

Human α-thrombin was crystallized in complex with specific peptide inhibitors of general sequence d-Phe-Pro-d-Arg-P1′-CONH2. The crystals belonged to the orthorhombic space group P212121 and diffracted to beyond 1.3 Å resolution.
Keywords: thrombin, blood coagulation, peptide inhibitors, noncovalent
Abstract
The serine protease thrombin plays a major role in thrombosis and haemostasis. This has driven interest in thrombin inhibitors as potential antithrombotic drugs. Here, the crystallization and preliminary crystallographic analysis of human α-thrombin in complex with three noncovalent peptide inhibitors of the general sequence d-Phe-Pro-d-Arg-P1′-CONH2 are reported. The crystals belonged to the orthorhombic space group P212121 and diffracted to beyond 1.3 Å resolution.
1. Introduction
Physical damage to the vascular system triggers the coagulation cascade, a series of activation reactions of precursor proteins and regulating factors circulating in the blood that ultimately prevent blood loss without compromising blood flow through either the uninjured or the damaged vessels.
α-Thrombin (EC 3.4.21.5), the last proteolytic enzyme of the coagulation cascade, arises from the cleavage of prothrombin by serine protease factor Xa in the context of the prothrombinase complex. Given its multiple roles in blood clotting, thrombin plays a major part in haemostasis (Stubbs & Bode, 1993 ▶), where it catalyses the conversion of soluble plasma fibrinogen to insoluble fibrin (Doolittle, 1984 ▶), which then forms the scaffold for the growth of the thrombus, but also cleaves and activates coagulation factors V, VIII, XI and XIII (Gailani & Broze, 1991 ▶; Muszbek et al., 1999 ▶; Kane & Davie, 1988 ▶) and stimulates platelet aggregation through activation of platelet receptors (Vu et al., 1991 ▶). The entire system must be strictly regulated to restrict clotting to the site of injury. As it diffuses away from the site of damage, thrombin associates with the endothelial surface receptor thrombomodulin, becoming an anticoagulant factor by activating protein C (Esmon, 1995 ▶).
Thrombosis-related disorders such as myocardial infarction, stroke and pulmonary embolism remain major causes of mortality and morbidity worldwide (Rauch et al., 2001 ▶). This has driven interest in thrombin inhibitors as potential antithrombotic drugs (Paoli et al., 2005 ▶). However, to date the discovery of safe, selective and orally available inhibitors has proven difficult to accomplish, therefore limiting their therapeutic use (Hauptmann, 2002 ▶; Kikelj, 2003 ▶).
In order to acquire and digest their blood meal, haematophagous animals must counteract the coagulation system of their hosts. This is often achieved by resorting to specific and highly selective thrombin inhibitors, of which only six have been structurally characterized to date (Macedo-Ribeiro et al., 2008 ▶; Richardson et al., 2000 ▶; Fuentes-Prior et al., 1997 ▶; van de Locht et al., 1995 ▶, 1996 ▶; Grütter et al., 1990 ▶), revealing six distinct modes of inhibition. Recently, a novel and sequence-unrelated thrombin inhibitor has been identified in the hard tick Haemophysalis longicornis (Iwanaga et al., 2003 ▶; Nakajima et al., 2006 ▶). The mode of inhibition of this cysteine-less molecule, termed chimadanin, is expected to provide essential clues for the design of improved synthetic anticoagulants.
Several small-molecule synthetic thrombin inhibitors have been described (Hauptmann, 2002 ▶; Kikelj, 2003 ▶) that mimic the binding modes of natural peptide ligands (for reviews on the inhibition modes of some natural anticoagulants, see Corral-Rodriguez et al., 2009 ▶, 2010 ▶). They can be broadly divided into two major groups: covalent electrophilic inhibitors (e.g. PPACK), which form a covalent tetrahedral intermediate upon nucleophilic attack of the activated carbonyl group of the P1 residue by Ser195 OG from the catalytic triad of the enzyme (Bode et al., 1989 ▶), and noncovalent inhibitors that interact with thrombin solely through hydrophobic and polar interactions.
Previous isothermal titration calorimetry (ITC) and structure–activity relationship (SAR) studies towards thrombin for peptides with the general sequence d-Phe-Pro-d-Arg-P1′-CONH2 (Clement et al., 2009 ▶; Clement & Philipp, 2006 ▶) identified these compounds as reversible thrombin inhibitors and potent anticoagulants. Furthermore, at concentrations of 5–15 times their K i (in an in vitro inhibition assay) these peptides were also able to completely inhibit thrombin-activated platelet aggregation (Clement et al., 2007 ▶). Here, we report the crystallization and preliminary crystallographic analysis of human α-thrombin in complex with three of these noncovalent peptide inhibitors with l-isoleucine (p3), l-cysteine (p4) or d-threonine (p6) at the P1′ position (Clement & Philipp, 2006 ▶).
2. Materials and methods
2.1. Purification of recombinant chimadanin
Chimadanin (GenBank accession No. BAE00177) was expressed in Escherichia coli BL21 CodonPlus (Stratagene) transformed with pET44-chimadanin (Iwanaga et al., 2003 ▶; Nakajima et al., 2006 ▶). Cells were grown in LB medium supplemented with 50 µl ml−1 ampicillin and 30 µl ml−1 chloramphenicol and were induced with IPTG (final concentration 1 mM) at mid-exponential growth (OD600 = 0.5). After 3 h at 310 K, the cells were harvested by centrifugation at 5000g at 277 K, resuspended in 20 mM bis-tris pH 6.3 (buffer A) supplemented with 50 µl ml−1 lysozyme, 10 µl ml−1 DNase and 5 mM MgCl2 and lysed by freezing and thawing. Cell debris was removed by centrifugation at 15 000g for 30 min at 277 K. The crude protein extract was filtered through a 0.22 µm pore low-protein-binding membrane and loaded onto a 5 ml Bio-Scale Mini UNOsphere Q Cartridge (Bio-Rad) pre-equilibrated with buffer A. Elution was performed with a linear NaCl gradient (0–0.4 M) in buffer A. Fractions were analysed by SDS–PAGE and probed for their ability to inhibit thrombin in an in vitro activity assay. Chimadanin-containing fractions were pooled, diluted tenfold in buffer A and loaded onto a UNO Q-1 column (Bio-Rad) pre-equilibrated with buffer A. The sample was eluted with a linear NaCl gradient (0–0.18 M) in buffer A. Fractions containing purified chimadanin were pooled and concentrated to 17 mg ml−1 on a centrifugal concentration device with a 3 kDa molecular-weight cutoff membrane.
2.2. Preparation of thrombin–chimadanin complexes
For complex preparation, 5 mg human α-thrombin (Haematologic Technologies, USA) was mixed with a 10% molar excess of purified chimadanin and incubated on ice for 1 h. The complex was separated from the isolated components by size-exclusion chromatography on a Superdex 75 HR10/30 column (GE Healthcare) equilibrated in 20 mM Tris pH 8.5, 150 mM NaCl. The complex-containing fractions were pooled and concentrated to 7.2 mg ml−1 on a centrifugal concentration device with a 10 kDa molecular-weight cutoff membrane.
2.3. Crystallization of human α-thrombin
Initial crystallization conditions were screened at 293 K using the sitting-drop method with commercial sparse-matrix crystallization screens. The drops contained identical volumes (2 µl) of complex solution (at 7.2 mg ml−1) and precipitant solution and were equilibrated against a 300 µl reservoir. Crystals were obtained after 2 d using 50 mM Tris pH 8.5, 50 mM Bicine, 30 mM sodium fluoride, 30 mM sodium bromide, 30 mM sodium iodide supplemented with 11.5%(v/v) MPD, 11.5%(w/v) PEG 1000 and 11.5%(w/v) PEG 3350 as precipitant.
The calculated Matthews coefficients considering the presence of one molecule of the protease (2.36 Å3 Da−1 for 36.7 kDa) or a single equimolar complex (1.97 Å3 Da−1 for 44.2 kDa) in the asymmetric unit raised doubts about the actual composition of the crystals. It was later verified that only thrombin had crystallized, with no evidence for the presence of chimadanin in the electron density.
2.4. Preparation of complexes of α-thrombin with synthetic inhibitors
The α-thrombin crystals obtained were subsequently used to prepare complexes with three synthetic inhibitors of general sequence d-Phe-Pro-d-Arg-P1′-CONH2 with l-isoleucine (p3), l-cysteine (p4) or d-threonine (p6) at the P1′ position.
Thrombin complexes were prepared by soaking thrombin crystals at 293 K in a pre-equilibrated (24 h) drop consisting of 2 µl crystallization solution and 1 µl of a 10 mM solution of the inhibitor (p3, p4 or p6) in water. For all inhibitors, soaking times of 3 and 48 h were tested. Although the longer soaking time with peptides p3 and p4 did not seem to negatively affect the crystals, they were severely damaged upon 48 h soaking with peptide p6. Therefore, the crystals used for data collection were soaked with peptides p3 or p4 for 48 h and with peptide p6 for 3 h. The crystallization solution contained 38.5% MPD/PEG 1000/PEG 3350 for p3, 36.5% MPD/PEG 1000/PEG 3350 for p4 or 34.5% MPD/PEG 1000/PEG 3350 for p6. The crystals were flash-cooled by plunging them into liquid nitrogen.
2.5. Data collection and processing
Diffraction data were collected using an ADSC Q210 detector on beamline ID14-EH1 at the European Synchrotron Radiation Facility (ESRF; Grenoble, France). For each complex a single cryocooled crystal was used and all data sets were measured in 1° oscillation steps.
For the thrombin–p3 complex three data sets were collected over a range of 150° with crystal-to-detector distances of 297.5, 227.3 and 141.3 mm and 1, 3 and 6 s exposures per frame, respectively.
For the thrombin–p4 complex a single data set was collected over a range of 150° with a 191.1 mm crystal-to-detector distance and 3 s exposure per frame.
For the thrombin–p6 complex two data sets were collected over a range of 270° with crystal-to-detector distances of 262.6 and 178.9 mm and 0.2 and 0.6 s exposures per frame, respectively. A third, high-resolution data set was collected over a range of 300° with a 114.8 mm crystal-to-detector distance and 6 s exposure per frame.
Diffraction data sets were processed with MOSFLM (Leslie, 1992 ▶) and scaled using SCALA (Evans, 2006 ▶) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). Data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the outermost shell.
| Thrombin complex | Unliganded | p3 | p4 | p6 |
|---|---|---|---|---|
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters (Å) | ||||
| a | 57.5 | 51.6 | 51.8 | 57.5 |
| b | 73.0 | 76.5 | 77.1 | 72.7 |
| c | 83.0 | 83.3 | 83.4 | 83.1 |
| Resolution range (Å) | 72.93–1.55 (1.63–1.55) | 51.6–1.47 (1.55–1.47) | 35.0–1.86 (1.96–1.86) | 47.3–1.28 (1.35–1.28) |
| Reflections measured | 500945 | 495888 | 166128 | 1002793 |
| Unique reflections | 50373 | 56417 | 28722 | 87023 |
| Completeness (%) | 98.1 (96.6) | 100.0 (100.0) | 99.9 (100) | 96.7 (93.4) |
| Multiplicity | 9.9 (7.6) | 8.8 (5.8) | 5.8 (5.6) | 11.5 (5.8) |
| Average mosaicity (°) | 0.20 | 0.39 | 0.88 | 0.15 |
| Rmerge† | 0.067 (0.349) | 0.061 (0.286) | 0.072 (0.181) | 0.048 (0.183) |
| Rp.i.m.‡ | 0.020 (0.135) | 0.018 (0.130) | 0.018 (0.130) | 0.011 (0.083) |
| 〈I/σ(I)〉 | 5.5 (2.2) | 8.0 (2.6) | 6.8 (4.1) | 8.7 (4.1) |
| Monomers per asymmetric unit | 1 | 1 | 1 | 1 |
| Matthews coefficient (Å3 Da−1) | 2.36 | 2.25 | 2.28 | 2.38 |
| Solvent content (%) | 47.9 | 45.3 | 46.1 | 48.2 |
R
merge = 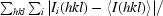
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
R
p.i.m. = 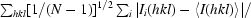 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
2.6. Structure solution
The structure of unliganded human α-thrombin was solved by molecular replacement with Phaser (McCoy et al., 2007 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) using the coordinates of the serine protease (PDB entry 1vzq; Schärer et al., 2004 ▶) as the search model. A partially refined model of unliganded human α-thrombin was subsequently used as the search model in the structural determination of the thrombin–p3, thrombin–p4 and thrombin–p6 complexes.
3. Results and discussion
3.1. Crystallization of thrombin–inhibitor complexes
Crystallization experiments were performed with an in vitro complex of human α-thrombin and recombinant chimadanin, a specific macromolecular thrombin inhibitor that was first isolated from the salivary gland of fed H. longicornis (hard tick; Iwanaga et al., 2003 ▶; Nakajima et al., 2006 ▶). Surprisingly, the crystals obtained only contained the unliganded protease, indicating that complex formation was not favoured under the crystallization conditions used, although thrombin itself was stabilized. The crystals grew to maximum dimensions of 50 × 185 × 50 µm (Fig. 1 ▶), belonged to the orthorhombic space group P212121 and diffracted to beyond 1.6 Å resolution on a synchrotron source (Fig. 2 ▶; Table 1 ▶), which prompted us to use them in the preparation of complexes of thrombin with small synthetic inhibitors. Although several orthorhombic thrombin crystals with similar unit-cell parameters have been reported previously, this is the first report of the crystallization of non-mutant unliganded human thrombin in this setting.
Figure 1.

Single crystals of human α-thrombin belonging to the orthorhombic space group P212121. The scale bar is 100 µm in length.
Figure 2.

X-ray diffraction images of crystals of unliganded thrombin (a) and of thrombin in complex with p3 (b), p4 (c) and p6 (d). The red circles correspond to resolution limits of 1.55 Å (a), 1.49 Å (b), 1.88 Å (c) and 1.29 Å (d).
For complex preparation, human α-thrombin crystals were incubated in solutions of synthetic peptides with the general formula d-Phe-Pro-d-Arg-Xaa-CONH2, where Xaa is either Ile (p3), Cys (p4) or d-Thr (p6) (Clement & Philipp, 2006 ▶; Clement et al., 2007 ▶). The crystals of the resulting complexes diffracted X-rays to 1.47 Å (p3), 1.86 Å (p4) and 1.28 Å (p6) resolution (Fig. 2 ▶). The data-collection and processing statistics are summarized in Table 1 ▶.
3.2. Structure solution
The molecular coordinates of the light chain and heavy chain of human α-thrombin from PDB entry 1vzq (Schärer et al., 2004 ▶) were used as a search model to solve the structure of the unliganded protease by molecular replacement. The model was subjected to alternating cycles of manual building with Coot (Emsley et al., 2010 ▶) and refinement with PHENIX (Adams et al., 2010 ▶) until an R factor of approximately 0.20 was reached. The coordinates of this partially refined model were then used as a search model to solve the structures of the three thrombin–inhibitor complexes by molecular replacement. The initial electron-density difference maps showed interpretable density for all inhibitors. The three-dimensional models are currently under refinement.
Acknowledgments
The pET44-chimadanin expression vector was a kind gift from Professor M. Onuma (Hokkaido University, Japan). We acknowledge the ESRF for the provision of synchrotron-radiation facilities and the ESRF staff for assistance in using beamline ID14-EH1. This work was funded in part by Fundação para a Ciência e a Tecnologia, Portugal through grants PTDC/BIA-PRO/70627/2006 and REEQ/564/B10/2005 (EU-FEDER and POCI 2010) and postdoctoral fellowship SFR/BPD/46722/2008 to ACF. MP was supported by a Fullbright Scholar Award.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bode, W., Mayr, I., Baumann, U., Huber, R., Stone, S. R. & Hofsteenge, J. (1989). EMBO J. 8, 3467–3475. [DOI] [PMC free article] [PubMed]
- Clement, C. C., Babinska, A., Kornecki, E. & Philipp, M. (2009). Adv. Exp. Med. Biol. 611, 579–580. [DOI] [PubMed]
- Clement, C. C., Babinska, A. & Philipp, M. (2007). FASEB J. 21, A1013.
- Clement, C. C. & Philipp, M. (2006). Understanding Biology Using Peptides. Proceedings of the Nineteenth American Peptide Symposium, edited by S. E. Blondelle, pp. 553–554. New York: Springer.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Corral-Rodriguez, M. A., Macedo-Ribeiro, S., Barbosa Pereira, P. J. & Fuentes-Prior, P. (2009). Insect Biochem. Mol. Biol. 39, 579–595. [DOI] [PubMed]
- Corral-Rodriguez, M. A., Macedo-Ribeiro, S., Barbosa Pereira, P. J. & Fuentes-Prior, P. (2010). J. Med. Chem. 53, 3847–3861. [DOI] [PubMed]
- Doolittle, R. F. (1984). Annu. Rev. Biochem. 53, 195–229. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Esmon, C. (1995). FASEB J. 9, 946–955. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fuentes-Prior, P., Noeske-Jungblut, C., Donner, P., Schleuning, W.-D., Huber, R. & Bode, W. (1997). Proc. Natl Acad. Sci. USA, 94, 11845–11850. [DOI] [PMC free article] [PubMed]
- Gailani, D. & Broze, G. Jr (1991). Science, 253, 909–912. [DOI] [PubMed]
- Grütter, M. G., Priestle, J. P., Rahuel, J., Grossenbacher, H., Bode, W., Hofsteenge, J. & Stone, S. R. (1990). EMBO J. 9, 2361–2365. [DOI] [PMC free article] [PubMed]
- Hauptmann, J. (2002). Eur. J. Clin. Pharmacol. 57, 751–758. [DOI] [PubMed]
- Iwanaga, S., Okada, M., Isawa, H., Morita, A., Yuda, M. & Chinzei, Y. (2003). Eur. J. Biochem. 270, 1926–1934. [DOI] [PubMed]
- Kane, W. H. & Davie, E. W. (1988). Blood, 71, 539–555. [PubMed]
- Kikelj, D. (2003). Pathophysiol. Haemost. Thromb. 33, 487–491. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 26
- Locht, A. van de, Lamba, D., Bauer, M., Huber, R., Friedrich, T., Kröger, B., Höffken, W. & Bode, W. (1995). EMBO J. 14, 5149–5157. [DOI] [PMC free article] [PubMed]
- Locht, A. van de, Stubbs, M. T., Bode, W., Friedrich, T., Bollschweiler, C., Höffken, W. & Huber, R. (1996). EMBO J. 15, 6011–6017. [PMC free article] [PubMed]
- Macedo-Ribeiro, S., Almeida, C., Calisto, B. M., Friedrich, T., Mentele, R., Stürzebecher, J., Fuentes-Prior, P. & Pereira, P. J. B. (2008). PLoS ONE, 3, e1624. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Muszbek, L., Yee, V. C. & Hevessy, Z. (1999). Thromb. Res. 94, 271–305. [DOI] [PubMed]
- Nakajima, C., Imamura, S., Konnai, S., Yamada, S., Nishikado, H., Ohashi, K. & Onuma, M. (2006). J. Vet. Med. Sci. 68, 447–452. [DOI] [PubMed]
- Paoli, G., Merlini, P. A. & Ardissino, D. (2005). Curr. Pharm. Des. 11, 3919–3929. [DOI] [PubMed]
- Rauch, U., Osende, J. I., Fuster, V., Badimon, J. J., Fayad, Z. & Chesebro, J. H. (2001). Ann. Intern. Med. 134, 224–238. [DOI] [PubMed]
- Richardson, J. L., Kroger, B., Hoeffken, W., Sadler, J. E., Pereira, P., Huber, R., Bode, W. & Fuentes-Prior, P. (2000). EMBO J. 19, 5650–5660. [DOI] [PMC free article] [PubMed]
- Schärer, K., Morgenthaler, M., Seiler, P., Diederich, F., Banner, D., Tschopp, T. & Obst-Sander, U. (2004). Helv. Chim. Acta, 87, 2517–2538.
- Stubbs, M. T. & Bode, W. (1993). Thromb. Res. 69, 1–58. [DOI] [PubMed]
- Vu, T.-K. H., Hung, D. T., Wheaton, V. I. & Coughlin, S. R. (1991). Cell, 64, 1057–1068. [DOI] [PubMed]