weights were summed across articles to obtain a total “author weight” in each date class. Darker colors in the figure above represent more recent time periods.
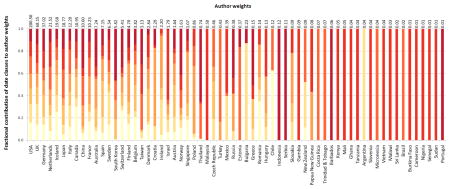
Analysis of GWA author weights reveals that the number of countries represented, and the representation of non-European countries such as China, South Korea, and Taiwan, has been increasing. A plot of the first two principal components of a matrix of country representation vectors (n1/n, n2/n, n3/n, …) appears on the right, with one point for each of the 473 articles. The vectors displayed for the six countries with the highest author weights represent the loadings of these countries for PC1 and PC2, describing the contributions of these countries to the first two principal components. The PCA plot identifies three main categories of articles—those with many UK authors, those with many US authors, and those with many authors elsewhere. Many of the most recent articles, represented by the darkest points, lie near the upper corner (“elsewhere”) or along the upper edge (collaborations between authors “elsewhere” and USA authors).
What challenges exist in GWA studies of non-European populations? Will the same results observed in Europeans be detected in diverse worldwide populations? Will causal variants have similar allele frequencies and disease risk in different populations? What factors will be the sources of differing results across groups? As the human genetics community diversifies the populations in which GWA studies are performed, the effort likely to be expended on this research program motivates careful consideration of the issues involved in designing the new wave of GWA studies and in interpreting their outcomes.
We argue that expansion of GWA studies to diverse populations is important not only for the ultimate goal of bringing medical advances resulting from genome science to the whole of the worldwide population, but also because use of diverse populations provides considerable scientific benefits in characterizing risk variants beyond what can be achieved with populations of European descent alone. We begin by reviewing factors that have contributed to the successes of GWA studies in Europeans. Next, we describe how consideration of diverse populations has the potential to build on these successes. We then discuss the challenges inherent in GWA studies in diverse populations, and the role of population-genetic modeling in investigating variation among GWA results across populations. We conclude with a discussion of how further development of genomic resources has the potential to improve prospects for GWA studies in diverse worldwide populations.
Successes in Europeans
Factors influencing the choice of study population
Owing to the expense involved in the execution of GWA studies, it was sensible to perform the first studies in a set of closely related populations for which shared resources could be used. As a first step, a focused effort in which GWA studies of many phenotypes were conducted largely in the same populations — and even in the same samples — had several advantages over a dispersed effort that would have considered a larger diversity of populations. The focus on fewer populations aided the development of standard single-nucleotide polymorphism (SNP) panels ascertained for informativeness in detecting common risk variants in those populations. It facilitated the use of shared controls in large studies of multiple phenotypes, reducing the effort required in sample collection and genotyping. Finally, it led to the collection by separate investigators of commensurable samples, enabling large meta-analyses with closely related populations.
Given these advantages of focusing on specific populations, populations of European descent were a natural choice for early GWA studies. Several European populations with a strong history of human-genetic research — such as the populations of Finland, Iceland, and Sardinia — are large enough that it was possible to conduct studies with large samples in the setting of a comparatively homogeneous population. In addition, extensive collaborations and long-term genetic studies had already been established involving investigators from European countries and from non-European countries with large populations of European descent, such as Australia, Canada, and the United States.
Population-genetic factors
Beyond these practical considerations that contributed to a focus on populations of European ancestry, specific population-genetic properties of the European population have facilitated the successes of GWA studies in groups of European origin. Allele-frequency variation across populations — a source of false-positive findings in association studies19,20,21 — is less pronounced in Europe than in other geographic regions22,23,24,25,26,27. Although large population-genetic studies have detected subtle geographic gradients in allele frequencies across the European continent28,29,30 as well as within individual countries31,32, well-designed GWA studies in Europeans have generally been able to control for the effects of underlying allele-frequency variation, and they generally have not identified false positives owing to population structure as a significant problem.
The comparatively low level of population structure has further contributed to GWA successes in Europeans through the utility of the HapMap CEU panel, the “CEPH European” collection of 30 European-American families genotyped at high density by the International Haplotype Map Project33,34. Early GWA studies used a tag-SNP approach 33,35,36, in which each SNP in a genome-wide subset of SNPs was tested for disease association. It was hoped that each true disease SNP not genotyped in a study would be “captured” through a minimal level of statistical association, or linkage disequilibrium (LD)37,38,39, with an informative nearby tag SNP included among the genotyped SNPs. The existence of a true disease SNP in an association study would then be detectable through separate associations of the disease SNP and the phenotype with the tag SNP.
In most cases, tag SNPs chosen with the HapMap CEU panel were indeed “portable” to studies of common variants in other Europeans36,40. Similarity of a target population to a reference panel on which tag SNPs were selected, along with general LD levels in the population, were identified as important determinants of the portability to a target population of tag SNPs selected using a reference population41. LD in Europeans is moderate compared to other populations42,43, so that Europeans are not disadvantaged in the tag-SNP approach by this variable. Further, portability is enhanced in Europeans owing to the low level of population structure and the resulting high level of genetic similarity of most European populations to the CEU sample41.
The combination of the various population-genetic factors with the pragmatic factors affecting the choice of study populations has uniquely favored European populations in GWA studies. These factors provide part of the explanation for two outcomes: (1) European GWA studies have produced many successes that can be replicated in different sets of individuals from the same European population as that in which the association was originally detected; (2) associations in one population of European descent are often replicable in other European populations, sometimes in groups that are quite geographically distant within the European continent.
The case for more populations
The advantages of European populations in GWA studies suggest that Europeans might productively be used for finding risk variants in non-Europeans. However, European populations contain only a subset of human genetic variation. Populations vary in terms of allele frequencies, biological adaptations, and other properties that affect the detectability and importance of risk variants, and several observations suggest that no single population is sufficient for fully uncovering the variants underlying disease in all populations.
First, risk variants can differ in their occurrence across populations. A high-risk variant might only occur in certain populations, as has recently been seen for a cardiomyopathy risk variant at MYBPC3 that has frequency ~4% in populations of the Indian subcontinent and that is rare or absent elsewhere44. Such variants differ substantially in their relevance to different groups.
Second, even if the same variant is present in diverse populations, allele frequencies might differ45,46, as has been seen at TCF7L2 and KCNQ1 in type 2 diabetes (Box 2). The particular histories of recombinations, mutations, and divergences of genealogical lineages in the various populations can influence the “mappability” of a variant, so that a variant might be more easily detectable in some populations than in others47,48 (Fig. 1). Populations with lower LD, in which correlations between genotypes extend over shorter distances along a chromosome, might be more suitable for finely localizing a risk variant once its genomic region has been identified, as the genomic distance from true risk variants of disease-associated markers is likely to be smaller in such populations49. Localization methods can potentially capitalize on LD differences across populations by identifying variants for which a causal relationship with disease underlies divergent patterns of association signals in a genomic region50.
Box 2. Common variants for type 2 diabetes.
In the last three years, large-scale genetic association studies have uncovered an impressive array of common variants that confer risk for type 2 diabetes (T2D) in populations of European origin131 and now also in East Asian populations13,14. GWA studies of T2D provide a microcosm of the variety of issues that arise in considering association results across populations.
In a study in Icelanders, Grant et al. 132 identified common alleles in TCF7L2 as associated with T2D, a finding that has been confirmed in many populations, including other Europeans133,134, West Africans135, East Asians136, South Asians137, and Mexican Americans138. These TCF7L2 SNP alleles appear to have the strongest effect on type 2 diabetes risk among common variants in Europeans. By analyzing data in Europeans and West Africans, Helgason et al. 135 narrowed the likely TCF7L2 candidate region using differences in association strength with several TCF7L2-region SNPs in these populations. Subsequent analysis of T2D association in East Asians suggests that while genetic effect sizes for these TCF7L2 variants are similar in East Asians, risk allele frequencies are substantially lower, so that much larger samples are required to identify the association139.
The first T2D GWA studies in East Asians identified T2D risk variants in KCNQ113,14. A recent metaanalysis in Europeans carried out by the DIAGRAM consortium detects this same signal with a similar effect size, but at a level not even approaching genomewide significance, owing to a much lower risk allele frequency (DIAGRAM Consortium, personal communication). Interestingly, this same meta-analysis identifies a second genome-wide significant T2D association signal ~150 kb from those discovered in East Asians.
These examples illustrate the value of carrying out large-scale genetic association studies in multiple populations to elucidate similarities and differences in genetic architecture and to help narrow candidate regions for identified disease-predisposing variants.
Figure 1. Differences in “mappability” of a risk variant between two populations with different LD patterns.

A disease mutation (orange) occurs on an ancestral chromosome that contains several marker alleles (green, purple, blue, yellow). Over time, recombination events (diamonds) break down the correlations between the disease mutation and the marker alleles. However, the recombination history differs for populations 1 and 2, separated by a barrier to gene flow (brown line). Consequently, if the purple or blue allele were examined in population 1, then a disease association might be found, but it might not be found in population 2. A similar situation applies for the yellow allele, with the roles of the populations reversed. The figure and caption are modified from Rosenberg and VanLiere121.
Third, diseases can have differences in prevalence across populations. While a large portion of this variation undoubtedly results from non-genetic factors, disease prevalence affects both the practicality of obtaining the large sample sizes required by GWA studies for detecting variants with small effects and the relevance to a population of the findings. A limited population focus risks underemphasis of diseases for which prevalence is high in non-European populations, or reduced power compared to potentially larger samples that could be obtained in higher-prevalence populations.
Fourth, risk variants can have different effect sizes in different populations, so that variation across populations can exist in the underlying determinants of the same disease51. The existence of these risk differences, such as for the APOE-ε4 allele in Alzheimer’s disease52, implies that the risk variants most relevant in a population might be most easily detected using samples from the population itself, rather than with other populations.
The case for employing diverse populations in GWA studies has recently been strengthened by the observation that the proportion of phenotypic variation explained by variants discovered through GWA is typically small53. GWA studies have focused on common variants, alleles that were typically present in ancestral African populations and that spread worldwide with ancient human migrations. Rare variants, which have not been examined to the same extent, provide one possible genetic source for unexplained phenotypic variation54,55,56. They might even be responsible for some association signals currently attributed to common variants47,57. Because rare variants are usually more recent in origin, not having had enough time to increase in frequency and become common, they are more likely to be geographically localized, so that separate populations are more likely to differ in their collections of rare alleles than in their collections of common alleles (Fig. 2).
Figure 2. Effect of frequency in Europe on the occurrence of an allele in other regions.

The figure illustrates that alleles that are more common in one group, in this case Europeans, are more likely to be present in other groups. It also shows that populations that are geographically closer to Europe, such as populations of the Middle East, tend to have more alleles shared with Europeans than more geographically distant populations, such as those of Oceania. The figure is based on the SNP data underlying Figure S21 of Jakobsson et al. 43, which uses 512,762 autosomal SNPs in indigenous populations from the Human Genome Diversity Panel140, and which standardizes sample sizes across groups by evaluating allele frequencies in samples of size 40.
These various reasons — differences in disease-allele frequency and LD patterns, phenotypic prevalence differences, differences in effect size, and differences in rare variants — provide the scientific motivation for GWA studies in diverse populations. Some variants that act in all populations might be more easily identifiable in certain groups owing to the properties of LD and allele frequency in those groups. For some phenotypes with low prevalence in Europeans, studies might be more practical in other groups49. In addition, use of multiple populations is the only way to uncover true biological variation in underlying risk variants, including biological variation resulting from differences across populations in the occurrence of rare risk alleles.
Challenges in non-Europeans
The properties of marker ascertainment, tag-SNP portability, and population structure that have been favorable to association mapping in Europeans instead pose challenges for studies in many non-European populations.
Marker ascertainment
Several investigations have found that the SNPs typically used in GWA studies are in various ways not representative58,59. They can have comparatively higher minor allele frequency (MAF) in Europeans and therefore higher expected heterozygosity than might be predicted on the basis of what is known about other types of markers that have less ascertainment bias (Fig. 3). These observations, which result from a likely focus on populations of European ancestry in the initial detection of SNPs, in turn affect the relative proportion of the genome suited to mapping in different populations. Because of ascertainment effects in the development of marker panels, the fraction of the genome that lies within a specified physical distance of at least one variable marker in a standard panel can vary across populations. Additionally, the LD statistic r2 that measures whether a locus is “covered” by a panel, typically on the basis of its maximal LD with some marker from the panel60, depends on marker allele frequencies61,62, with intermediate-MAF markers having greater potential to produce high r2 values with markers at a range of other minor allele frequencies63. Thus, ascertainment bias producing many low-MAF markers in a population can lead to decreased potential to detect phenotypically important alleles across the full range of possible allele frequencies, ultimately reducing the genome-wide utility in the population of standard marker panels.
Figure 3. Excess SNP variability in Europeans resulting from ascertainment bias.

The y-axis depicts mean heterozygosity across loci in 443 individuals from 29 populations, on the basis of 512,762 autosomal SNPs from an Illumina genotyping panel43. The x-axis depicts mean heterozygosity in the same individuals, on the basis of 783 autosomal microsatellite markers141,142. Because individual microsatellites, unlike SNPs, are highly variable, microsatellite ascertainment is less dependent on the initial ascertainment sample than is SNP ascertainment143. Thus, the imperfect correlation of SNP heterozygosity with microsatellite heterozygosity might reflect ascertainment bias in the SNP set. This figure is similar to Figure 3 of Conrad et al. 41.
Tag-SNP portability
Ascertainment issues might have contributed to a decreased level of tag-SNP portability seen in some non-European populations compared to what might have been predicted on the basis of their LD levels41. Although evaluations of tag-SNP portability have generally found that tag SNPs chosen from the HapMap are indeed portable to most non-European populations36,40, they have identified low-LD populations and intermediate-LD indigenous populations genetically distant from HapMap reference panels to be among those populations in which tag SNPs capture the fewest non-tag SNPs41. Tag-SNP portability can potentially be improved in populations genetically intermediate between the primary HapMap populations by using a mixture strategy to select SNPs for genotyping panels. In this approach, tag SNPs are selected to be informative for a mixture of haplotypes drawn from multiple HapMap groups, rather than from a single group64,65. However, this mixture strategy does not solve the problem of low portability in sub-Saharan African populations, whose LD levels are considerably lower than those of other populations43,49,66.
Genotype imputation
Recently, tag-SNP analyses have been augmented by a genotype-imputation approach, in which data analysis is not restricted to SNPs that have actually been genotyped. In imputation-based GWA studies7,8,67, densely genotyped reference individuals, typically from the HapMap Project, provide information for predicting the genotypes at SNP positions measured in the reference data but not in the study sample. These predicted genotypes are then tested for disease association. Imputation is possible because two haplotypes that are identical for a set of nearby markers are likely to share the intervening chromosomal stretch identically by descent. Thus, if one of the two haplotypes is genotyped more densely than the other, then genotypes at unmeasured positions in the more sparsely genotyped haplotype can be predicted by copying the genotypes from the more densely genotyped haplotype. Genome-wide imputation of study haplotypes proceeds by locally copying the most appropriate reference haplotypes in a probabilistic manner.
In imputation studies, the reduced portability of tag SNPs previously observed in populations genetically intermediate among reference groups and in African populations has taken the form of reduced imputation accuracy for these populations68, and consequently, reduced statistical power for imputation-based association mapping69. The accuracy of imputation depends largely on the same two factors that influence portability in the tag-SNP case. First, the overall level of LD in a study population reflects the distance over which the genotypic correlations that permit imputation extend, so that imputation is more accurate in high-LD populations68. Second, imputation accuracy is influenced by the level of genetic relationship of the study population to the reference population8,68, which affects the utility of the haplotypes copied from the reference population in imputing genotypes in the study population. In an assessment of imputation accuracy in 29 populations worldwide, similarly to the tag-SNP case, imputation accuracy based on HapMap reference panels was highest in European populations closely related to the HapMap CEU panel, and lowest in African populations and populations genetically intermediate between the panels68 (Fig. 4). Use of mixture panels as reference data in imputation algorithms can improve imputation accuracy for GWA studies in genetically intermediate populations, but imputation in low-LD African populations continues to pose a particular challenge49.
Figure 4. Genotype imputation accuracy in 29 populations, with and without external reference panels.

Imputation accuracy is plotted as a function of LD measured by mean r2 at a distance of 10 kb in a genome-wide dataset43. Genotypes in a genome-wide study are hidden and then imputed, with two different designs. In the shaded region, genotypes in each population are imputed without an external reference panel, so that the information for imputing “missing” genotypes comes from other individuals in the population. In the unshaded region, genotypes in the population are imputed using an external reference panel, chosen optimally among 36 mixtures of the HapMap CEU (European American), CHB+JPT (Chinese and Japanese), and YRI (Yoruba) panels. Color coding for populations follows that of Fig. 3. The regression lines exclude the African populations, and they have coefficients of determination 0.003 (external reference) and 0.953 (internal reference). The figure shows that imputation accuracy based on an internal reference is highly correlated with LD. However, imputation accuracy based on an external reference is not correlated with LD (and instead depends on the composition of the particular reference panels available). The figure is based on the data in scenarios 1, 3, and 6 in Table 1 of Huang et al. 68.
Admixed populations
In the effort to improve the potential of GWA studies for diverse human populations, African populations are not the only populations that pose significant challenges. Tag-SNP and imputation studies have found that indigenous populations genetically intermediate between reference groups are among those that require special consideration. In these cases, the challenges result largely from the way in which genomic resources have been developed, rather than from intrinsic population properties. A second form of intermediate population exists, however, in which the challenges are in fact intrinsic.
In admixed populations, individual genomes can be viewed as mosaics of ancestry segments, with different segments arising from different “parental” populations that participated in an admixture process. Admixed populations often have high variation across individuals in the proportions of ancestry from the various source groups70,71,72, and in the same way that use of multiple subgroups of a larger population in an association study can give rise to false-positive associations, variation in admixture proportions can also produce spurious association of genotypes and phenotypes through their separate associations with ancestry73.
Heterogeneity of admixture has posed a barrier to association mapping in admixed populations. These populations have instead been considered with other designs, such as admixture mapping, in which genomic segments with excess ancestry from a high-prevalence parental population are identified as potential locations for risk variants74,75,76,77. The utility of admixture mapping, which has had some success in mapping loci for traits with strong differences in phenotypic distribution between parental populations78,79,80,81, has relied on its relative efficiency. Whereas GWA has typically used tens to hundreds of thousands of markers, admixture mapping requires only a few thousand markers for estimating the ancestry of genomic segments82,83,84,85. However, as GWA designs have improved, the efficiency of GWA now exceeds that of admixture mapping over a broad range of possible values for model parameters86. Future analyses in admixed populations might rely on a combination of GWA and admixture-mapping principles, considering unusual local ancestry estimates jointly with association signals. In addition, because it requires fewer markers, admixture mapping might continue to be valuable in genomic regions poorly covered by typical GWA marker sets.
In the imputation context, it has been largely unclear whether genotypes in an admixed population can be most accurately imputed using a mixture of reference panels from the parental populations, or using a comparable reference panel from the admixed population itself. Numerous techniques are now available for inferring ancestry blocks along the genome87,88,89,90,91, and one recent approach uses imputation accuracy as a basis for evaluating inference of ancestry blocks92. This set of developments now offers the possibility of improving imputation in admixed populations by integrating the inference of admixture and missing genotypes93, either by locally imputing from parental reference panels along the genome (Fig. 5), or by concurrently imputing genotypes and inferring ancestry. Although evaluations in admixed populations of the performance of different imputation approaches have not yet utilized local ancestry94, these advances suggest that the intrinsic challenges of working with admixed populations in GWA studies can be surmounted or at least reduced.
Figure 5. Imputation in admixed populations.

Admixture segments are estimated in each individual sampled from a GWA study. Consider reference haplotypes from two separate panels (red and blue boxes). Separately for each admixture segment of a haplotype, alleles are imputed using reference haplotypes from the same population as the inferred source. Within a source population, a haplotype might have alleles imputed from multiple reference haplotypes, as depicted on the left with both haplotypes from the same (blue) source population serving as imputation templates. If admixture estimates for a segment are uncertain, then conditional imputations at a site given each of the possible source populations for the segment can be weighted by the probabilities of those sources.
Population-genetic modeling
We have seen that information on the population-genetic properties of individual populations and sets of populations is useful in understanding the features and limitations of GWA studies in diverse populations. Important roles for population-genetic data and modeling have been part of the planning for GWA studies from the early stages3,95; modeling efforts can now help in addressing concerns about the similarities and differences among GWA results in separate populations.
Population-genetic models begin from the perspective that the factors that affect the genealogical descent of a disease mutation — such as migrations, changes in population size, natural selection, and local recombination landscape — ultimately affect the distribution of the mutation across individuals in the present. Because the full genetic history of the human population is unknown, population-genetic models based on relatively few parameters can be used instead to simulate plausible histories, to examine the properties of risk variants simulated under the models, and to evaluate strategies for detecting these variants. Many of these models use the coalescent framework 96,97, which provides a flexible, computationally efficient, and theoretically grounded approach that can simulate one or more populations retrospectively, back in time from the present.
New population-genetic simulation tools that account for shared descent among individuals, both through the coalescent and through forward-time approaches98,99,100,101, now provide an improved basis for GWA modeling. Simulation programs have incorporated newly appreciated phenomena, such as recombination hotspots102, as well as approximations and computational advances that improve the potential for simulating large genomic regions103,104,105,106. Human population-genetic data have been recently used to calibrate evolutionary models107,108,109,110,111,112; further advances in human population genetics offer the potential to make these models increasingly detailed and therefore increasingly relevant for GWA applications.
A primary use of a population-genetic perspective in the GWA context has been in predicting expected patterns of disease variation113,114,115. However, GWA statistical analysis tools have not yet fully taken advantage of this perspective. From a population-genetic standpoint, all individuals have some degree of relationship through their shared descent in the complete human pedigree. In standard GWA analyses, however, in which alleles that are more common in cases than in controls are identified by testing contingency tables locally along the genome, an implicit assumption is that the genotypes of separate individuals can be treated as independent random variates. Approximating separate individuals as independent has been productive as a first approximation, but more information is potentially available by accounting for correlation among individuals owing to shared descent. Fine-mapping association methods designed for localization of risk variants do seek to consider this shared descent116,117,118,119,120. These methods have been informative on a small scale, and a current challenge is to extend them for large datasets.
Similar independence approximations are made in GWA replication analyses, which check for close relationships among sampled individuals but otherwise treat separate studies of nonoverlapping samples as independent. A genealogical perspective suggests that replication studies are in fact pseudoreplication studies, owing to potential correlations among outcomes that could arise from shared genealogy. From this viewpoint, particularly in small populations, separate association studies identifying the same risk variant in a population might not provide the same degree of confirmation as replicate studies in a context in which events truly are independent121. As in the analysis of individual GWA studies, the independence assumption has provided a sensible initial strategy for replication studies, but unlike in the case of genealogical dependence within studies, approaches that account for dependence between studies have not yet been considered. The magnitude in real populations of the pseudoreplication effect — the degree to which separate association studies provide the same outcome as a result of shared ancestry of study participants — is unknown, so that it remains uncertain how likely a replication study is to detect a risk variant under the hypothesis that the variant has the same disease effect in all populations; the probability of pseudoreplicating a false positive across populations is also unknown. Although efforts have been devoted to statistical issues of replication in relation to sample size and measured effect size122,123, studies of the population genetics of replication are in their infancy. As the frequency of replication studies continues to increase, methods for evaluating intrinsic correlations between study outcomes and their effects on interpretations of replication studies would provide a useful development.
Prospects
GWA studies have dramatically increased the number of variants known for numerous complex diseases. They have been remarkably successful for identifying targets of exploration, often suggesting unforeseen directions for research on disease mechanisms. Especially for those working on diseases for which few if any genetic variants were previously known, GWA studies have provided a true quantum advance for studies of human biology. At the same time, they have established complex genetic diseases to be incontestably complex, caused by many variants, with mostly small effects unsuited to immediate risk prediction and clinical use. These results have understandably triggered a series of reflections on the magnitude of the contributions of GWA studies in general4,124,125,126,127. Clearly, genome-wide association is relatively new, and its full contribution will only become clear as the biological properties of the variants it uncovers are further investigated. Looking forward, the GWA field is now diversifying its emphasis, with attention shifting not only to diverse populations, but also to structural variation, interaction effects, rare sequence variation, and molecular assays of identified variants.
We and others128,129 have argued that use of diverse populations will be an essential component of the next phase of GWA work, and we have discussed the benefits that arise from the consideration of GWA studies in diverse populations. Not least among these benefits is that in the long-term, as knowledge gained from GWA becomes relevant to medicine, reducing differences across populations in the understanding of underlying genetic variation can help to avoid unintentionally promoting health disparities. Many GWA studies in diverse populations are now ongoing or are imminent, largely using the same approaches as current and past GWA studies in populations of European origin. To achieve their maximal potential, these studies will profit from deeper investigation of such issues as imputation, admixture, and replication, as we have described.
The current GWA strategy of using preselected markers to search for risk variants common in human populations is giving way to a paradigm of using whole-genome sequence approaches that can search for rare disease-risk variants as well. Future GWA studies —and some studies now in progress — will incorporate partial or complete genome sequences on some or all of the study participants. For many of the same reasons that GWA studies to date have emphasized populations of European descent, early sequence studies might also have a European focus. As we have seen, however, rare risk variants whose detection is a priority of sequence-based studies are likely to be more geographically restricted than the common variants currently of interest. Consequently, it will be even more important in sequence-based GWA studies than in current studies of common variants that multiple populations be considered.
The 1000 Genomes Project, a large-scale community effort to produce genome sequence data on ~2000 diverse individuals, will facilitate sequence-based GWA studies in diverse populations, serving as the analogous public resource for sequence-based GWA studies that the HapMap provided for tag-SNP GWA studies. With sequencing, concerns about population biases in marker ascertainment are likely to subside. Further, the larger number of individuals in the 1000 Genomes Project compared to the initial 270 individuals in the HapMap permits examination of a wider diversity of samples. Thus, forthcoming genomic resources already under development are expected to improve the conditions for examination of diverse populations in GWA studies.
At the same time, it must be remembered that the worldwide human population and its distribution of disease-risk variation represent the singular outcome of an evolutionary experiment, and that large portions of this experiment continue to remain untapped for their potential to contribute to the modern enterprise of human genetics. Each new genetic resource expands the consideration of human diversity, but necessarily provides an incomplete picture of its full extent. Thus, many opportunities exist for identifying new aspects of genetic variation to examine for future resources, as well as for creative application of worldwide populations in risk-variant discovery, characterization of known variants, and the population-genetic modeling and statistical designs that will facilitate these efforts. As technological barriers to the production of genomic data continue to fall, it can be hoped that the community will accept the challenge of capitalizing on the full range of human diversity for the next wave of investigations of the variants that underlie human genetic disease.
Highlighted references
1 An informative overview of key issues in the field of genome-wide association.
6 An investigation of the properties of GWA findings in the National Human Genome Research Institute catalog of published genome-wide association studies.
30 This article and the seven that precede it provide extensive genome-wide analyses of population structure in individual geographic regions.
44 An example of a high-risk complex disease variant absent in Europe but with a nontrivial frequency in a non-European population.
49 A review that focuses on particular challenges for GWA studies in Africa.
50 This simulation study argues that fine mapping of causal variants is improved by joint analysis of multiple populations. The study provides an approach for selecting multi-population samples for following up GWA discoveries.
69 This article and the article that precedes it provide detailed analyses of genotype imputation in diverse populations.
Acknowledgments
We thank L. Hindorff for detailed information on the NHGRI catalog of GWA studies, the DIAGRAM Consortium for use of prepublication data, J. Li and S. Zöllner for helpful discussions, and N. Patterson and an anonymous reviewer for comments on a draft of the manuscript. We are grateful to M. DeGiorgio, M. Jakobsson, S. Reddy, and P. Scheet for assistance with Box 1 and with figure preparation. Support was provided by NIH grants DK062370, GM081441, HG000376, and HL090564, and by grants from the Burroughs Wellcome Fund and the Alfred P. Sloan Foundation.
Glossary
- GENOME-WIDE ASSOCIATION STUDY
A study design in which many markers spread across a genome are genotyped, and tests of statistical association with a phenotype are performed locally along the genome
- GENOTYPE IMPUTATION
Probabilistic prediction of genotypes that have not actually been measured experimentally
- PRINCIPAL COMPONENT
A composite variable that summarizes the variation across a larger number of variables, each represented by a column of a matrix
- LOADING
In a principal components analysis, a quantity that represents the contribution of one of the original variables (columns of the data matrix) to one of the principal components
- SNP
A nucleotide site at which two or more variants exist in a population. Most SNPs in GWA studies are biallelic
- TAG SNP
A SNP chosen from a larger set of available SNPs for use in an association study. Tag SNPs are generally selected on the basis of favorable linkage disequilibrium properties; to be precise we do not require as part of the definition that they be selected using properties of LD.
- LINKAGE DISEQUILIBRIUM
A statistical association in the occurrence of alleles at separate loci
- PORTABILITY OF TAG SNPS
The utility of SNPs chosen as tags in one population for use as tags in another population
- MINOR ALLELE FREQUENCY
The frequency of the less frequent allele at a biallelic genetic locus
- EXPECTED HETEROZYGOSITY
The probability for a locus that two alleles drawn from its allele-frequency distribution are distinct
- ASCERTAINMENT BIAS
A distortion in results owing to use of a subsample that, in a systematic manner, fails to properly represent a larger sample
- MICROSATELLITE
A type of genetic marker in which individuals vary in their number of tandemly repeated copies of a short DNA unit
- ADMIXED POPULATION
A population formed recently from the mixing of two or more groups whose ancestors had long been separated
- COALESCENT
A specific stochastic process that describes the relationship among genetic lineages sampled in a population
- RECOMBINATION HOTSPOT
A region of the genome in which the per-generation recombination rate is substantially elevated above the genome-wide average
- CONTINGENCY TABLE
A table of observations of two or more variables whose statistical relationship is of interest. For each variable, a contingency table places each observation into one of a series of categories
Biographies
Noah Rosenberg earned his PhD in biological sciences in 2001 from Stanford University and completed his postdoctoral training at the University of Southern California. He is now Associate Professor of Human Genetics, Biostatistics, and Ecology & Evolutionary Biology at the University of Michigan. Research in his laboratory focuses on human evolutionary genetics, population-genetic theory, mathematical phylogenetics, and the connection between human evolutionary history and the search for disease genes.
Lucy Huang is a bioinformatics PhD student at the University of Michigan. She earned her BS in mathematics at the University of Chicago and her MS in biostatistics at the University of Michigan. Her PhD research focuses on genotype imputation, admixture, and the population genetics of association mapping.
Ethan Jewett is a bioinformatics PhD student at the University of Michigan. He earned his BA in physics at Reed College and his MA in education and MS in applied & interdisciplinary mathematics at the University of Michigan. His PhD research focuses on mathematical modeling in population genetics and phylogenetics.
Zachary Szpiech is a bioinformatics PhD student at the University of Michigan. He earned his BS in mathematics and MS in bioinformatics at the University of Michigan. His PhD research focuses on human population genetics, with an emphasis on the properties of private alleles.
Ivana Jankovic is a molecular & cellular biology undergraduate at the University of Michigan. Upon completion of her undergraduate degree, she will begin training as an MD/PhD student.
Michael Boehnke earned his PhD in biomathematics in 1983 at the University of California, Los Angeles. He is now the Richard G. Cornell Distinguished University Professor of Biostatistics and Director of the Center for Statistical Genetics at the University of Michigan. Dr. Boehnke and his research group develop statistical methods for human-genetic studies and apply those methods for understanding complex diseases such as type 2 diabetes and bipolar disorder. Their current research is focused on genome-wide association studies and targeted and whole-genome sequencing studies.
References
- 1.McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 2.Frazer KA, et al. Human genetic variation and its contribution to complex traits. Nature Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 3.Altshuler D, et al. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manolio TA, et al. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halperin E, Stephan DA. SNP imputation in association studies. Nature Biotechnol. 2009;4:349–351. doi: 10.1038/nbt0409-349. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, et al. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bakker PIW, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeggini E, Ioannidis JPA. Meta-analysis in genome-wide association studies. Pharmacogenomics. 2009;10:191–201. doi: 10.2217/14622416.10.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Barcelo MM, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung’s disease. Proc Natl Acad Sci. 2009;106:2694–2699. doi: 10.1073/pnas.0809630105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang XJ, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nature Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 13.Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature Genet. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 14.Yasuda K, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature Genet. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 15.Cho YS, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nature Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, et al. Alpha-T-catenin (CTNNA3) gene was identified as a risk variant for toluene diisocyanate-induced asthma by genome-wide association analysis. Clin Exp Allergy. 2009;39:203–212. doi: 10.1111/j.1365-2222.2008.03117.x. [DOI] [PubMed] [Google Scholar]
- 17.Lowe JK, et al. Genome-wide association studies in an isolated founder population from the Pacific island of Kosrae. PLoS Genet. 2009;5:e1000365. doi: 10.1371/journal.pgen.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JG, et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm. 2009;6:634–641. doi: 10.1016/j.hrthm.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 20.Ziv E, Burchard EG. Human population structure and genetic association studies. Pharmacogenomics. 2003;4:431–441. doi: 10.1517/phgs.4.4.431.22758. [DOI] [PubMed] [Google Scholar]
- 21.Tiwari HK, et al. Review and evaluation of methods for correcting for population stratification with a focus on underlying statistical principles. Hum Hered. 2008;66:67–86. doi: 10.1159/000119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg NA, et al. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, et al. Genetic variation and population structure in Native Americans. PLoS Genet. 2007;3:2049–2067. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedlaender JS, et al. The genetic structure of Pacific Islanders. PLoS Genet. 2008;4:e19. doi: 10.1371/journal.pgen.0040019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The HUGO Pan-Asian SNP Consortium. Mapping human genetic diversity in Asia. Science. 2009;326:1541–1545. doi: 10.1126/science.1177074. [DOI] [PubMed] [Google Scholar]
- 26.Reich D, et al. Reconstructing Indian population history. Nature. 2009;461:489–494. doi: 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tishkoff SA, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heath SC, et al. Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet. 2008;16:1413–1429. doi: 10.1038/ejhg.2008.210. [DOI] [PubMed] [Google Scholar]
- 29.Lao O, et al. Correlation between genetic and geographic structure in Europe. Curr Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 30.Novembre J, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jakkula E, et al. The genome-wide patterns of variation expose significant substructure in a founder population. Am J Hum Genet. 2008;83:787–794. doi: 10.1016/j.ajhg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price AL, et al. The impact of divergence time on the nature of population structure: an example from Iceland. PLoS Genet. 2009;5:e1000505. doi: 10.1371/journal.pgen.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson CS, et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu CC, et al. On transferability of genome-wide tagSNPs. Genet Epidemiol. 2008;32:89–97. doi: 10.1002/gepi.20269. [DOI] [PubMed] [Google Scholar]
- 37.Nordborg M, Tavaré S. Linkage disequilibrium: what history has to tell us. Trends Genet. 2002;18:83–90. doi: 10.1016/s0168-9525(02)02557-x. [DOI] [PubMed] [Google Scholar]
- 38.Slatkin M. Linkage disequilibrium — understanding the evolutionary past and mapping the medical future. Nature Rev Genet. 2008;9:477–485. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weir BS. Linkage disequilibrium and association mapping. Annu Rev Genomics Hum Genet. 2008;9:129–142. doi: 10.1146/annurev.genom.9.081307.164347. [DOI] [PubMed] [Google Scholar]
- 40.Xing J, et al. HapMap tagSNP transferability in multiple populations: general guidelines. Genomics. 2008;92:41–51. doi: 10.1016/j.ygeno.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad DF, et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nature Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 42.Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ‘race’ and medicine. Nature Genet. 2004;36:S21–S27. doi: 10.1038/ng1438. [DOI] [PubMed] [Google Scholar]
- 43.Jakobsson M, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 44.Dhandapany PS, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nature Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myles S, et al. Worldwide population differentiation at disease-associated SNPs. BMC Med Genomics. 2008;1:22. doi: 10.1186/1755-8794-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adeyemo A, Rotimi C. Genetic variants associated with complex human diseases show wide variation across multiple populations. Public Health Genomics. 2010;13:72–79. doi: 10.1159/000218711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008;17:R156–R165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teo YY, et al. Power consequences of linkage disequilibrium variation between populations. Genet Epidemiol. 2009;33:128–135. doi: 10.1002/gepi.20366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teo YY, et al. Methodological challenges of genome-wide association analysis in Africa. Nature Rev Genet. 2010;11:149–160. doi: 10.1038/nrg2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaitlen N, et al. Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet. 2010;86:23–33. doi: 10.1016/j.ajhg.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang H. Confronting ethnicity-specific disease risk. Nature Genet. 2006;38:13–15. doi: 10.1038/ng0106-13. [DOI] [PubMed] [Google Scholar]
- 52.Tang MX, et al. The APOE-ε4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. J Am Med Assoc. 1998;279:751–755. doi: 10.1001/jama.279.10.751. [DOI] [PubMed] [Google Scholar]
- 53.Maher B. The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 54.Bodmer W, Bonilla C. Common and rare variants in multi-factorial susceptibility to common diseases. Nature Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iles MM. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genet. 2008;4:e33. doi: 10.1371/journal.pgen.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schork NJ, et al. Common vs. rare allele hypotheses for complex diseases. Curr Op Genet Devel. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dickson SP, et al. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen R. Population genetic analysis of ascertained SNP data. Hum Genomics. 2004;1:218–224. doi: 10.1186/1479-7364-1-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark AG, et al. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nature Genet. 2006;38:659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- 61.Wray NR. Allele frequencies and the r2 measure of linkage disequilibrium: impact on design and interpretation of association studies. Twin Res Hum Genet. 2005;8:87–94. doi: 10.1375/1832427053738827. [DOI] [PubMed] [Google Scholar]
- 62.Eberle MA, et al. Frequency-matching SNPs reveals extended linkage disequilibrium in genic regions. PLoS Genet. 2006;2:1319–1327. doi: 10.1371/journal.pgen.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.VanLiere JM, Rosenberg NA. Mathematical properties of the r2 measure of linkage disequilibrium. Theor Pop Biol. 2008;74:130–137. doi: 10.1016/j.tpb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pemberton TJ, et al. Using population mixtures to optimize the utility of genomic databases: linkage disequilibrium and association study design in India. Ann Hum Genet. 2008;72:535–546. doi: 10.1111/j.1469-1809.2008.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egyud MRL, et al. Use of weighted reference panels based on empirical estimates of ancestry for capturing untyped variation. Hum Genet. 2009;125:295–303. doi: 10.1007/s00439-009-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Browning SR. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum Genet. 2008;124:439–450. doi: 10.1007/s00439-008-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang L, et al. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet. 2009;84:235–250. doi: 10.1016/j.ajhg.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang L, et al. The relationship between imputation error and statistical power in genetic association studies in diverse populations. Am J Hum Genet. 2009;85:692–698. doi: 10.1016/j.ajhg.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang S, et al. Geographic patterns of genome admixture in Latin American mestizos. PLoS Genet. 2008;4:e1000037. doi: 10.1371/journal.pgen.1000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silva-Zolezzi I, et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc Natl Acad Sci USA. 2009;106:8611–8616. doi: 10.1073/pnas.0903045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bryc K, et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci USA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosenberg NA, Nordborg M. A general population-genetic model for the production by population structure of spurious genotype-phenotype associations in discrete, admixed, or spatially distributed populations. Genetics. 2006;173:1665–1678. doi: 10.1534/genetics.105.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKeigue PM. Prospects for admixture mapping of complex traits. Am J Hum Genet. 2005;76:1–7. doi: 10.1086/426949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reich D, Patterson N. Will admixture mapping work to find disease genes? Phil Trans R Soc Lond B. 2005;360:1605–1607. doi: 10.1098/rstb.2005.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith MW, O’Brien SJ. Mapping by admixture linkage disequilibrium: advances, limitations and guidelines. Nature Rev Genet. 2005;6:623–632. doi: 10.1038/nrg1657. [DOI] [PubMed] [Google Scholar]
- 77.Seldin MF. Admixture mapping as a tool in gene discovery. Curr Op Genet Devel. 2007;17:177–181. doi: 10.1016/j.gde.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu X, et al. Admixture mapping for hypertension loci with genome-scan markers. Nature Genet. 2005;37:177–181. doi: 10.1038/ng1510. [DOI] [PubMed] [Google Scholar]
- 79.Freedman ML, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci USA. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reich D, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nalls MA, et al. Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am J Hum Genet. 2008;82:81–87. doi: 10.1016/j.ajhg.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smith MW, et al. A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tian C, et al. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am J Hum Genet. 2006;79:640–649. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Price AL, et al. A genomewide admixture map for Latino populations. Am J Hum Genet. 2007;80:1024–1036. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tian C, et al. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet. 2007;80:1014–1023. doi: 10.1086/513522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Risch N, Tang H. Whole genome association studies in admixed populations. Am J Hum Genet. 2006;79:S254. [Google Scholar]
- 87.Falush D, et al. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoggart CJ, et al. Design and analysis of admixture mapping studies. Am J Hum Genet. 2004;74:965–978. doi: 10.1086/420855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang H, et al. Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sankararaman S, et al. Estimating local ancestry in admixed populations. Am J Hum Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Price AL, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paşaniuc B, et al. Imputation-based local ancestry inference in admixed populations. Lect N Bioinformat. 2009;5542:221–233. [Google Scholar]
- 93.Paşaniuc B, et al. Inference of locus-specific ancestry in closely related populations. Bioinformatics. 2009;25:i213–i221. doi: 10.1093/bioinformatics/btp197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shriner D, et al. Practical considerations for imputation of untyped markers in admixed populations. Genet Epidemiol. 2010;34:258–265. doi: 10.1002/gepi.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kruglyak L. The road to genome-wide association studies. Nature Rev Genet. 2008;9:314–318. doi: 10.1038/nrg2316. [DOI] [PubMed] [Google Scholar]
- 96.Hein J, et al. Gene Genealogies, Variation and Evolution. Oxford University Press; 2005. [Google Scholar]
- 97.Wakeley J. Coalescent Theory. Roberts & Company; 2008. [Google Scholar]
- 98.Peng B, et al. Forward-time simulations of human populations with complex diseases. PLoS Genet. 2007;3:407–420. doi: 10.1371/journal.pgen.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chadeau-Hyam M, et al. Fregene: simulation of realistic sequence-level data in populations and ascertained samples. BMC Bioin-formatics. 2008;9:364. doi: 10.1186/1471-2105-9-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hernandez RD. A flexible forward simulator for populations subject to selection and demography. Bioinformatics. 2008;24:2786–2787. doi: 10.1093/bioinformatics/btn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Padhukasahasram B, et al. Exploring population genetic models with recombination using efficient forward-time simulations. Genetics. 2008;178:2417–2427. doi: 10.1534/genetics.107.085332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hellenthal G, Stephens M. msHOT: modifying Hudson’s ms simulator to incorporate crossover and gene conversion hotspots. Bioinformatics. 2007;23:520–521. doi: 10.1093/bioinformatics/btl622. [DOI] [PubMed] [Google Scholar]
- 103.McVean GAT, Cardin NJ. Approximating the coalescent with recombination. Phil Trans R Soc Lond B. 2005;360:1387–1393. doi: 10.1098/rstb.2005.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marjoram P, Wall JD. Fast “coalescent” simulation. BMC Genet. 2006;7:16. doi: 10.1186/1471-2156-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liang L, et al. GENOME: a rapid coalescent-based whole genome simulator. Bioinformatics. 2007;23:1565–1567. doi: 10.1093/bioinformatics/btm138. [DOI] [PubMed] [Google Scholar]
- 106.Chen GK, et al. Fast and flexible simulation of DNA sequence data. Genome Res. 2009;19:136–142. doi: 10.1101/gr.083634.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marth GT, et al. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schaffner SF, et al. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Voight BF, et al. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci USA. 2005;102:18508–18513. doi: 10.1073/pnas.0507325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:972–979. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fagundes NJR, et al. Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci USA. 2007;104:17614–17619. doi: 10.1073/pnas.0708280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.DeGiorgio M, et al. Explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proc Natl Acad Sci USA. 2009;106:16057–16062. doi: 10.1073/pnas.0903341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 115.Di Rienzo A. Population genetics models of common diseases. Curr Op Genet Devel. 2006;16:630–636. doi: 10.1016/j.gde.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 116.Liu JS, et al. Bayesian analysis of haplotypes for linkage disequilibrium mapping. Genome Res. 2001;11:1716–1724. doi: 10.1101/gr.194801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morris AP, et al. Fine-scale mapping of disease loci via shattered coalescent modeling of genealogies. Am J Hum Genet. 2002;70:686–707. doi: 10.1086/339271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zöllner S, Pritchard JK. Coalescent-based association mapping and fine mapping of complex trait loci. Genetics. 2005;169:1071–1092. doi: 10.1534/genetics.104.031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Minichiello MJ, Durbin R. Mapping trait loci by use of inferred ancestral recombination graphs. Am J Hum Genet. 2006;79:910–922. doi: 10.1086/508901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kimmel G, et al. Association mapping and significance estimation via the coalescent. Am J Hum Genet. 2008;83:675–683. doi: 10.1016/j.ajhg.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rosenberg NA, VanLiere JM. Replication of genetic associations as pseudoreplication due to shared genealogy. Genet Epidemiol. 2009;33:479–487. doi: 10.1002/gepi.20400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gorroochurn P, et al. Non-replication of association studies: “pseudo-failures” to replicate? Genet Med. 2007;9:325–331. doi: 10.1097/gim.0b013e3180676d79. [DOI] [PubMed] [Google Scholar]
- 123.Zöllner S, Pritchard JK. Overcoming the winner’s curse: estimating penetrance parameters from case-control data. Am J Hum Genet. 2007;80:605–615. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 125.Hirschhorn JN. Genomewide association studies — illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 126.Kraft P, Hunter DJ. Genetic risk prediction — are we there yet? N Engl J Med. 2009;360:1701–1703. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- 127.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cooper RS, et al. Genome-wide association studies: implications for multiethnic samples. Hum Mol Genet. 2008;17:R151–R155. doi: 10.1093/hmg/ddn263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Need AC, Goldstein DB. Next generation disparities in human genomics: concerns and remedies. Trends Genet. 2009;25:489–494. doi: 10.1016/j.tig.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 130.Hindorff LA, et al. [Accessed February 25, 2010];A catalog of published genome-wide association studies. www.genome.gov/gwastudies/
- 131.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grant SF, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 133.Groves CJ, et al. Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes. 2006;55:2640–2644. doi: 10.2337/db06-0355. [DOI] [PubMed] [Google Scholar]
- 134.Scott LJ, et al. Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes. 2006;55:2649–2653. doi: 10.2337/db06-0341. [DOI] [PubMed] [Google Scholar]
- 135.Helgason A, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nature Genet. 2007;39:218–225. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 136.Luo Y, et al. Meta-analysis of the association between SNPs in TCF7L2 and type 2 diabetes in East Asian population. Diabetes Res Clin Pr. 2009;85:139–146. doi: 10.1016/j.diabres.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 137.Chandak GR, et al. Common variants in the TCF7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia. 2007;50:63–67. doi: 10.1007/s00125-006-0502-2. [DOI] [PubMed] [Google Scholar]
- 138.Lehman DM, et al. Haplotypes of transcription factor 7-like 2 (TCF7L2) gene and its upstream region are associated with type 2 diabetes and age of onset in Mexican Americans. Diabetes. 2007;56:389–393. doi: 10.2337/db06-0860. [DOI] [PubMed] [Google Scholar]
- 139.Tan JT, et al. Polymorphisms identified through genome-wide association studies and their associations with type 2 diabetes in Chinese, Malays, and Asian-Indians in Singapore. J Clin Endocrinol Metab. 2010;95:390–397. doi: 10.1210/jc.2009-0688. [DOI] [PubMed] [Google Scholar]
- 140.Cann HM, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 141.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rosenberg NA, et al. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 2005;1:660–671. doi: 10.1371/journal.pgen.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rogers AR, Jorde LB. Ascertainment bias in estimates of average heterozygosity. Am J Hum Genet. 1996;58:1033–1041. [PMC free article] [PubMed] [Google Scholar]