

Abstract
Many protein-protein interactions are mediated through the recognition of β-turn secondary structures. Consequently, small-molecule β-turn mimetics are invaluable as probes for assessing bioactive ligand conformations, establishing pharmacophoric requirements, and pursuing rational drug design. While effective drug scaffolds have been developed to precisely position up to four functionalities primarily in two-dimensions, an analogous rigid scaffold capable of predictably juxtaposing four amino acid side chains in three-dimensions has required the use of pentameric or larger cyclopeptides. Diverse approaches have been taken in efforts to constrain peptides into turn conformations,[1] but one strategy that has not been broadly explored is the use of cyclic tetrapeptides.[2,3] Cyclic tetrapeptides offer an attractive platform to mimic protein turn regions due to their appropriate size, shape, and synthetic modularity, but these structures remain largely unexplored due to poor synthetic efficiency in constructing the strained 12-membered ring, an inability to control cis-trans geometry of backbone amides, and the apparent requirement to sacrifice one of four amino acid residues to incorporate a proline or other turn-forming residue.[2,4,5]
Keywords: drug design, cyclic peptides, somatostatin, structure-activity relationship, triazole
Here we report the syntheses and analyses of two classes of 13-and 14-membered ring pseudo-tetrapeptides[5–7] containing either one or two triazole moieties, respectively, and describe the design, syntheses, structural analyses, and somatostatin (SST) receptor binding activities of a library of all 16 possible stereoisomeric compounds incorporating the somatostatin pharmacophore. These studies exploit the 1,4-disubstituted 1,2,3-triazole as a trans peptide bond surrogate.[7–10] Structural analysis of the diastereomeric library using NMR spectroscopy indicated that each of the peptide scaffolds adopts a distinct, rigid, conformationally homogeneous turn-like structure.[11] The three-dimensional pharmacophoric display of the compounds is systematically altered by varying the stereochemistry around the otherwise constitutionally identical scaffolds, yielding both compounds with broad-spectrum activity against the five human SST receptor subtypes (pan-somatostatins) and compounds with receptor selectivity. Our studies therefore provide a basis set of scaffolds having subtle but predictable differences in their spatial display of amino acid side chains that are useful for rational, structurally-informed design of bioactive agents.
As part of a research program investigating the incorporation of triazoles into linear and cyclic peptide architectures,[9] we synthesized compounds 1–4, which can be divided into two classes that differ by the presence of either one (class I) or two (class II) backbone triazole moieties. Encouraged by the relatively facile cyclization of these 13- and 14-membered rings via triazole ring formation, and motivated by their potential for predictable side chain display, we undertook high resolution structural analyses of these peptides. In all cases, the obtained structures revealed a rigid rectangular scaffold that superimposed well on idealized β-turn motifs (Figure S2). Importantly, the 1H NMR spectra for all four compounds were sharp and consistent with a single solution species present on the NMR time scale. Although the observed NOEs involving the triazole C-H atom indicate that the triazole populates multiple rotameric conformations in fast exchange on the NMR time scale, this variability has virtually no impact on the position of the rest of the atoms in the backbone or the Cα-Cβ vectors (see Supporting Information for the synthesis of compounds 1–4).
We wondered if the observed conformational homogeneity for 1–4 would hold for the entire series of 16 possible stereoisomeric pseudo-tetrapeptides of a given sequence. If so, structural determination of the eight diastereomers in a given enantiomeric series (from which the eight mirror image counterparts could be easily modeled) would provide a basis set of conformationally predictable, three-dimensional scaffolds amenable to drug design. Accordingly, we set out to synthesize the 16 possible stereoisomeric pseudo-tetrapeptides incorporating the pharmacophoric residues of somatostatin-14 (SRIF-14, Phe7-Trp8-Lys9-Thr10), a well-studied ligand known to bind its cognate receptors using a β-turnmotif.[12]
To synthesize the 16 stereoisomeric structures, we prepared two sets of dipeptides, each comprising four diastereomers (compounds 5a–5d and 6a–6d), from which all 16 required diastereomeric tetramers could be prepared (Scheme 1). Macrocyclizations of the crude linear azido alkynes via copper (I) mediated [3+2] Huisgen dipolar cycloaddition proceeded in yields ranging from 31–90%, as determined by HPLC (isolated yields of 20–59% following deprotection, based on the dipeptide starting materials). Alternative attempts to synthesize compounds of type 7 via macrolactamization resulted in racemization and poor cyclization yields, consistent with previous reports.[7] Inclusion of the copper chelating ligand tris-(benzyltriazolylmethyl)amine (TBTA)[13] during the [3+2] macrocyclization step favored formation of the desired cyclic tetramer over formation of the cyclic octamer species that results from head-to-tail dimerization of two tetrapeptide substrates.[14] An attempt to facilitate macrocyclization by heating to 70 °C (for compound 7a) resulted in complex 1H NMR spectra that were ascribed to either multiple product conformations or racemization. On the other hand, cyclizations conducted at room temperature yielded conformationally homogeneous compounds that remained homogeneous after being heated to 70 °C (for compound 7d).
Scheme 1.

Synthetic strategy for the pseudo-tetrapeptide library. Compounds 5a–d, compounds 6a–d, and compounds 7a–h differ only at the marked (asterisk) chiral centers. R = Cbz or 2-Cl-Cbz. HATU = 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluoro-phosphate, TBTA = tris-(benzyltriazolylmethyl)amine.
With the 16 stereoiso meric somatostatin analogs in hand, we first assessed the extent of their conformational homogeneity. Encouragingly, all 16 compounds gave sharp 1H NMR spectra consistent with a single conformation on the NMR time scale, so we proceeded to determine NMR solution structures using the program CNS[15] for the eight compounds in one enantiomeric series (Figure 2). Each of the eight diastereomers exhibited a distinct and conformationlly homogeneous structure, which is somewhat remarkable considering the conformational heterogeneity previously noted in cyclic tetrapeptides.[2,4,5] Not surprisingly, it appears that the relative chirality of the four stereogenic centers, exerts the most dominant influence on the amide backbone orientation and the distinct three-dimensional side chain display adopted by each scaffold.
Figure 2.

NMR solution structures (d6-DMSO) and schematic diagrams for eight diastereomeric pseudo-tetrapeptide scaffolds. Stereocenter configurations are labeled according to the amino acid starting materials from which they are derived. For each structure shown, the corresponding enantiomer was also synthesized and assayed for hSSTR1–5 binding.
The library of the 16 stereoisomeric pseudo-tetrapeptides were screened for binding activity to the five human somatostatin receptor subtypes (hSSTR1–5) (Table 1). Despite the fact that all 16 compounds are constitutionally identical (having the same sequence of amino acids), a range of bioactivities was observed, including a total lack of binding even at concentrations up to 10 μM (7g), selectivity for hSSTR1 (7h) or hSSTR4 (ent-7f), and broad-spectrum activity mimicking that of somatostatin (ent-7g). The hSSTR2 receptor appeared to have the most stringent binding requirements for these compounds, whereas hSSTR4 bound nearly all of the compounds with some affinity.
Table 1.
hSSTR1–5 binding affinities (IC50, nM) for the heterocyclic pseudo-tetrapeptides.
| peptide | IC50 (nM)[a] |
||||
|---|---|---|---|---|---|
| hSSTR1 | hSSTR2 | hSSTR3 | hSSTR4 | hSSTR5 | |
| SRIF-28[b] | 1.8 ± 0.5 | 2.0 ± 0.3 | 2.4 ± 0.7 | 3.3 ± 0.3 | 2.4 ± 0.7 |
| 7a | >10,000 | >10,000 | 1,000 ± 100 | 2,600 ± 200 | 2,100 ± 400 |
| 7b | >10,000 | >10,000 | >10,000 | 1,600 ± 200 | >10,000 |
| 7c | 5,800 ± 500 | >10,000 | >10,000 | >10,000 | >10,000 |
| 7d | >10,000 | >10,000 | 6,700 ± 1,500 | 5,600 ± 500 | >10,000 |
| 7e | 2,100 ± 200 | >10,000 | 4,200 ± 1,600 | 570 ± 60 | 5,000 ± 1,500 |
| 7f | >10,000 | >10,000 | >10,000 | 2,900 ± 300 | >10,000 |
| 7g | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| 7h | 110 ± 10 | 1,200 ± 200 | 7,700 ± 1,100 | 2,200 ± 300 | 3,800 ± 800 |
| ent-7a | >10,000 | >10,000 | 2,800 ± 600 | 980 ± 60 | 5,100 ± 400 |
| ent-7b | 4,100 ± 1,200 | >10,000 | >10,000 | 5,000 ± 700 | 6,100 ± 600 |
| ent-7c | >10,000 | 3,300 ± 200 | 5,400 ± 1,300 | 4,700 ± 1,000 | >10,000 |
| ent-7d | 180 ± 4 | 1,300 ± 400 | 730 ± 200 | 790 ± 100 | 1,600 ± 300 |
| ent-7e | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| ent-7f | >10,000 | >10,000 | >10,000 | 230 ± 50 | >10,000 |
| ent-7g | 170 ± 20 | 160 ± 50 | 67 ± 30 | 140 ± 10 | 170 ± 50 |
| ent-7h | >10,000 | >10,000 | 3,300 ± 400 | 2,900 ± 700 | >10,000 |
Compounds were tested in triplicate. The IC50 values derived from competitive radioligand displacement assays reflect the affinities of the peptides for the cloned somatostatin receptors using the nonselective 125I-[Leu8, DTrp22, Tyr25] SRIF-28 as the radioligand.
For the chemical structure of SRIF-28, see ref. 10d.
To clarify the three-dimensional pharmacophoric determinants of affinity and specificity for these compounds, we next sought to identify structural similarities among the peptides having comparable activities. Because each scaffold is conformationally rigid, the backbone atoms and the Cα-Cβ vectors can be used with relatively high confidence in structural analyses. To assess the hSSTR4 binding profile, we overlayed all 16 compounds onto ent-7g (the highest affinity hSSTR4 binder) using the Cβ atoms of the Trp and Lys side chains (known to be the most important components of the pharmacophore) and the Cα and Cβ atoms of the Phe side chain (because the highest affinity hSSTR4 binders all contain a conserved D-Phe chirality). After removing scaffolds with RMSD values above a cutoff of 0.7 Å (see Table S1 for all RMSD values), we qualitatively observed clustering into three conformational families (Figure 3a). Interestingly, the three observed groupings correspond to the set of peptides that bind multiple hSSTR receptors (ent-7d and ent-7g, blue atoms in Figure 3a), the peptides that are most selective for hSSTR4 (7b, 7e, ent-7a, and ent-7f, yellow atoms in Figure 3a), and peptides that bound with low affinity (7d, 7g, ent-7c, and ent-7h, pink atoms in Figure 3a). In other words, when constrained by the overlay to place the pharmacophoric Phe, Lys, and Trp residues in a particular region of space (much as the receptor binding site would require a particular spatial arrangement of side chains), the rigidity of the scaffolds enforces different orientations that correlate with their observed binding characteristics.
Figure 3.

Pairwise fittings of pseudo-tetrapeptides correlating structure with bioactivity. a) All 16 compounds were overlayed onto ent-7g (highest affinity hSSTR4 binder) using pairfits of the Trp, Lys, and Phe Cβ atoms and the Phe Cα atom. Compounds with an RMSD higher than 0.7 Å are not shown; compounds ent-7g and ent-7d (blue) bind multiple receptors including hSSTR4; compounds ent-7f, 7e, ent-7a, and 7b (gold) are specific for hSSTR4; compounds 7d, 7g, ent-7c, and ent-7h (pink) did not bind or bound with low affinity. Sticks are shown for the triazole atoms and the Cα and Cβ atoms of Phe, Trp, and Lys. b) Overlay of the 16 compounds onto ent-7f (most selective hSSTR4 binder) (pink). An RMSD cutoff of 0.55 Å left compounds 7b (gold), 7e (blue), and ent-7a (white), which represent the hSSTR4-selective peptides. c) Overlay of the 16 compounds using pairfits of the Cα atoms from all four residues and Trp and Lys Cβ atoms onto 7h (most selective hSSTR1 binder) (gold). An RMSD cutoff of 0.3 Å left only molecules ent-7g (blue) and ent-7d (white), which correspond to the only other high affinity, albeit less selective, binders of hSSTR1.
To better understand the requirements for selective binding to hSSTR4, a different overlay of the 16 compounds onto ent-7f (the most selective hSSTR4 binder) was carried out. When the Thr Cα atom and additional backbone atoms (Lys carbonyl carbon and Trp amine) were used along with the fit atoms described above, discarding scaffolds above an RMSD cutoff of 0.55 Å (see Table S1 for all RMSD values) left the four most selective hSSTR4 ligands (7b, 7e, ent-7a, and ent-7f) (Figure 3b). The most notable similarities of these compounds are that all four contain a D-Phe residue and all four have Lys and Trp residues of the same chirality (both L- or both D-). The requirement that additional Thr and backbone atoms be included in the fit to ensure that the lowest RMSD compounds corresponded to the most selective hSSTR4 ligands suggests that avoiding steric clashes near the Thr residue in the receptor binding pocket is an additional important factor in selective binding to hSSTR4. The RMSD of these compounds from ent-7f follows the same trend as their affinity for the hSSTR4 receptor (ent-7f < 7e < ent-7a < 7b).
A similar structural analysis was conducted for hSSTR1-binding ligands. The 16 diastereomers were overlayed onto 7h (the most selective hSSTR1 binder) using the Cα atoms of all four residues and the Cβ atoms of Trp and Lys (Figure 3c). After discarding molecules with an RMSD cutoff of greater than 0.3 Å (see Table S1), the remaining peptides (ent-7g and ent-7d) represent the only other high affinity, albeit less selective, binders of the hSSTR1 receptor. Considering the bioactivities and the atoms required for this fit, it appears that the four Cα positions and the Cα-Cβ vectors of Lys and Trp are the major determinants of hSSTR1 affinity for our compounds, while the Phe and Thr side chains influence receptor selectivity.
We hope that the well-defined diastereomeric structures described here will serve as a basis set from which future structure-based drug design studies can be initiated. Furthermore, by determining the “negative structural image”[16] of receptor binding pockets, the use of small libraries of scaffolds having systematic and predictable differences in their spatial display of amino acid side chains could be useful in delineating the three-dimensional pharmacophoric requirements for receptor binding and selectivity, especially in cases where high-resolution structural data are not readily available.
Supplementary Material
Figure 1.
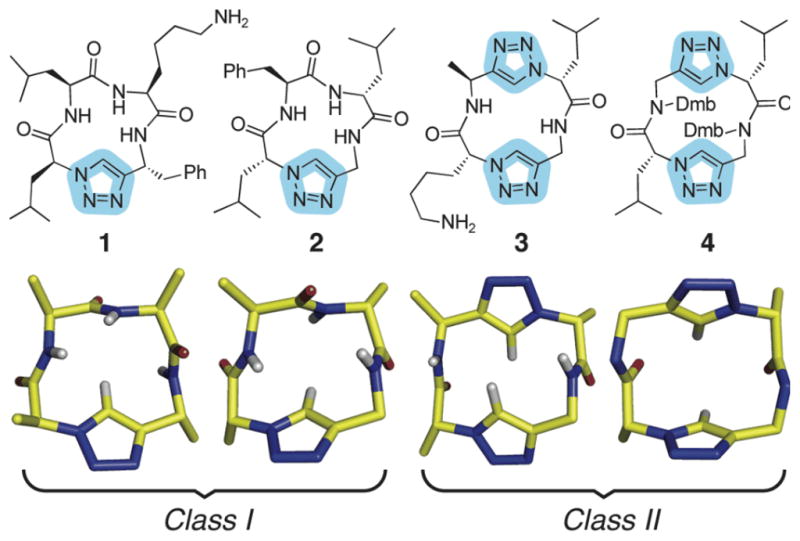
Chemical and molecular structures of representative members of two classes of cyclic pseudo-tetrapeptide scaffolds. Structures were determined using multidimensional NMR (1–3) or X-ray crystallography (4). Some atoms have been omitted for clarity. Dmb = 2,4-dimethoxybenzyl.
Acknowledgments
We thank the NASA Earth and Space Science Fellowship Program (Grant NNX07AR35H) (J.M.B.), the Netherlands Organization for Scientific Research (NWO) for a travel grant (J.V.M.), and Dr. L. J. Leman for assistance with the manuscript. This work was supported in part by a grant from National Institute of General Medical Sciences (GM52190) and the Skaggs Institute for Chemical Biology.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author. Coordinates for the NMR structures reported in this manuscript have been deposited at the BMRB databank (www.bmrb.wisc.edu, accession numbers 20036 20043).
Contributor Information
Mr. John M. Beierle, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA)
Dr. W. Seth Horne, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA)
Prof. Dr. Jan H. van Maarseveen, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA).
Beatrice Waser, Division of Cell Biology and Experimental Cancer Research University of Berne Berne, Switzerland 3010.
Prof. Dr. Jean Claude Reubi, Division of Cell Biology and Experimental Cancer Research University of Berne Berne, Switzerland 3010
Prof. Dr. M. Reza Ghadiri, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA).
References
- 1.For selected reviews, see: Reissmann S, Imhof D. Curr Med Chem. 2004;11:2823. doi: 10.2174/0929867043364135.Li P, Roller PP, Xu J. Curr Org Chem. 2002;6:411.MacDonald M, Aube J. Curr Org Chem. 2001;5:417.Kessler H, Gratias R, Hessler G, Gurrath M, Mueller G. Pure Appl Chem. 1996;68:1201.Kessler H. Angew Chem, Int Ed Engl. 1982;21:512.
- 2.a) Shute RE, Kawai M, Rich DH. Tetrahedron. 1988;44:685. [Google Scholar]; b) Kawai M, Rich DH. Tetrahedron Lett. 1983;24:5309. [Google Scholar]
- 3.Kawai M, Jasensky RD, Rich DH. J Am Chem Soc. 1983;105:4456. [Google Scholar]
- 4.a) Tamamura H, et al. J Med Chem. 2005;48:3280. doi: 10.1021/jm050009h. [DOI] [PubMed] [Google Scholar]; b) Loiseau N, Gomis JM, Santolini J, Delaforge M, Andre F. Biopolymers. 2003;69:363. doi: 10.1002/bip.10339. [DOI] [PubMed] [Google Scholar]; c) Meutermans WDF, Bourne GT, Golding SW, Horton DA, Campitelli MR, Craik D, Scanlon M, Smythe ML. Org Lett. 2003;5:2711. doi: 10.1021/ol034907o. [DOI] [PubMed] [Google Scholar]; d) Horton DA, Bourne GT, Smythe ML. Molecular Diversity. 2002;5:289. doi: 10.1023/a:1021365402751. [DOI] [PubMed] [Google Scholar]; e) Terada Y, Kawai M, Rich DH. Int J Pept Protein Res. 1989;33:3. doi: 10.1111/j.1399-3011.1983.tb02098.x. [DOI] [PubMed] [Google Scholar]; f) Kawai M, Pottorf RS, Rich DH. J Med Chem. 1986;29:2409. doi: 10.1021/jm00161a046. [DOI] [PubMed] [Google Scholar]
- 5.Glenn MP, Kelso MJ, Tyndall JDA, Fairlie DP. J Am Chem Soc. 2003;125:640. doi: 10.1021/ja029205t. [DOI] [PubMed] [Google Scholar]
- 6.a) Montero A, Beierle JM, Olsen CA, Ghadiri MR. J Am Chem Soc. 2009 doi: 10.1021/ja809508f. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seebach D, et al. Helv Chim Acta. 2008;91:1736. [Google Scholar]; c) Norgren AS, Buettner F, Prabpai S, Kongsaeree P, Arvidsson PI. J Org Chem. 2006;71:6814. doi: 10.1021/jo060854n. [DOI] [PubMed] [Google Scholar]; d) Schumann F, Mueller A, Koksch M, Mueller G, Sewald N. J Am Chem Soc. 2000;122:12009. [Google Scholar]
- 7.Bock VD, Perciaccante R, Jansen TP, Hiemstra H, van Maarseveen JH. Org Lett. 2006;8:919. doi: 10.1021/ol053095o. [DOI] [PubMed] [Google Scholar]
- 8.a) Horne WS, Olsen CA, Beierle JM, Montero A, Ghadiri MR. Angew Chem, Int Ed. 2009 doi: 10.1002/anie.200805900. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. J Med Chem. 2008;51:3437. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]; c) Bock VD, Speijer D, Hiemstra H, van Maarseveen JH. Org Biomol Chem. 2007;5:971. doi: 10.1039/b616751a. [DOI] [PubMed] [Google Scholar]; d) Turner RA, Oliver AG, Lokey RS. Org Lett. 2007;9:5011. doi: 10.1021/ol702228u. [DOI] [PubMed] [Google Scholar]; e) Appendino G, Bacchiega S, Minassi A, Cascio Maria G, De Petrocellis L, Di Marzo V. Angew Chem, Int Ed. 2007;46:9312. doi: 10.1002/anie.200703590. [DOI] [PubMed] [Google Scholar]; f) Angell YL, Burgess K. Chem Soc Rev. 2007;36:1674. doi: 10.1039/b701444a. [DOI] [PubMed] [Google Scholar]; g) Choi WJ, Shi ZD, Worthy KM, Bindu L, Karki RG, Nicklaus MC, Fisher RJ, Burke TR. Bioorg Med Chem Lett. 2006;16:5265. doi: 10.1016/j.bmcl.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Angelo NG, Arora PS. J Am Chem Soc. 2005;127:17134. doi: 10.1021/ja056406z. [DOI] [PubMed] [Google Scholar]; i) Brik A, Alexandratos J, Lin YC, Elder JH, Olson AJ, Wlodawer A, Goodsell DS, Wong CH. ChemBioChem. 2005;6:1167. doi: 10.1002/cbic.200500101. [DOI] [PubMed] [Google Scholar]; j) Angell Y, Burgess K. J Org Chem. 2005;70:9595. doi: 10.1021/jo0516180. [DOI] [PubMed] [Google Scholar]
- 9.a) van Maarseveen JH, Horne WS, Ghadiri MR. Org Lett. 2005;7:4503. doi: 10.1021/ol0518028. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Horne WS, Yadav MK, Stout CD, Ghadiri MR. J Am Chem Soc. 2004;126:15366. doi: 10.1021/ja0450408. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Horne WS, Stout CD, Ghadiri MR. J Am Chem Soc. 2003;125:9372. doi: 10.1021/ja034358h. [DOI] [PubMed] [Google Scholar]
- 10.Trisubstituted 1,2,3-triazoleds have veen employed as amide bond surrogates in ghrelin mimics: Moulin A, Demange L, Ryan J, M’Kadmi C, Galleyrand JC, Martinez J, Fehrentz JA. Bioorg Med Chem Lett. 2008;18:164. doi: 10.1016/j.bmcl.2007.10.113.Moulin A, et al. J Med Chem. 2008;51:689. doi: 10.1021/jm701292s.Moulin A, et al. J Med Chem. 2007;50:5790. doi: 10.1021/jm0704550.
- 11.For a discussion of conformational homogeneity, see: Chatterjee J, Mierke D, Kessler H. J Am Chem Soc. 2006;128:15164. doi: 10.1021/ja063123d.
- 12.a) Nikiforovich GV, Marshall GR, Achilefu S. Chem Biol Drug Design. 2007;69:163. doi: 10.1111/j.1747-0285.2007.00493.x. [DOI] [PubMed] [Google Scholar]; b) Grace CRR, Durrer L, Koerber SC, Erchegyi J, Reubi JC, Rivier JE, Riek R. J Med Chem. 2005;48:523. doi: 10.1021/jm049518u. [DOI] [PubMed] [Google Scholar]; c) Grace CRR, Koerber SC, Erchegyi J, Reubi JC, Rivier J, Riek R. J Med Chem. 2003;46:5606. doi: 10.1021/jm030246p. [DOI] [PubMed] [Google Scholar]; d) Weckbecker G, Lewis I, Albert R, Schmid H, Hoyer D, Bruns C. Nat Rev Drug Disc. 2003;2:999. doi: 10.1038/nrd1255. [DOI] [PubMed] [Google Scholar]; e) Hirschmann R, et al. J Med Chem. 1998;41:1382. doi: 10.1021/jm9800346. [DOI] [PubMed] [Google Scholar]; f) Mattern RH, Tran TA, Goodman M. J Med Chem. 1998;41:2686. doi: 10.1021/jm970392t. [DOI] [PubMed] [Google Scholar]; Rohrer SP, et al. Science. 1998;282:737. doi: 10.1126/science.282.5389.737. [DOI] [PubMed] [Google Scholar]; g) Melacini G, Zhu Q, Osapay G, Goodman M. J Med Chem. 1997;40:2252. doi: 10.1021/jm960851a. [DOI] [PubMed] [Google Scholar]; h) Melacini G, Zhu Q, Goodman M. Biochemistry. 1997;36:1233. doi: 10.1021/bi962497o. [DOI] [PubMed] [Google Scholar]; i) Kessler H, Bats JW, Griesinger C, Koll S, Will M, Wagner K. J Am Chem Soc. 1988;110:1033. [Google Scholar]; j) Gilon C, et al. J Med Chem. 1998;41:919. doi: 10.1021/jm970633x. [DOI] [PubMed] [Google Scholar]; k) Kessler H, Bernd M, Damm I. Tetrahedron Lett. 1982;23:4685. [Google Scholar]; l) Veber DF, et al. Nature. 1981;292:55. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]; m) von Roedern EG, Lohof E, Hessler G, Hoffmann M, Kessler H. J Am Chem Soc. 1996;118:10156. [Google Scholar]
- 13.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 14.Punna S, Kuzelka J, Wang Q, Finn MG. Angew Chem, Int Ed. 2005;44:2215. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]
- 15.Brunger AT, et al. Acta Crystallogr D, Biol Crystallogr. 1998;D54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 16.a) Fukunishi Y, Kubota S, Kanai C, Nakamura H. J Comput-Aided Mol Des. 2006;20:237. doi: 10.1007/s10822-006-9047-1. [DOI] [PubMed] [Google Scholar]; b) DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babe LM, Craik CS. Proc Natl Acad Sci U S A. 1990;87:6644. doi: 10.1073/pnas.87.17.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.