

Summary
Neisseria gonorrhoeae is a human-specific organism that is not usually exposed to UV light or chemicals but is likely to encounter reactive oxygen species during infection. Exposure of N. gonorrhoeae to sublethal hydrogen peroxide revealed that the ng1427 gene was up regulated 6-fold (Stohl et al., 2005). N. gonorrhoeae was thought to lack an SOS system, although NG1427 shows amino acid sequence similarity to the SOS response regulator LexA from E. coli. Similar to LexA and other S24 peptidases, NG1427 undergoes autoproteolysis in vitro, which is facilitated by either the gonococcal or E. coli RecA proteins or high pH, and autoproteolysis requires the active and cleavage site residues conserved between LexA and NG1427. NG1427 controls a three gene regulon: itself; ng1428, a Neisseria-specific, putative integral membrane protein; and recN, a DNA repair gene known to be required for oxidative damage survival. Full NG1427 regulon de-repression requires RecA following methyl methanesulfonate or mitomycin C treatment, but is largely RecA-independent following hydrogen peroxide treatment. NG1427 binds specifically to the operator regions of the genes it controls, and DNA-binding is abolished by oxidation of the single cysteine residue encoded in NG1427. We propose that NG1427 is inactivated independently of RecA by oxidation.
Introduction
Neisseria gonorrhoeae (Gc) is a Gram negative diplococcal bacterium and the sole causative agent of the sexually transmitted infection gonorrhea. Gc is an obligate human pathogen that infects the genitourinary tract and associates with mucosal epithelial cells. In females, gonococcal cervicitis can cause pelvic inflammatory disease leading to permanent fallopian tube scarring, resulting in sterility and ectopic pregnancy (Edwards & Apicella, 2004). Symptomatic gonococcal infection leads to a purulent exudate consisting of polymorphonuclear leukocytes (PMNs) and exfoliated epithelial cells (Shafer & Rest, 1989).
PMNs can kill bacteria by the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Shafer & Rest, 1989, Roos et al., 2003) generated by the NADPH oxidase enzyme and the nitric-oxide generating iNOS enzyme (Blackford et al., 1994), and through the use of non-oxidative agents such as antimicrobial peptides and proteases (Segal, 2005). The PMN oxidative burst results in the generation of superoxide anion (O2•−), hydrogen peroxide (H2O2), and hypochlorous acid (HClO), all of which are known to have antimicrobial activities (Root et al., 1975, Thomas, 1979). The iNOS enzyme generates nitric oxide that interacts with O2•− to generate peroxynitrite (ONOO−) (Radi et al., 1991a). H2O2 can lead to lipid peroxidation (Fang, 2004) and the DNA lesion 8-oxo-guanine, leading to G-T transversions (Henle et al., 1996). HClO and ONOO− can target DNA and intracellular proteins as well as cell-membrane components (Hayatsu et al., 1971, Albrich et al., 1981, Winterbourn et al., 1992, Yermilov et al., 1995, Radi et al., 1991b, Radi et al., 1991a). Viable Gc are associated with activated PMNs in purulent exudates, suggesting that Gc is able to resist killing by PMNs (Shafer & Rest, 1989). Gc express opacity proteins (Opa) (Heckels, 1981) that can mediate attachment of bacteria to PMNs (Chen & Gotschlich, 1996). These Opa proteins can stimulate the PMN oxidative burst (Simons et al., 2005), and Opa− Gc induce less of an oxidative burst than Opa+ Gc (Rest et al., 1982). Since human volunteers infected with Opa− Gc shed mostly Opa+ Gc (Schmidt et al., 2000), it is likely that this organism is experiencing oxidative radicals in the human host. Whereas actively growing Gc have been shown to abrogate the PMN oxidative burst (Criss & Seifert, 2008), dead Gc have been shown to potentiate the burst (Criss & Seifert, 2008) and both live and dead Gc are present during infection. These observations support the hypothesis that Gc has a transcriptional network in place that acts to resist oxidative killing in order to survive ROS challenge and persist in its human host.
While some of the individual proteins that protect Gc from oxidative damage have been characterized (Seib et al., 2006), it is not known how Gc regulates the response to oxidative products. To determine how Gc responds to oxidative products, the Gc transcriptome profile of strain FA1090 was determined following H2O2 exposure (Stohl et al., 2005). 75 genes were up regulated following H2O2 treatment and 80 genes were down regulated. One of the three up regulated genes predicted to encode transcriptional regulators was ng1427 (up regulated 6-fold after H2O2 treatment), which has sequence similarity to the SOS repressor LexA.
The SOS response system is a well characterized regulatory network in E. coli consisting of approximately 40 genes whose function is increase the cell’s ability to undergo recombinational repair, excise damaged nucleotides, and to inhibit cell division until DNA damage is repaired (Kelley, 2006). LexA is a transcriptional repressor which controls the SOS regulon by binding to a consensus sequence (Wertman & Mount, 1985) that is present in the promoters of SOS genes including recA. The SOS response is activated when DNA damage results in an interruption of DNA replication, leading to the presence of stalled replication forks, and the existence of ssDNA gaps at stalled replication forks. In vitro, the RecA protein becomes activated by binding to single-stranded DNA and a nucleoside triphosphate (Craig & Roberts, 1980). Binding of the activated RecA to LexA leads to LexA autoproteolysis (Gudas & Pardee, 1975, Little et al., 1980, Little et al., 1981) and subsequent derepression of the SOS regulon. Genes of the E. coli SOS response include uvrA, uvrB, and uvrD (Little & Mount, 1982), recA, and lexA itself (Brent & Ptashne, 1980). The conserved LexA binding site is not present in the Gc genome, and no gene encoding a homolog to LexA has been previously described in Neisseria (Davidsen & Tonjum, 2006). Additionally, following treatment of Gc with DNA damage-inducing agents such as UV light or methyl methanesulfonate (MMS), the transcription of genes such as recA, uvrA, and uvrB are not up regulated (Black et al., 1998). Interestingly, although Gc does not up regulate DNA repair genes such as recA, uvrA, or uvrB following DNA damage, the up regulation of the putative LexA ortholog ng1427 and numerous other genes by hydrogen peroxide suggested that a SOS-like system may exist in Gc. In support of this suggestion, we find that NG1427 acts as a repressor of a three gene regulon and that NG1427 retains RecA-facilitated autoproteolysis, but that NG1427 can be inactivated independently of RecA by oxidation.
Results
NG1427 is a transcriptional repressor
A bioinformatics approach was taken to characterize the NG1427 protein in Gc Strain FA1090 (accession #AE004969). Utilizing the NCBI Conserved Domain Database (http://www.ncbi.nlm.nih.gov/protein?db=cdd), NG1427 showed similarity to the SF clan of serine peptidases (Barrett & Rawlings, 1995), specifically the S24 peptidase family (Little et al., 1994). The canonical member of the S24 peptidase family, the SOS-response repressor LexA has 22% amino acid identity and 37% amino acid similarity to the NG1427 protein (Fig. 1A) and a similar arrangement of catalytic domains (Fig. 1B). The dissimilarity in the N-terminal region where the putative DNA binding domain of NG1427 would be located was expected since the SOS binding box in not found in Gc and NG1427 would likely have a different set of amino acids to bind to a unique promoter sequence. NG1427 encodes the conserved S24 cleavage site (A112/G113) and the conserved serine/lysine catalytic dyad (S154 and K190).
Figure 1. NG1427 is a transcriptional repressor that shares similarity to E. coli LexA.
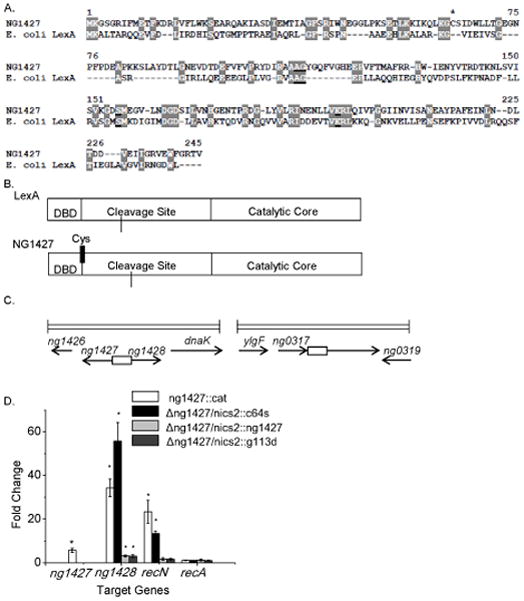
A: Amino acid sequence alignment comparing NG1427 and E. coli LexA. S24 peptidase cleavage and active sites are underlined and conserved residues are highlighted. Asterisk denotes single cysteine residue found in NG1427. B: Cartoon depiction of NG1427 and LexA protein domains. “DBD,” refers to the DNA binding domain. Location of single cysteine (Cys) in NG1427 is marked with black line. C: Diagram of the ng1427/ng1428 and recN chromosomal loci in FA1090 Gc. Boxes denote intergenic regions containing putative promoters of ng1427/ng1428 or recN. D: Fold change of target gene expression in ng1427::cat, Δng1427/nics2::c64s, Δng1427/nics2::g113d, and Δng1427/nics2::ng1427, relative to the parental strain as measured by qPCR with omp3 used as a reference gene. Asterisks denote p<0.05 as calculated by Student’s t-test relative to negative control (recA). Error bars +/− s.e.m. All data shown are the result of at least 3 biological replicates (n=3).
To determine the function of NG1427, a Gc loss-of-function mutant was created by deleting approximately 45% of the coding sequences and inserting a chloramphenicol-resistance cassette (cat) in its place (FA1090 1-81-S2nv ng1427::cat). Microarray analysis was used to compare gene expression differences between ng1427::cat and the parent strain. In the ng1427::cat mutant three genes showed increased transcript levels relative to the parent strain: the ng1427 gene itself, ng1428, and recN. No genes showed decreased transcript levels (data not shown). The ng1427 transcript was increased 3.6-fold in ng1427::cat relative to the parental strain. Although the ng1427 transcript was interrupted by the cat cassette, the intact 5’ end of the transcript hybridized to the microarray, an observation we exploited for real-time quantitative PCR (qPCR) analysis (see below). The ng1428 gene showed 16.5-fold higher transcripts in the mutant and encodes a Neisseria-specific gene, predicted to be an integral membrane protein. The NCBI Conserved Domain Database did not detect any conserved domains in ng1428. The genes ng1427 and ng1428 are adjacent and divergently transcribed (Fig. 1C). The final gene of the NG1427 regulon, recN, showed a 9.8-fold increase in transcripts in ng1427::cat relative to the parental strain. The RecN gene product has been implicated in Gc DNA repair processes (Skaar et al., 2002) and contributes to Gc survival after exposure to ROS and PMNs (Stohl et al., 2005). These results show that the LexA homolog NG1427 encodes a self-repressing transcriptional repressor, but the only gene affected by NG1427 that has previously been found in an SOS regulon is recN.
To verify the microarray results, qPCR was performed on RNA isolated from exponentially growing ng1427::cat and parental Gc. qPCR analysis of basal gene expression from ng1427::cat and parental Gc corroborated the microarray data, confirming that NG1427 represses the three-gene regulon of ng1427, ng1428, and recN, hereafter referred to as the “NG1427 regulon” (Fig. 1D, white bars). Analysis of recA gene transcripts confirmed that recA is not controlled by NG1427 (Fig. 1D). These results confirm that ng1427 encodes a self-repressing transcriptional repressor, but that the only gene controlled by NG1427 that has previously been found in an SOS regulon is recN. To affirm that de-repression of the NG1427 regulon is due to the absence of ng1427, a wild-type copy of ng1427 was expressed under control of its native promoter in an unlinked locus in the Gc chromosome (Mehr et al., 2000). In strain Δng1427/nics2::ng1427 the repression of ng1428 and recN was restored to near, but not identical to, parental levels, illustrating that the transcriptional effects we detected were due solely to the ng1427 mutation (Fig. 1D, gray bars). ng1427 expression was measured by qPCR in the complemented strains but due to the fact that ng1427 was not expressed from the wild-type locus in the complemented strains these data were not included in Fig. 1. The data regarding the Δng1427/nics2::g113d and Δng1427/nics2::c64s are discussed below. Taken together, the microarray and qPCR analysis shows that ng1427 encodes a transcriptional repressor of a three-gene regulon.
NG1427 undergoes RecA-mediated autoproteolysis
Since NG1427 is predicted to be a S24 peptidase like LexA, which requires RecA for autoproteolysis and SOS regulon de-repression, we directly tested whether NG1427 undergoes RecA-mediated autoproteolysis. NG1427-HIS was purified as a recombinant protein from E. coli. Incubation of NG1427-HIS with either activated Gc or E. coli RecA promoted autoproteolysis (Figure 2A, lanes 5, 6, and 8). When we performed this assay under the same conditions but substituting E. coli LexA for NG1427-HIS, LexA was cleaved much faster than NG1427-HIS under the same conditions (data not shown). Interestingly, we observed that NG1427-HIS underwent limited autoproteolysis even in the absence of RecA (Fig 2A, lanes 1 and 2), despite purification of the protein from E. coli cells lacking RecA and in the presence of protease inhibitors. We also found that NG1427-HIS underwent limited autoproteolysis immediately during purification and that this autoproteolysis was pH-sensitive, a shared trait between this protein and E. coli LexA (Little, 1984) (data not shown). To demonstrate that, as with LexA, activated RecA facilitated the autoproteolysis of NG1427-HIS, the reaction was also performed in the absence of RecA (mock-treated with RecA storage buffer) (Figure 2A, lane 2) and in the absence of ssDNA (lane 4). Both of these conditions resulted in less proteolysis of NG1427-HIS compared to the amount of proteolysis observed in the complete reaction (lanes 5). We found that pre-treatment of NG1427-HIS with 15mM of the oxidizing agent H2O2 or the reducing agent DTT did not alter the ability or extent of RecA- promoted NG1427-HIS autoproteolysis when added to the cleavage reaction (data not shown). We also observed no increase in RecA-promoted NG1427-HIS autoproteolysis when we incubated with a three-fold increase of RecA and ssDNA in the cleavage reaction. These results suggest that there remains a population of NG1427-HIS that is recalcitrant to RecA-promoted autoproteolysis for reasons that remain unclear.
Figure 2. NG1427 undergoes RecA-mediated autoproteolysis in vitro, but limited in vivo autoproteolysis following oxidative treatment.
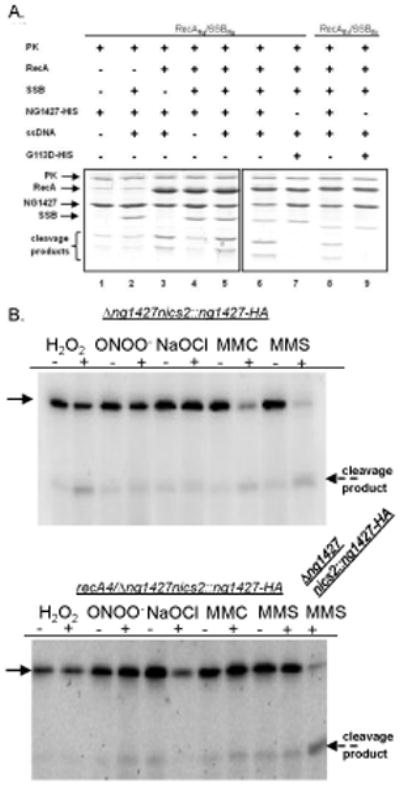
A: Examination of RecA-facilitated autoproteolysis of NG1427 in vitro. Purified NG1427-HIS or NG1427-G113D-HIS protein was incubated with either RecANg or RecAEc protein in the presence of the appropriate matched SSB protein, ssDNA, and an ATP regeneration system for 45 min. When a protein component (e.g. RecA or SSB) (lanes 2 and 3, respectively) was omitted as a control, the reaction was mock-treated with the storage buffer for the protein in question; when ssDNA was omitted, the reaction was mock-treated with TE storage buffer. Reactions were stopped and visualized on a 17% SDS-PAGE stained with Coomassie Brilliant Blue. The two NG1427 cleavage products are visible at the bottom of the gel. Gels are boxed to indicate that they were run on separate days. PK indicates pyruvate kinase.B: Examination of NG1427-HA turnover in vivo following treatment with oxidative and non-oxidative DNA damaging agents. Gc expressing NG1427-HA were treated with H2O2, ONOO−, NaOCl, MMC, or MMS, and cell lysates were immunoblotted for the HA tag. Thick arrow points to full-length NG1427-HA protein, dotted arrow denotes NG1427-HA C-terminal cleavage product with HA tag. Upper blot is NG1427-HA turnover in RecA+ cells, lower blot is NG1427-HA turnover in RecA−cells.
To determine whether the conserved S24 peptidase cleavage site of NG1427 was the site of RecA-facilitated cleavage, the residue was mutated (NG1427-G113D), and NG1427-G113D-HIS protein was purified from E. coli. NG1427-G113D-HIS and NG1427-HIS proteins were incubated with purified Gc or E. coli RecA. Incubation of NG1427-HIS with either activated Gc or E. coli RecA resulted in efficient autoproteolysis (Fig. 2A, lanes 6 and 8), whereas the NG1427-G113D-HIS mutant remained intact (Fig. 2A, lanes 7 and 9), verifying that Gly113 is required for proteolytic cleavage.
To determine whether NG1427 undergoes RecA-facilitated autoproteolysis in Gc, the ng1427 mutant was complemented with a 3’ hemagglutinin epitope (HA)-tagged version of ng1427, expressed under control of the ng1427 promoter. This tagged complement construct was also introduced into a ng1427 mutant also carrying a recA loss-of-function allele, recA4, (Seifert, 1997). We monitored autoproteolysis of the NG1427-HA protein following treatment of the liquid grown Gc with oxidants, the DNA cross linking agent mitomycin C (MMC), or the DNA alkylation agent MMS. There was a slight increase in NG1427-HA breakdown product when NG1427-HA was exposed to H2O2, and minimal, if any, turnover of NG1427-HA turnover occurred following exposure the ROS agents ONOO−, or the hypochlorous-acid-generating chemical sodium hypochlorite (NaOCl) in the presence or absence of RecA (Fig. 2B). This suggested that either these agents are not causing DNA damage in vivo to activate RecA, or that NG1427-HA turnover is not mediated by RecA. In contrast, RecA-mediated turnover of NG1427-HA was nearly complete following treatment of Gc with MMC or MMS (Fig. 2B). These results suggest that DNA damage by MMC and MMS is leading to activation of RecA, causing turnover of NG1427 in the bacterial cell.
The NG1427 regulon is de-repressed independently of RecA following H2O2 treatment
Given the lack of RecA-mediated autoproteolysis of NG1427 following H2O2 treatment, we tested the RecA dependence for regulon de-repression using a strain carrying the IPTG-inducible recA gene, recA6 (Seifert, 1997). Gc were treated with H2O2 in the presence and absence of IPTG, and expression levels of the NG1427 regulon were measured relative to a reference gene, omp3. We observed that treatment with H2O2 caused de-repression of the NG1427 regulon, irrespective of RecA expression (Fig. 3A). To confirm the RecA-independent observation, NG1427 regulon de-repression following H2O2 treatment was determined in the recA4 loss-of-function strain background. NG1427 regulon de-repression still occurred after H2O2 treatment in recA4 Gc (Fig. 3B, dark gray bars, dark gray striped bars) at a level similar to that of the parent (Fig. 3B, white bars, white gridded bars). No increase in NG1427 regulon gene expression was observed in the ng1427::cat strain after H2O2 treatment (Fig. 3B, light gray bars, light gray gridded bars), as these genes were already fully de-repressed. These data show that NG1427 regulon de-repression in response to H2O2 requires NG1427, but is independent of RecA.
Figure 3. NG1427 is inducible following treatment with DNA damaging agents H2O2, and does not require RecA.
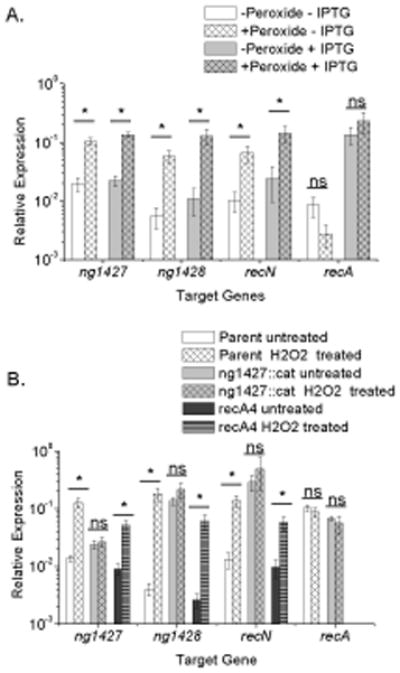
All data show relative expression of each target gene calculated using a control transcript omp3 for each condition tested. Error bars are +/− s.e.m, Asterisks denote p<0.05 as calculated by Student’s t-test. A: H2O2 treatment induces the NG1427 regulon in a recA-independent manner. Induction of the NG1427 regulon following treatment with 5 mM H2O2 was measured in recA6 Gc with or without IPTG induction. ng1427 transcript was measured with the “ng1427 probe” for each strain. B: The absence of RecA does not affect de-repression of the NG1427 regulon following H2O2 treatment. Induction of the NG1427 regulon following peroxide treatment was measured in parental, ng1427::cat, and recA4 Gc. ng1427 transcript was measured with the “ng1427 qPCR probe” for parental and recA4 strains, and the “ng1427::cat 5’ end qPCR probe” for the ng1427::cat strain. All data show a minimum of 3 biological replicates (n=3).
NG1427 requires RecA for full regulon de-repression following DNA damage
Given that RecA facilitated NG1427 autoproteolysis, we tested whether the NG1427 regulon was induced by MMS or MMC treatment and whether this required RecA. De-repression of the ng1427 regulon was observed in the parent strain in response to MMS and MMC (Fig. 4A, white bars, white gridded bars; Fig 4B, white bars, white gridded bars). De-repression of the NG1427 regulon was not seen in the ng1427::cat strain (Fig. 4A,light gray bars, light gray gridded bars; Fig 4B, light gray bars, light gray gridded bars) as the regulon was fully de-repressed. A slight, but inconsistent up regulation of the NG1427 regulon that was less than the increase of transcript levels observed in the parental strain occurred in recA4 Gc following MMS and MMC treatment, suggesting an additional minor RecA-independent pathway of NG1427 regulon regulation may exist (Fig. 4A and 4B, dark gray bars, dark gray striped bars). These results suggest that, similar to LexA in E. coli, treatment with these DNA damaging agents activates the RecA co-protease within Gc to promote cleavage of NG1427 and induction of the regulon. The regulon was not induced by heat shock (data not shown), suggesting that the induction of the NG1427 regulon is specific to DNA damaging agents. Additionally, the NG1427 regulon was was not greatly affected following treatment with ONOO− (Fig. 5A) and NaOCl (Fig. 5B) in either RecA+ or RecA- cells. Therefore, NG1427 responds to different DNA damaging agents by de-repression of the NG1427 three-gene regulon in a RecA-dependent manner following non-oxidative DNA damaging agent treatment, and in a RecA-independent manner following treatment with H2O2.
Figure 4. NG1427 requires RecA for full regulon induction following treatment with DNA damaging agents MMC and MMS.
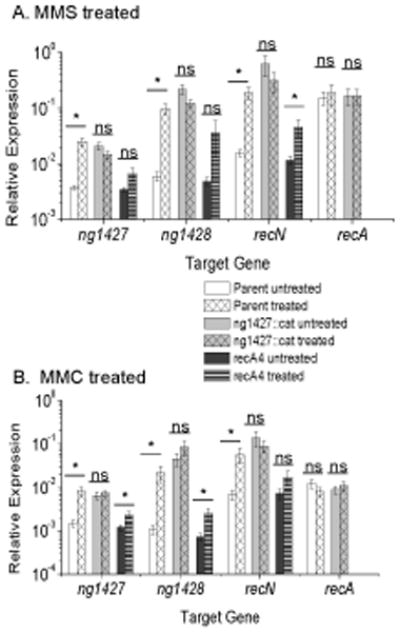
All data show relative expression of each target gene calculated using a control transcript omp3 for each condition tested. Error bars are +/− s.e.m, Asterisks denote p<0.05 as calculated by Student’s t-test. A: Full de-repression of the NG1427 regulon following MMS treatment requires expression of RecA. Induction of the NG1427 regulon after exposure to 0.02% (v/v) MMS was measured in parental, ng1427::cat, and recA4 Gc. ng1427 transcript was measured with the “ng1427::cat 5’ end qPCR probe” for all strains. B: Full de-repression of the NG1427 regulon following MMC treatment requires expression of RecA. Induction of the NG1427 regulon after exposure to 25 ng/ml conc. MMC was measured in parental, ng1427::cat and recA4 Gc. ng1427 transcript was measured with the “ng1427::cat 5’ end qPCR probe” for all strains. All data show a minimum of 3 biological replicates (n=3).
Figure 5. Treatment with ONOO− and NaOCl does not meaningfully affect the NG1427 regulon.
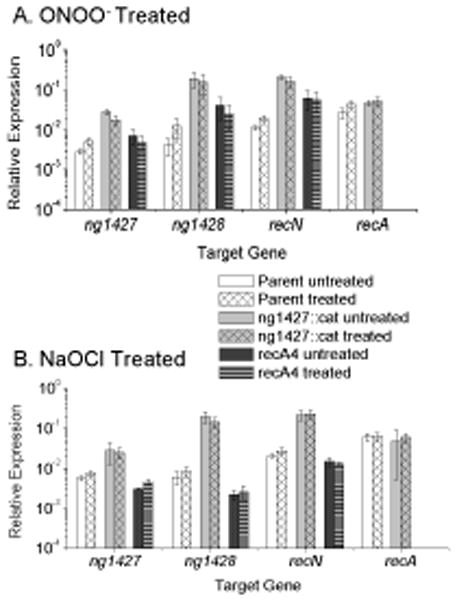
All data show relative expression of each target gene calculated using a control transcript omp3 for each condition tested. Error bars are +/− s.e.m, Asterisks denote p<0.05 as calculated by Student’s t-test. A: ONOO− treatment does not strongly induce de-repression of the NG1427 regulon. Induction of the NG1427 regulon following exposure to 3 mM conc. ONOO− was measured in parental, ng1427::cat and recA4 Gc. ng1427 transcript was measured with the “ng1427::cat 5’ end qPCR probe” for all strains. B: NaOCl treatment does not strongly induce de-repression of the NG1427 regulon. Induction of the NG1427 regulon following exposure to 1.7 mM conc. NaOCl was measured in parental, ng1427::cat and recA4 Gc. ng1427 transcript was measured with the “ng1427::cat 5’ end qPCR probe” for all strains. All data show a minimum of 3 biological replicates (n=3).
Mutation of the single cysteine of NG1427 (C64) affects DNA binding and RecA-mediated turnover of NG1427
One notable difference between NG1427 and the E. coli LexA protein is the presence of a single cysteine residue near the DBD domain of NG1427 (Fig. 1A, 1B). Cysteine residues can become modified by ROS to form sulfenic acid (Winterbourn & Hampton, 2008), leading to an inter- or intramolecular disulfide bond with other cysteine amino acid residues (Thornton, 1981). We postulated that the C64 residue was becoming oxidized during H2O2 treatment, leading to inability of NG1427 to bind DNA. To test whether disruption of C64 altered the ability of NG1427 to bind DNA, a C64S mutant of NG1427 was constructed and introduced into an ectopic site on the chromosome of ng1427::cat using the NICS complementation system to generate the strain Δng1427/nics2::c64s. The C64S complement construct did not restore the ng1427::cat transcriptional phenotype (Fig. 1D, black bars), as the NG1427 regulon remained de-repressed following expression of c64s. Since disruption of residue C64 prevented complementation of ng1427::cat, the presence of this cysteine residue must be required for required for NG1427 repressor activity.
To test whether mutation of the C64 residue influenced RecA-enhanced autoproteolysis, an HA-tagged version of the NG1427-C64S mutant was constructed and introduced into the ng1427 mutant by complementation. Treatment of cells with H2O2, ONOO−, NaOCl, MMC, or MMS incurred minimal turnover of NG1427-C64S-HA relative to the untreated samples (Fig. 6), suggesting that the C64 residue is important for RecA-mediated turnover.
Figure 6. Disrupting cysteine 64 (Cys64) prevents RecA-mediated NG1427 autoproteolysis.
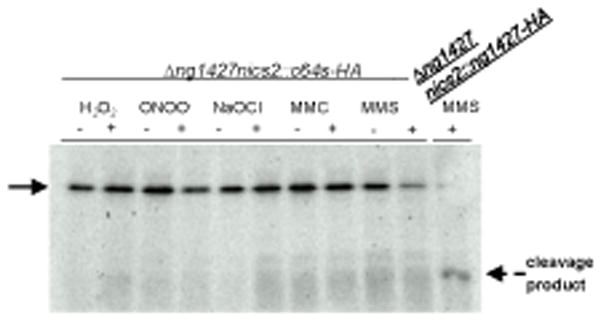
Western analysis of strain NG1427-C64S-HA following treatment with either H2O2,−ONOO−, NaOCl, MMC, or MMS. Treatment of NG1427-HA was done in parallel and is included as a positive control Arrow points to full-length protein, dotted arrow denotes NG1427-HA C-terminal cleavage product with HA tag.
NG1427 binds specifically to promoter regions, and oxidatively treated NG1427 is unable to bind DNA
To directly test whether oxidation of the C64 residue would inhibit NG1427 repressor activity, we utilized an electrophoretic mobility shift assay (EMSA) to detect DNA binding of NG1427 to the ng1427-ng1428 intergenic region and the recN promoter region. First, purified non-cleavable NG1427-G113D-HIS protein was incubated with the putative operator regions of ng1427/ng1428 and recN. NG1427-G113D-HIS was utilized due to its stability under all conditions tested and because g113d was able to restore the repression phenotype of ng1427::cat to near parental levels in vivo (Fig. 1D, dark gray bars). Competition EMSAs demonstrated that NG1427-G113D was able to bind specifically to the putative promoter regions of the NG1427 regulon and not to DNA carrying a viral transcription factor (EBNA) target sequence (Fig. 7, lanes 9–11, 20–22). The NG1427-G113D protein bound the ng1427/ng1428 intergenic region at higher affinity than the recN intergenic region (Fig. 7, lanes 14–16) as determined by diminution of bound probe and increase of free probe. This binding could be abrogated by the addition of oxidative agents to the reaction. Abrogation of DNA binding by NG1427-G113D was seen at approximately 3 mM H2O2 (Fig 8A) and binding activity was restored with the addition of DTT to the reaction mixture (data not shown). With ONOO−, the loss of binding occurred at 0.2mM (Fig. 8C), and with NaOCl the abrogation of DNA binding occurred at 2 μM (Fig. 8D). There was no effect on DNA binding with MMS (Fig. 8B) or degraded ONOO− (data not shown). We attempted to measure the rates of oxidation mediated by H2O2, or ONOO−, but could only determine that abrogation of DNA binding of NG1427-G113D occurred in less than 30 seconds of exposure regardless of the dose of oxidant used (data not shown).
Figure 7. NG1427 binds specifically to the putative promoter regions of the NG1427 regulon.
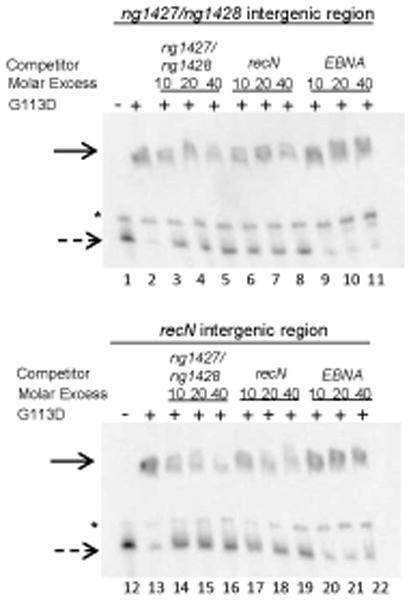
2 nM biotin-labeled DNA consisting of the ng1427/ng1428 intergenic region or the upstream recN intergenic region was incubated with 100 nM of NG1427-G113D and unlabelled competitor DNA in molar excess denoted. “EBNA,” Epstein-Barr nuclear antigen target DNA, is an irrelevant piece of DNA used as a negative control. Reactions were then separated by electrophoresis on a 5% acrylamide Tris-HCl gel. The solid arrow denotes NG1427 bound DNA. The dotted arrow denotes unbound DNA. The asterisk denotes a non-specific band found in all reactions.
Figure 8. NG1427 cannot bind DNA following oxidative treatment.
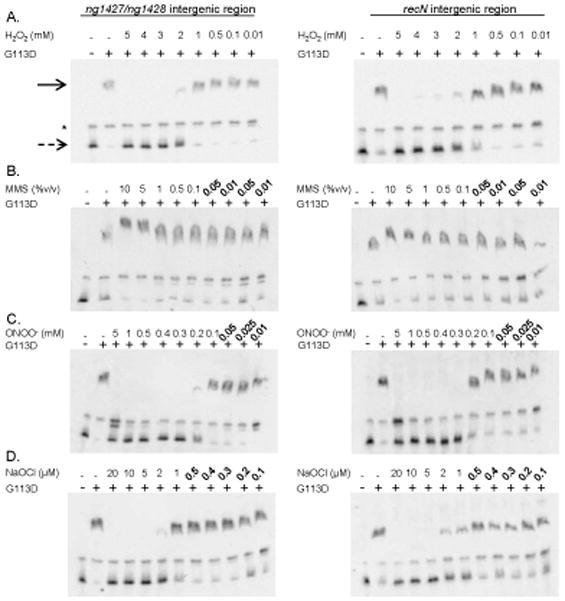
− 2 nM biotin-labeled DNA consisting of the ng1427/ng1428 intergenic region or the upstream recN intergenic region was incubated with 100nM of NG1427-G113D. Where indicated, NG1427-G113D was either left untreated or treated with a varying range of concentrations of A: H2O2, B: MMS, C: ONOO−, or D: NaOCl and incubated for 15 minutes following addition of DNA. The reaction was run on a 5% acrylamide Tris-HCl gel, transferred to a nylon membrane, and detected with streptavadin-HRP. The solid arrow denotes NG1427-G113D-bound DNA. The dotted arrow denotes unbound DNA. The asterisk denotes a non-specific band found in all reactions
NG1427 is directly modified by oxidative chemicals
While we were able to infer oxidation of NG1427 by abrogation of DNA binding, we wished to determine whether the protein was becoming directly oxidized by our oxidizing agents. Again, NG1427-G113D-HIS was utilized due to its stability under all conditions tested and because it was able to restore near, but not identical, parental levels of NG1427 regulon repression in vivo (Fig 1D, dark gray bars). Oxidation of NG1427-G113D was detected using maleimide-PEG11-biotin (Pierce) to label unmodified cysteine residues, and labeling was detected by both a mobility shift and detection of the biotin by streptavidin-HRP. There was a loss of labeling on NG1427-G113D-HIS by maleimide-PEG11-biotin following treatment with all the oxidative agents (Fig. 9A). Additionally, we observed via Coomassie-stained protein gel the formation of a higher molecular weight structure that corresponded to a NG1427-G113D dimer, most likely created by an intermolecular disulfide-bond (Fig. 9A). This higher molecular weight structure disappeared when samples were treated with the reducing agent β-mercaptoethanol (Fig. 9C). Additionally, there appeared to be a loss of binding of maleimide-PEG11-biotin following treatment with a reducing agent, suggesting less-efficient binding was occurring following reduction of the label (Fig. 9D). Lack of detection of biotin labeling in a higher molecular weight form of the protein following H2O2 and NaOCl treatment (Fig. 9B) showed that there were no free sulfhydryl groups available to bind the malemide-PEG11-biotin, confirming that the dimer was due to disulfide bonding. Detection of the biotin label in the higher molecular weight form of NG1427-G113D following treatment with ONOO− despite β-mercaptoethanol treatment suggests that this higher molecular weight structure is not due to the formation of the disulfide-bond (Fig. 9C, 9D). This insensitivity of the higher mobility form of the protein to reduction was unexpected, but formation of non-reducible protein aggregates following treatment with ONOO− has been observed previously (Soszynski & Bartosz, 1996). These results demonstrate that NG1427-G113D is clearly becoming directly and reversible oxidized by H2O2 and NaOCl.
Figure 9. NG1427 is directly and reversibly oxidized.
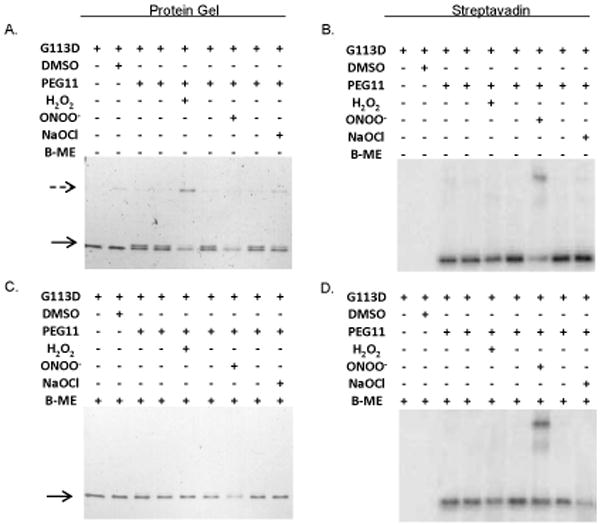
A: Direct visualization of the oxidation of NG1427. 100 nM of NG1427-G113D-HIS was incubated with H2O2, ONOO−, or NaOCl for 15 minutes, then incubated with maleimide-PEG11-biotin. The reactions were then visualized on a non-reducing 4–20% SDS-PAGE gel stained with Coomassie Brilliant Blue. B: Determination of putative NG1427-G113D-HIS dimers free sulfhydryls groups. An equal volume aliquot from (A) was run out on a non-reducing 4–20% SDS-Page gel, transferred to a PVDF membrane, and detected with streptavadin-HRP. C: Examination of reversibility of disulfide-dimers of NG1427-G113D. 100 nM of NG1427-G113D-HIS was incubated with H2O2, ONOO−, or NaOCl for 15 minutes then incubated with maleimide-PEG11-biotin. B-mercaptoethanol (B-ME) was then added to the reactions and were subsequently then visualized on a 4–20% SDS-PAGE gel stained with Coomassie Brilliant Blue. D: Detection of NG1427-G113D dimers following reduction. An equal volume aliquot of B-ME treated samples from (C) were run out on 4–20% SDS-Page gel, transferred to a PVDF membrane, and detected with streptavadin-HRP. The dotted arrow denotes the formation of the higher-molecular weight NG1427-G113D-HIS band, and the lower arrow denotes the mobility shift of NG1427-G113D-HIS following treatment with maleimide-PEG11-biotin.
Discussion
We have identified a LexA ortholog in the obligate human pathogen N. gonorrhoeae, NG1427, which undergoes a RecA-independent de-repression of its regulon following oxidation. NG1427 was the most highly up regulated putative transcriptional regulator in Gc following treatment with H2O2 (Stohl et al., 2005). Analysis of the transcriptome of Gc revealed that NG1427 is a transcriptional repressor that controls a three gene regulon of itself, ng1428 and recN. The small size of the NG1427 regulon is consistent with other characterized Gc regulons, which vary from 3–12 genes in size (Kidd et al., 2005, Seib et al., 2007, Wu et al., 2006).
Although the in vitro data clearly shows that activated RecA can facilitate the autoproteolysis of NG1427, full de-repression of the NG1427 regulon following oxidative DNA damage does not require RecA activity. This is the first fully-characterized S24 peptidase transcriptional repressor to have a RecA-independent mode of regulation. There was a slight but inconsistent increase of NG1427 regulon expression measured in the recA4 strain following MMS and MMC treatment suggesting a minor, oxidation- and RecA-independent pathway for NG1427 regulon activation may exist. It is interesting that other oxidatively-damaging, thiol-modifying agents such as ONOO− or NaOCl did not strongly affect the NG1427 regulon even though they are quite potent at inactivating the DNA-binding activity of NG1427 in vitro. Either these oxidants are not as stable as H2O2 in Gc, or there may be an intermediate required for NG1427 inactivation with the bacterial cell that is responsive to hydrogen peroxide but not the other oxidants.
NG1427 possesses a single cysteine, C64, near the DNA binding domain. The –SH group on Cys residues can be modified by reactive oxygen species to form sulfenic acid (Winterbourn & Hampton, 2008). Several transcriptional regulators such as OxyR, FurR and OhrR function by using Cys residues as sensors for an oxidative environment (Chen et al., 2008, Katona et al., 2004, Fuangthong & Helmann, 2002, Zheng et al., 1998, Paget & Buttner, 2003). A reactive oxygen species-mediated modification of the -SH group on C64 to a sulfenic acid could be altering protein structure. Indeed, mutation of C64 results in an inability of this protein to restore repression of the NG1427 regulon and to undergo RecA-mediated turnover within Gc cells. Moreover, we detected direct oxidation of NG1427-G113D by the presence of free sulfhydryl groups and formation of disulfide bond- mediated dimers in vitro. We were unable to purify C64 mutants in order to more directly test the oxidation of the Cys residue in NG1427- disruption of this particular residue causes the protein insolubility (data not shown). This suggests that C64 is important for proper folding and structure of NG1427. The G113D complement construct was used in an attempt to demonstrate that a non-cleavable NG1427 protein could undergo RecA-independent regulon de-repression following H2O2 treatment. However, we had minimal induction from the NICS locus following treatment with a variety of agents using a range of complementation constructs (data not shown), yet we still observed restoration of the transcriptional repression phenotype (Fig. 1D, data not shown). This observation is intriguing and is a current focus of ongoing study. There are no cysteine residues found in the LexA proteins of E. coli or B. subtilis, but a subset of bacterial species including Pseudomonas aeruginosa, P. fluorescens, Vibrio splendidus, and Azotobacter vinelandii do encode a single cysteine, suggesting these repressors may also be directly regulated by oxidation.
It is possible that NG1427 senses ROS in the presence of activated innate immune cells and that up regulation of the three gene regulon provides a survival advantage that our in vitro assays cannot detect. However, since the genes found in this regulon are almost entirely conserved in the sequenced genus Neisseria (ng1428 is not present in, or is not significantly conserved in, N. elongata, N. subflava, N. mucosa, or N. oral taxon 014), and both pathogenic and nonpathogenic members of the species carry NG1427 orthologs with the conserved cysteine, it is likely that this regulon is important for human colonization and not pathogenesis per se. It should be noted that the commensal Neisseria probably interact with the innate immune system as they establish and maintain colonization. It is unknown whether Gc or the other Neisseria encounter the type of host-derived DNA damage that cause replication fork stalls that would lead to RecA activation and NG1427 autoproteolysis. Thus, it is possible that the regulation of the NG1427 regulon through oxidation is the primary mechanism for transcriptional control and that the autoproteolysis is a residual function that is retained due to structural requirements. Also, given the slight up regulation observed in the absence of RecA and non-oxidative damage, there might exist yet another mechanism for NG1427 regulon control.
Previous investigation into the Gc response to DNA-damaging agents concluded that there was no LexA-ortholog encoded by Gc (Black et al., 1998, Campbell & Yasbin, 1984). While NG1427 is a functional LexA homolog, it is not participating in a “classical” SOS system since it does not regulate recA. LexA represses SOS genes by binding to a consensus sequence (Wertman & Mount, 1985), conventionally referred to as SOS boxes. This SOS binding box consensus sequence can vary from species to species. In E. coli and B. subtilis, the SOS box consists of a palindromic repeat of CTGTA-N6-TACAG and GAAC-N4-GTTC respectively (Erill et al., 2007), although there are reports of divergent SOS boxes in Alphaproteobacteria that are characterized by an odd-numbered spacer and a direct repeat of GTTC-N7-GTTC (Erill et al., 2007). However, the intergenic regions upstream of ng1427/ng1428 and recN do not encode any conserved LexA binding site, and analysis using MEME/MAST (http://meme.sdsc.edu/meme4_3_0/intro.html) did not reveal an alternative putative NG1427 binding site. The different levels of gene expression changes within the NG1427 regulon after induction suggests there may be variability in the strength of NG1427 binding to its operators.
It has not been clearly defined what defines a bacterial SOS system. NG1427 is a functional homologue of LexA that is auto regulatory, but it does not control recA expression or the expression any DNA repair gene except recN. In addition, NG1427 does not affect Gc’s survival to a variety of DNA damaging or redox perturbing agents in any appreciable manner under the laboratory growth conditions used, either when deleted or over expressed (Fig. S1). recN was previously shown to be required for Gc survival against oxidative damage and PMN-mediated killing (Stohl et al., 2005), but deletion of ng1428 does not provide a significant phenotype compared to the parental strain of Gc following treatment with H2O2 (Fig. S1A). We therefore hypothesize that basal expression of recN is sufficient to protect Gc from oxidative damage under laboratory growth conditions and that there are redundant processes that mask Δng1427 and Δng1428 survival phenotypes. Based on the additional regulation of NG1427 by oxidation and the major differences between the Gc and E. coli regulons, we conclude that the Neisseria encode an SOS-like regulon.
Experimental Procedures
Bacterial strains, growth conditions, and chemicals
Gc strains were grown at 37°C on Gc medium base (GCB; Difco) plus Kellogg supplement I (22.2 mM glucose, 0.68 mM glutamine, 0.45 mM cocarboxylase) and II [1.23 mM Fe(NO3)3] (Kellogg et al., 1963) at 37°C in 5% CO2 or in Gc liquid (GCBL) medium [1.5% proteose peptone No. 3 (Difco), 0.4% K2HPO4, 0.1% KH2PO4, 0.1% NaCl] with Kellogg supplements I, II and 0.042% sodium bicarbonate. GCBL always contained 0.042% sodium bicarbonate, except for ONOO− assays. Liquid-grown Gc strains were prepared as follows: Gc was grown from freezer stocks for approximately 20h and ~10 colonies were passaged onto GCB. After 12h, colonies were collected with a Dacron swab (Puritan), resuspended in GCBL, grown for 12h, diluted to OD550≅0.3, grown for 2.5–3h, and diluted to OD550≅0.06. This culture was then grown to an OD600 of approximately 0.5. All FA1090 strains used in this study contained the 1-81-S2 variant pilE sequence (Seifert et al., 1994) and which, other than the recA6 strain used in this paper, contain a transposon mutation upstream of the pilE locus that blocks antigenic variation (1-81-S2 non-varying or 1-81-S2nv) (Sechman et al., 2005). The E. coli plasmids pSmart (Lucigen) and pBlunt (Invitrogen) were used for cloning, and were propagated using E.coli TOP10 cells (Invitrogen).
DNA manipulations and analysis
Standard procedures for cloning, Southern blot analysis, and restriction digest were performed as described (Sambrook et al., 1989). Plasmid DNA from E. coli, genomic DNA from Gc, and PCR products were isolated using Qiagen kits. All modifying and restriction enzymes were obtained from New England Biolabs (NEB), unless otherwise indicated, and used as specified. Gonococcal pilE sequences were determined as described (Stohl & Seifert, 2001). Sequencing reactions were performed using a commercial vendor (Seqwright). Analysis of DNA sequences was performed using VectorNTI (Invitrogen).
Construction of the Insertion/Deletion Mutants ng1427::cat and ng1428::tetM
An insertion/deletion mutation of ng1427 was created by amplifying the 5’ and 3’ ends of ng1427 along with flanking regions using ng1427 5, 6, 7, and 8 primers (Table S1). ng1427 primers 6 and 7 encoded a PmeI site, which allowed the insertion of a chloramphenicol (Cam) resistance cassette (cat). The resulting construct, ng1427::cat, lacks 311 bp from the interior of the 708bp ng1427 gene. This construct was transformed into FA1090 1-81-S2nv and integrated into the chromosome by homologous recombination, and transformants were selected for growth on GCB containing 0.75 μg/ml Cam. PCR and Southern blot analysis was used to verify ng1427::cat insertion. The ng1428::tetM mutant was constructed by removing 200 bp internal to ng1428 by amplifying the 5’ and 3’ ends of the gene, along with flanking regions, with NG1428 1, 2H, 3H, and 4 primers. Subsequent cloning into pBlunt and subsequent digest with XbaI and BamHI allowed insertion of a tetracycline (Tet) resistance cassette TetM (Seifert, 1997), resulting in ng1428::tetM. The ng1428::tetM construct was sequenced, transformed into FA1090 1-81-S2nv, integrated into the chromosome by homologous recombination, and screened using antibiotic selection of 0.75 μg/ml of Tet, and confirmed by PCR.
Point Mutation Generation
NG1427-G113D and NG1427-C64S were created using the Stratagene QuikChange Mutagenesis protocol. PCR was performed using the mutagenic primers with the pBlunt/ng1427 construct as a template. Following completion of the PCR, the template circular DNA was digested using DpnI for 90 minutes at 37°C. The remaining DNA was cloned into TOP10 E. coli cells (Invitrogen) and screened on antibiotic media. Plasmids were extracted by miniprep (Qiagen) and sequenced.
Protein Purification
ng1427 was amplified with primers NG1427 Ab2.2 and NG1427 nostop2 that allowed cloning into the IPTG-inducible pET28(a) Protein Expression Plasmid (Novagen), which introduced a C-terminal HIS tag (NG1427-HIS). Plasmids were selected on Kan antibiotic, sequenced to validate the correct insertion of ng1427, and transformed into BLR(DE3) expression cells (Novagen).
To isolate the NG1427-HIS protein, 1 ml of overnight culture of pET28-NG1427-HIS in LB-Kan was inoculated into 500 ml of LB/Kan. The culture was grown with vigorous shaking at 37°C for 3 hours, then treated with 1 mM IPTG for another 3 hours. The culture was pelleted at 10,000g for 10 minutes at 4°C. The cell pellet was directly mixed with 2 mM PMSF and 2x Protease Cocktail Inhibitor (Pierce), resuspended in 10 ml of Binding Buffer (Novagen), sonicated on ice for 15 seconds with 60 seconds between sonications, and pelleted for 30 minutes at 10,000g at 4°C. The supernatant was passed through a 0.45 micron filter and applied to a Novagen Quick-Bind Column. Washed NG1427-HIS was eluted from the column in 10ml of Elution Buffer (Novagen). The protein eluate was diluted 1:300 in 25 mM Tris, 150 mM NaCl, 20% glycerol pH 8.3, dialyzed with a Slide-a-Lyzer (Pierce), snap-frozen in dry-ice/ethanol, and stored at −80°C. This protocol was repeated for the NG1427-G113D and point mutation, except without PMSF and protease inhibitor treatment. Purity of the protein was assayed by loading purified protein onto a 17% SDS-PAGE, subjecting to electrophoresis, and staining with Coomassie Brilliant Blue.
Complementation
Complementation of the ng1427::cat mutation was performed using the NICS system (Mehr et al., 2000) which inserts a functional copy of the gene at an unlinked chromosomal locus. ng1427 was placed under endogenous promoter control (nics2) and placed into the plasmid pGCC2 using the IGR1427 and Comp3’ primers. The IPTG-inducible ng1427 and g113d complement constructs (nics4) were cloned into the pGCC4 plasmid using primers PacI-NG1427-CF and FseI-NG1427-CR. PCR products and plasmids were digested using PacI and FseI and purified using the Qiaquick kit (Qiagen) and subsequently ligated. After selection on 250 μg/ml Erm colonies were screened for plasmids bearing the insert by PCR. Complement constructs were sequenced to verify no additional mutations had been introduced, transformed into ng1427::cat Gc, selected on antibiotics, and verified by PCR.
Microarrays
Parental and ng1427::cat Gc were grown to mid-log phase in amended GCBL with the RNA extracted as described below. RNA quality was determined by 2100 Bioanalyzer (Agilent) by the Northwestern University Genomics Core and by spectrophotometer. Six biologically independent samples of RNA consisting of three samples from each of the Parental and ng1427::cat Gc were processed and analyzed by Nimblegen (Madison, WI, USA).
Quantitative PCR Primer/Probe Design
A fluorescent probe hybridization approach was used for the quantitative PCR (qPCR) analysis of gene expression in Gc. For ng1427, ng1428, and recN probes, the Beacon Design program (http://www.premierbiosoft.com/molecular_beacons/index.html) was used to determine the best primer/probe set for each gene. Each probe has a 5’-FAM fluorescent tag and a 3’-TAM quencher. For recA, ng1427 5’ end, and omp3, probes, the Integrated DNA Technology Sci-Tools website (http://www.idtdna.com/scitools/scitools.aspx) was used to determine the best primer/probe set for each gene. Each probe was diluted to a storage stock of 300 μM in DEPC-treated H2O prior to use.
RNA extraction and cDNA synthesis
Gc were grown in liquid culture to mid-log phase (Stohl et al. submitted). For production of cDNA for examination of basal gene expression, 1 ml aliquots of mid-log phase Gc were taken. To examine the role of H2O2, ONOO−, NaOCl,MMC, or MMS on Gc gene expression, 1 ml aliquots of mid-logarithmic phase Gc were diluted 1:10 in amended GCBL. An untreated culture was maintained, and the experimental culture was treated with either 5 mM H2O2, 3 mM ONOO− (Fisher) in GCBL lacking sodium bicarbonate, 1.7 mM NaOCl (Sigma), 25 ng/ml MMC (Sigma), or 0.02% v/v MMS (Sigma). After incubation with shaking at 37°C for 15 minutes (H2O2, ONOO−, NaOCl), 30 minutes (MMC), or 60 minutes (MMS), 1 volume of Gc culture was added immediately to 2 volumes RNAprotect Bacteria Reagent (Qiagen). RNA extraction was performed using the RNeasy Mini kit (Qiagen), with an in-solution DNase digestion step at the end of the extraction. Purity and concentration of RNA was assayed by spectrophotometer. cDNA was synthesized as described (Stohl et al., 2005) with Superscript III (Invitrogen).
qPCR
qPCR was performed using gene-specific fluorescent probes, with samples not treated with reverse-transcriptase and a non-template sample as negative controls. Each reaction contained 12.5 μl of iQ Supermix (Bio-Rad), 0.15 μl (300 nM final conc) primer, 2.5 μl probe (final conc 0.3 μM), 4.7 μl H2O, and 5 μl of 1/100 diluted cDNA. Reactions were run using the iQ5 Real-Time PCR Detection System (Bio-Rad). Reaction conditions were as follows: 1 cycle of 95°C for 3:00 minutes; 40 cycles of 95°C for 00:10 minutes, 58°C for 00:30 minutes. Omp3, a gene encoding an outer-membrane protein not regulated by ROS, was used as a control (Stohl et al., 2005). ng1427 expression in ng1427::cat was measured with the “ng1427::cat 5’ end qPCR” primer/probe set that detects ng1427 transcript upstream of the ng1427 insertion/deletion mutation and in other strains where noted. ng1427 expression in other strains was measured with “NG1427 qPCR” primer/probe set that detects full-length ng1427 transcript where noted. Fold changes were determined using the comparative 2ΔΔCT while relative message expression was calculated using the 2ΔCT method (Schmittgen & Livak, 2008).
RecA-promoted in vitro NG1427 cleavage assay
This assay was adapted from the established LexA repressor cleavage assay (Gruenig et al., 2008). Reactions were carried out at 37oC in solutions containing 25 mM Tris-OAc (80% cation, pH 7.4) 1 mM DTT, 5% (w/v) glycerol, 3 mM postassium glutamate, 3 mM Mg(OAc)2, and an ATP regeneration system (2 mM phosphoenolpyruvate, 10 units/mL pyruvate kinase). The RecANg or RecAEc proteins (3 μM) were preincubated with 9 μM φX174 circular ssDNA (NEB) for 5 minutes. ATP (3 mM) and the appropriate matched SSB protein (SSBEc or SSBNg) was added to 0.9 μM and the reactions were incubated a further 5 minutes. NG1427 protein or the NG1427-G113D protein (3 μM) was added to initiate the reaction, which continued for 45 minutes. Laemmli protein sample buffer (250 mM Tris-Cl pH 6.8, 4% SDS, 20% w/v glycerol, 10% β–mercaptoethanol, and 0.1% w/v bromophenol blue) was added to stop the reaction. Samples were subjected to SDS-PAGE electrophoresis on 17% acrylamide gels and stained with Coomassie Brilliant Blue to visualize protein.
EMSA
The intergenic regions of ng1427/ng1428 and recN were amplified with primers specific to the intergenic regions via PCR by KOD polymerase (Novagen). The resulting PCR products were purified using Qiaquick kit (Qiagen) and labeled with biotin using the Biotin 3’ End DNA Labeling Kit (Pierce). Purified NG1427-G113D was added at 100 nM final concentration to a binding buffer containing a final concentration of 35 mM Tris, 20.25 mM NaCl, 150 mM KCl, 1 mM DTT, 50 ng/μl Poly (dI-dC), 0.05% NP-40, 2.7% glycerol, 5 mM MgCl2; pH 7.5 and incubated for 20 minutes at 25°C. 40 fmol (final concentration 2 nM) of biotin-labeled DNA was then added and incubated for an additional 20 minutes at 25°C. Where indicated H2O2, NaOCl, ONOO−, degraded ONOO− (Fisher)or MMS was added and incubated for 15 minutes at 25°C. The EMSA reaction was run out on a 5% PAGE gel (Bio-Rad) in chilled 0.5X TBE buffer and detected according to the LightShift Chemiluminescent EMSA Kit (Pierce) instructions.
Western Blot
Gc were grown to mid-log phase in liquid culture. Following a 1:10 dilution into amended GCBL (the ONOO− sample lacking Sodium Bicarbonate), the cultures were incubated for 15minutes (5 mM H2O2, 3 mM ONOO−, 1.7 mM NaOCl), 30 minutes (25 ng/μl MMC) or 60 minutes (0.2% v/v MMS) at 37°C, or left untreated After 30 seconds, spectinomycin was added to a final concentration of 20 ng/ml to inhibit protein synthesis. An aliquot was then taken from each culture, spun down at ~15,000g for one minute, and stored on wet ice. The cell pellets were resuspended in 125 μl PBS, and quantified with BCA (Pierce). 3 μg of total cellular protein was loaded onto a 20% SDS-PAGE gel and subjected to electrophoresis. Western blot analysis was performed as previously described (Helm et al., 2007), with 1:5000 anti-HA primary antibody (Roche) and 1:5000 horseradish peroxidase-conjugated anti-rat secondary antibody (Boehringer Mannheim). The signal was recorded using a chemilluminescent imager (Bio-Rad).
Detection of oxidized cysteine
NG1427-G113D was diluted 1/10 in protein storage buffer (25 mM Tris, 150 mM NaCl, 20% glycerol) with a pH of 7.0. NG1427-G113D was then added to a final concentration of 100 nM, and where applicable, 15 mM H2O2, 5 mM ONOO− , or 20 μM NaOCl was added to the reaction. After a 15 minute incubation, 25-molar excess (2.5μM) of maleimide-PEG11-biotin in DMSO was added to the reaction and incubated for 16 hours at 25°C. The reactions were then split into two, with 1 μl of β–mercaptoethanol added to one 25 μl reaction and the other aliquot left untreated. 5X protein loading buffer lackingβ–mercaptoethanol was added, and samples were loaded onto 4–20% PAGE gels (Bio-Rad). Protein gels were stained with Coomassie Brilliant Blue. For detection of the biotin label, the proteins were transferred to a PVDF membrane (Millipore) and detected according to the LightShift® Chemiluminescent EMSA Kit instructions.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr. Ludmilla Shuvalova for her assistance in purifying the NG1427-G113D-HIS protein, and Paul M. Duffin for reading of the manuscript. Additionally, we would like to acknowledge Dr. Mike Cox for providing E. coli RecA, SSB, LexA proteins. This work was supported by NIH grants R01AI044239, R01AI055977 and R37 AI033493 to H.S.S.
References
- Albrich JM, McCarthy CA, Hurst JK. Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proc Natl Acad Sci U S A. 1981;78:210–214. doi: 10.1073/pnas.78.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND. Families and clans of serine peptidases. Arch Biochem Biophys. 1995;318:247–250. doi: 10.1006/abbi.1995.1227. [DOI] [PubMed] [Google Scholar]
- Black CG, Fyfe JA, Davies JK. Absence of an SOS-like system in Neisseria gonorrhoeae. Gene. 1998;208:61–66. doi: 10.1016/s0378-1119(97)00653-7. [DOI] [PubMed] [Google Scholar]
- Blackford JA, Jr, Antonini JM, Castranova V, Dey RD. Intratracheal instillation of silica up-regulates inducible nitric oxide synthase gene expression and increases nitric oxide production in alveolar macrophages and neutrophils. Am J Respir Cell Mol Biol. 1994;11:426–431. doi: 10.1165/ajrcmb.11.4.7522485. [DOI] [PubMed] [Google Scholar]
- Brent R, Ptashne M. The lexA gene product represses its own promoter. Proc Natl Acad Sci U S A. 1980;77:1932–1936. doi: 10.1073/pnas.77.4.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LA, Yasbin RE. Mutagenesis of Neisseria gonorrhoeae: absence of error-prone repair. J Bacteriol. 1984;160:288–293. doi: 10.1128/jb.160.1.288-293.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Xu G, Zhao Y, Tian B, Lu H, Yu X, Xu Z, Ying N, Hu S, Hua Y. A novel OxyR sensor and regulator of hydrogen peroxide stress with one cysteine residue in Deinococcus radiodurans. PLoS One. 2008;3:e1602. doi: 10.1371/journal.pone.0001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Gotschlich EC. CGM1a antigen of neutrophils, a receptor of gonococcal opacity proteins. Proc Natl Acad Sci U S A. 1996;93:14851–14856. doi: 10.1073/pnas.93.25.14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig NL, Roberts JW. E. coli recA protein-directed cleavage of phage lambda repressor requires polynucleotide. Nature. 1980;283:26–30. doi: 10.1038/283026a0. [DOI] [PubMed] [Google Scholar]
- Criss AK, Seifert HS. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol. 2008;10:2257–2270. doi: 10.1111/j.1462-5822.2008.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidsen T, Tonjum T. Meningococcal genome dynamics. Nat Rev Microbiol. 2006;4:11–22. doi: 10.1038/nrmicro1324. [DOI] [PubMed] [Google Scholar]
- Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev. 2004;17:965–981. doi: 10.1128/CMR.17.4.965-981.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erill I, Campoy S, Barbe J. Aeons of distress: an evolutionary perspective on the bacterial SOS response. FEMS Microbiol Rev. 2007;31:637–656. doi: 10.1111/j.1574-6976.2007.00082.x. [DOI] [PubMed] [Google Scholar]
- Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenig MC, Renzette N, Long E, Chitteni-Pattu S, Inman RB, Cox MM, Sandler SJ. RecA-mediated SOS induction requires an extended filament conformation but no ATP hydrolysis. Mol Microbiol. 2008;69:1165–1179. doi: 10.1111/j.1365-2958.2008.06341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudas LJ, Pardee AB. Model for regulation of Escherichia coli DNA repair functions. Proc Natl Acad Sci U S A. 1975;72:2330–2334. doi: 10.1073/pnas.72.6.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayatsu H, Pan S, Ukita T. Reaction of sodium hypochlorite with nucleic acids and their constituents. Chem Pharm Bull (Tokyo) 1971;19:2189–2192. doi: 10.1248/cpb.19.2189. [DOI] [PubMed] [Google Scholar]
- Heckels JE. Structural comparison of Neisseria gonorrhoeae outer membrane proteins. J Bacteriol. 1981;145:736–742. doi: 10.1128/jb.145.2.736-742.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm RA, Barnhart MM, Seifert HS. pilQ Missense mutations have diverse effects on PilQ multimer formation, piliation, and pilus function in Neisseria gonorrhoeae. J Bacteriol. 2007;189:3198–3207. doi: 10.1128/JB.01833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henle ES, Luo Y, Gassmann W, Linn S. Oxidative damage to DNA constituents by iron-mediated fenton reactions. The deoxyguanosine family. J Biol Chem. 1996;271:21177–21186. [PubMed] [Google Scholar]
- Katona LI, Tokarz R, Kuhlow CJ, Benach J, Benach JL. The fur homologue in Borrelia burgdorferi. J Bacteriol. 2004;186:6443–6456. doi: 10.1128/JB.186.19.6443-6456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley WL. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Mol Microbiol. 2006;62:1228–1238. doi: 10.1111/j.1365-2958.2006.05444.x. [DOI] [PubMed] [Google Scholar]
- Kellogg DS, Jr, Peacock WL, Deacon WE, Brown L, Pirkle CI. Neisseria gonorrhoeae. I. Virulence genetically linked to clonial variation. Journal of Bacteriology. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SP, Potter AJ, Apicella MA, Jennings MP, McEwan AG. NmlR of Neisseria gonorrhoeae: a novel redox responsive transcription factor from the MerR family. Mol Microbiol. 2005;57:1676–1689. doi: 10.1111/j.1365-2958.2005.04773.x. [DOI] [PubMed] [Google Scholar]
- Little JW. Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci U S A. 1984;81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW, Edmiston SH, Pacelli LZ, Mount DW. Cleavage of the Escherichia coli lexA protein by the recA protease. Proc Natl Acad Sci U S A. 1980;77:3225–3229. doi: 10.1073/pnas.77.6.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW, Kim B, Roland KL, Smith MH, Lin LL, Slilaty SN. Cleavage of LexA repressor. Methods Enzymol. 1994;244:266–284. doi: 10.1016/0076-6879(94)44022-0. [DOI] [PubMed] [Google Scholar]
- Little JW, Mount DW. The SOS regulatory system of Escherichia coli. Cell. 1982;29:11–22. doi: 10.1016/0092-8674(82)90085-x. [DOI] [PubMed] [Google Scholar]
- Little JW, Mount DW, Yanisch-Perron CR. Purified lexA protein is a repressor of the recA and lexA genes. Proc Natl Acad Sci U S A. 1981;78:4199–4203. doi: 10.1073/pnas.78.7.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehr IJ, Long CD, Serkin CD, Seifert HS. A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics. 2000;154:523–532. doi: 10.1093/genetics/154.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget MS, Buttner MJ. Thiol-based regulatory switches. Annu Rev Genet. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991a;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991b;266:4244–4250. [PubMed] [Google Scholar]
- Rest RF, Fischer SH, Ingham ZZ, Jones JF. Interactions of Neisseria gonorrhoeae with human neutrophils: effects of serum and gonococcal opacity on phagocyte killing and chemiluminescence. Infection & Immunity. 1982;36:737–744. doi: 10.1128/iai.36.2.737-744.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos D, van Bruggen R, Meischl C. Oxidative killing of microbes by neutrophils. Microbes Infect. 2003;5:1307–1315. doi: 10.1016/j.micinf.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Root RK, Metcalf J, Oshino N, Chance B. H2O2 release from human granulocytes during phagocytosis. I. Documentation, quantitation, and some regulating factors. J Clin Invest. 1975;55:945–955. doi: 10.1172/JCI108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Maniatis T, Fritsch EF. Molecular cloning : a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 1989. [Google Scholar]
- Schmidt KA, Deal CD, Kwan M, Thattassery E, Schneider H. Neisseria gonorrhoeae MS11mkC opacity protein expression in vitro and during human volunteer infectivity studies. Sex Transm Dis. 2000;27:278–283. doi: 10.1097/00007435-200005000-00008. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Sechman EV, Rohrer MS, Seifert HS. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;57:468–483. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Wu HJ, Kidd SP, Apicella MA, Jennings MP, McEwan AG. Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol Mol Biol Rev. 2006;70:344–361. doi: 10.1128/MMBR.00044-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Wu HJ, Srikhanta YN, Edwards JL, Falsetta ML, Hamilton AJ, Maguire TL, Grimmond SM, Apicella MA, McEwan AG, Jennings MP. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol Microbiol. 2007;63:54–68. doi: 10.1111/j.1365-2958.2006.05478.x. [DOI] [PubMed] [Google Scholar]
- Seifert HS. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene. 1997;188:215–220. doi: 10.1016/s0378-1119(96)00810-4. [DOI] [PubMed] [Google Scholar]
- Seifert HS, Wright CJ, Jerse AE, Cohen MS, Cannon JG. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J Clin Invest. 1994;93:2744–2749. doi: 10.1172/JCI117290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer WM, Rest RF. Interactions of gonococci with phagocytic cells. Annu Rev Microbiol. 1989;43:121–145. doi: 10.1146/annurev.mi.43.100189.001005. [DOI] [PubMed] [Google Scholar]
- Simons MP, Nauseef WM, Apicella MA. Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect Immun. 2005;73:1971–1977. doi: 10.1128/IAI.73.4.1971-1977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar EP, Lazio MP, Seifert HS. Roles of the recJ and recN genes in homologous recombination and DNA repair pathways of Neisseria gonorrhoeae. J Bacteriol. 2002;184:919–927. doi: 10.1128/jb.184.4.919-927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soszynski M, Bartosz G. Effect of peroxynitrite on erythrocytes. Biochim Biophys Acta. 1996;1291:107–114. doi: 10.1016/0304-4165(96)00052-9. [DOI] [PubMed] [Google Scholar]
- Stohl EA, Criss AK, Seifert HS. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol. 2005;58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Seifert HS. The recX gene potentiates homologous recombination in Neisseria gonorrhoeae. Mol Microbiol. 2001;40:1301–1310. doi: 10.1046/j.1365-2958.2001.02463.x. [DOI] [PubMed] [Google Scholar]
- Thomas EL. Myeloperoxidase, hydrogen peroxide, chloride antimicrobial system: nitrogen-chlorine derivatives of bacterial components in bactericidal action against Escherichia coli. Infect Immun. 1979;23:522–531. doi: 10.1128/iai.23.2.522-531.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JM. Disulphide bridges in globular proteins. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- Wertman KF, Mount DW. Nucleotide sequence binding specificity of the LexA repressor of Escherichia coli K-12. J Bacteriol. 1985;163:376–384. doi: 10.1128/jb.163.1.376-384.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, van den Berg JJ, Roitman E, Kuypers FA. Chlorohydrin formation from unsaturated fatty acids reacted with hypochlorous acid. Arch Biochem Biophys. 1992;296:547–555. doi: 10.1016/0003-9861(92)90609-z. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Seib KL, Srikhanta YN, Kidd SP, Edwards JL, Maguire TL, Grimmond SM, Apicella MA, McEwan AG, Jennings MP. PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol Microbiol. 2006;60:401–416. doi: 10.1111/j.1365-2958.2006.05079.x. [DOI] [PubMed] [Google Scholar]
- Yermilov V, Rubio J, Ohshima H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995;376:207–210. doi: 10.1016/0014-5793(95)01281-6. [DOI] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.