

Abstract
BACKGROUND AND PURPOSE
1,4-Naphthoquinones exhibit antiplatelet activity both in vivo and in vitro. In the present study, we investigated the antiplatelet effect of a novel naphthoquinone derivative NP-313, 2-acetylamino-3-chloro-1,4-naphthoquinone and its mechanism of action.
EXPERIMENTAL APPROACH
We measured platelet aggregation, Ca2+ mobilization, thromboxane B2 formation and P-selectin expression and examined several enzymatic activities. Furthermore, we used the irradiated mesenteric venules in fluorescein sodium–treated mice to monitor the antithrombotic effect of NP-313 in vivo.
KEY RESULTS
NP-313 concentration-dependently inhibited human platelet aggregation induced by collagen, arachidonic acid, thapsigargin, thrombin and A23187. NP-313 also inhibited P-selectin expression, thromboxane B2 formation and [Ca2+]i elevation in platelets stimulated by thrombin and collagen. NP-313 at 10 µM inhibited cyclooxygenase, thromboxane A2 synthase, and protein kinase Cα, whereas it did not affect phospholipase A2 or phospholipase C activity. In the presence of indomethacin and an adenosine 5-diphosphate scavenger, NP-313 concentration-dependently inhibited thrombin- and A23187-induced [Ca2+]i increase through its inhibitory effects on Ca2+ influx, rather than blocking Ca2+ release from intracellular stores. NP-313 also inhibited thapsigargin-mediated Ca2+ influx through store-operated calcium channel but had no effect on Ca2+ influx through store-independent calcium channel evoked by the diacylglycerol analogue 1-oleoyl-2-acetyl-sn-glycerol. Nevertheless, it had little effect on cyclic AMP and cyclic GMP levels. Also, intravenously administered NP-313 dose-dependently inhibited the thrombus occlusion of the irradiated mesenteric vessels of fluorescein-pretreated mice.
CONCLUSIONS AND IMPLICATIONS
Taken together, these results indicate that NP-313 exerts its antithrombotic activity through dual inhibition of thromboxane A2 synthesis and Ca2+ influx through SOCC.
Keywords: antiplatelet, naphthoquinone, store-operated calcium channel, thromboxane A2 synthesis
Introduction
Myocardial infarction and ischaemic stroke are the leading causes of morbidity and mortality. The crucial role of platelet activation in thrombogenesis is well known. Antiplatelet agents have been developed as potential therapies for both the treatment and prevention of cardiovascular diseases (Bhatt and Topol, 2003; Jackson and Schoenwaelder, 2003). Upon vessel injury, platelets rapidly adhere to the newly exposed subendothelial matrix, such as collagen and von Willebrand factor, and undergo shape change, spreading and release of both adenosine 5-diphosphate (ADP) and thromboxane A2 (TXA2), reinforcing platelet adhesion and aggregation (Walsh, 2004).
The major intracellular stimulus involved in platelet aggregation is an increase in cytosolic free calcium concentration (Rink and Sage, 1990). As in most non-excitable cells, the increase in platelet [Ca2+]i induced by various agonists involves both influx of extracellular calcium through plasma membrane calcium channels and mobilization of intracellular calcium from the dense tubular system (Berridge et al., 2003; Berridge, 2004). The key elements involved in Ca2+ signalling include the activation of surface receptors that lead to the stimulation of phospholipase C (PLC), resulting in the hydrolysis of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to release inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds to the inositol 1,4,5-triphosphate receptors (IP3R) on intracellular stores, releasing Ca2+, and the consequential depletion of Ca2+ stored within the dense tubular system serves as the primary trigger for activation of store-operated Ca2+ channels (SOCC), which mediate Ca2+ entry (Putney et al., 2001). Besides store-operated Ca2+ entry (SOCE), Hassock et al. (2002) have suggested that another SOCE-independent mechanism also contributes to Ca2+ entry in platelets and is activated by DAG. It has been reported that agents with inhibitory effects on the cytosolic Ca2+ mobilization in platelets may suppress platelet aggregation and thrombus growth (Kang et al., 2001; Jin et al., 2004; 2005b; Lee et al., 2005), whereas few studies have further investigated whether these agents have a differential effect on the Ca2+ channels involved in modulating intracellular Ca2+ mobilization in human platelets.
The stimulation of platelets with different stimuli results in early activation of Ca2+-dependent cytosolic phospholipase A2 (PLA2), which hydrolyzes membrane phospholipids leading to arachidonic acid (AA) release. The liberated AA is metabolized via the cyclooxygenase (COX) pathway to form prostaglandin endoperoxide, PGH2, which sequentially, by the action of prostacyclin synthase and TXA2 synthase, is converted to prostaglandins (Walsh, 2004; Jin et al., 2005b) and TXA2 (Arita et al., 1989) respectively.
The important role of TXA2 synthesis in activation of platelets is underlined by the well-known clinical efficacy of aspirin related to its COX enzyme inhibition. Nevertheless, there is evidence indicating that there are subpopulations that do not respond to the antithrombotic action of aspirin (Szczeklik et al., 2005). Thus, the interest in developing other antithrombotic drugs such as TXA2 modulators raised (Dogne et al., 2004; 2006).
Compounds with the chemical structure of a 1,4-naphthoquinone backbone have been shown to have a wide variety of pharmacological effects including anticancer and antiplatelet activities (Rodriguez et al., 1995; Lien et al., 2002; Verma, 2006). In the present study, a series of 2,3-disubstituted 1,4-naphthoquinones were synthesized and tested for their inhibitory activities on platelet aggregation. Among them, 2-acetylamino-3-chloro-1,4-naphthoquinone (NP-313), a newly synthesized naphthoquinone derivative, was found to possess differential inhibitory effect on platelet aggregation induced by various stimulators. We investigated its antiplatelet effects and found that in addition to having inhibitory activity on TXA2 synthesis, it also showed an inhibitory effect on SOCC involved in modulating intracellular Ca2+ mobilization. Furthermore, i.v. administration of NP-313 dose-dependently protected mice against platelet plug formation with only a slight effect on haemostasis.
Methods
Reagents and animals
NP-313 (2-acetylamino-3-chloro-1,4-naphthoquinone, molecular weight, 249 Da, Figure 1) was synthesized and provided by Dr Jin-Cherng Lien and Sheng-Chu Kuo (China Medical University, Taichung, Taiwan), and its purity (>95%) was confirmed by 1H-NMR analysis. Collagen (bovine tendon type I), thrombin, 4-bromo-A23187, thapsigargin, or 1-oleoyl-2-acetyl-sn-glycerol (OAG), indomethacin, fluorescein sodium, Fura-2 acetoxymethyl ester (Fura 2-AM), BSA, PGE1, nitroglycerin (NTG), dimethyl sulphoxide (DMSO), heparin, ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetra acetic acid (EGTA) and ethylene diamine tetraacetic acid (EDTA), creatine phosphate (CP), creatine phosphokinase (CPK) and phorbol-12-myristate-13-acetate (PMA) were purchased from Sigma Chem. (St. Louis, MO, USA). AA, PGH2 and cPLA2 assay kit was from Cayman Chemical (Ann Arbor, MI, USA). Fluorescein isothiocyanate–labelled monoclonal antibody to P-selectin (fluorescein isothiocyanate (FITC)-labelled anti-CD62P) was obtained from Becton-Dickson (Franklin Lakes, NJ, USA). Cyclic AMP (cAMP), cyclic GMP (cGMP) and TXB2 assay kit were purchased from Amersham (Buckinghamshire, HP, UK). Lactate dehydrogenase (LDH) assay kit was purchased from Randox (Crumlin, Co. Antrim, UK). Drug and channel nomenclature conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2009).
Figure 1.
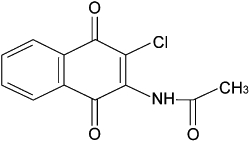
Chemical structure of NP-313, 2-acetylamino-3-chloro-1,4-naphthoquinone.
Male ICR mice were used in animal studies. The animals were maintained on a 12 h light/dark cycle under controlled temperature (20 ± 1°C) and humidity (55 ± 5%). Animals were given continuous access to food and water. All procedures involving animal experiments were approved by the Institutional Animal Care and Use Committee at College of Medicine, National Taiwan University.
Preparation of washed human platelets and platelet-rich plasma
Human platelet suspension was prepared according to the method described previously (Mustard et al., 1972). Blood samples, freshly obtained from healthy volunteers who had taken no medications during the preceding 2 weeks, were treated with acid citrate/dextrose in a volume ratio of 9:1. After centrifugation at 120×g, 25°C for 9 min, platelet-rich plasma (PRP) was transferred into another plastic tube and incubated with heparin (6.4 U·mL−1) as well as PGE1 (1 µM). Platelets were spun down by centrifugation at 500×g, 25°C for 8 min and subsequently washed twice with Tyrode's solution (NaH2PO4, 0.4 mM; NaCl, 136.9 mM; KCl, 11.9 mM; NaHCO3, 11.9 mM; CaCl2, 2 mM; MgCl2, 2.1 mM; glucose, 11.1 mM; BSA, 3.5 mg·mL−1; pH 7.35−7.4). The washed platelets were finally suspended in Tyrode's solution, and the platelet count was adjusted to 3.75 × 108 platelets·mL−1. For PRP preparation, whole blood was anticoagulated with sodium citrate (3.8%, w/v) in a volume ratio of 9:1 and centrifuged at 150×g, 25°C for 9 min.
Measurement of platelet aggregation
Platelet aggregation was measured turbidimetrically with a Lumi-aggregometer (Payton Scientific, Buffalo, NY, USA) at 37°C while being stirred (900 rpm) (Born and Cross, 1963). Washed human platelet suspension was prewarmed to 37°C for 2 min, and NP-313 was added 3 min before the addition of platelet aggregation activator. Platelet aggregation, measured as the change in light transmission, was recorded for 8 min after the addition of aggregation agonist. The extent of inhibition of platelet aggregation is expressed as percentage of inhibition using the following equation: X (%) = (1 –B/A) × 100%, where A is the maximum aggregation rate of vehicle-treated platelets, and B is the maximum aggregation of sample-treated platelets.
Flow cytometric analysis of P-selectin expression
The washed platelet suspension (3 × 108 mL−1) containing tirofiban (100 ng·mL−1) was preincubated with NP-313 for 3 min at 37°C and activated with the platelet activators for 10 min at 37°C. Then the sample was further incubated with FITC-labelled anti-CD62P in the dark for 15 min at room temperature. The platelet suspension was immediately assayed by fluorescence-activated cell sorter (FACScan System, Becton-Dickinson, San Jose, CA, USA) using excitation and emission wavelength of 488 and 525 nm respectively. Data were collected from 10 000 platelets per experimental group. The level of P-selectin expression was expressed as mean fluorescence intensity.
Measurement of TXB2 formation
This was performed according to a previously described method (Chang et al., 1998). The platelet suspension (3 × 108 mL−1) was incubated with NP-313 or DMSO for 3 min and then treated with platelet activators. At 6 min after the addition of agonists, indomethacin (50 µM) and EDTA (2 mM) were added to terminate the reaction. The platelet suspensions were centrifuged at 14 000×g for 2 min, and the TXB2 concentrations were measured using a competitive enzyme immunoassay (EIA) kit according to the instructions of the manufacturer.
TXA2 synthase activity assay
The TXA2 synthase activity was assayed as previously described (Son et al., 2004). Briefly, indomethacin (50 µM)-pretreated platelet suspension was incubated with NP-313 imidazole or DMSO at 37°C for 3 min prior to the addition of 5 µM PGH2. At 5 min after the addition of PGH2, the incubation was terminated by the addition of cooling EGTA (2 mM) and centrifuged at 12 000×g at 4°C for 4 min. The amount of TXB2 in the supernatant was assayed by a commercial EIA kit according to the manufacturer's instructions. TXA2 synthase activity is reflected by the production of TXB2.
Biochemical assays of COX-1, PLA2, PLC and PKCα activities
These enzymatic assays were performed by MDS Pharma Services utilizing standard protocols. Briefly, COX-1 (human platelet) activity was EIA quantified by measuring the PGE2 level converted from AA. PLA2 activity (pig pancreas) was measured by quantifying [14C]-oleate release from 1-palmitoyl-2-[1-14C] oleoyl-l-3-phosphatidylcholine. PLC activity was determined by measuring the chromogen of phosphatidylcholine release from 1, 2-dihexanoyl sn-glycerol-3-phosphocholine. PKCα activity was measured as phosphorylation of histone ([P32]-histone) stimulated by diacylglycerol in the presence of phosphatidylserine, Mg2+ and Ca2+.
Estimation of cAMP and cGMP formation
Platelet suspension (3 × 108 mL−1) was warmed to 37°C for 1 min, then NP-313, PGE1 or NTG was added and incubated for 2 min at 37°C. Incubation was stopped by the addition of 10 mM EDTA and followed by boiling the mixture for 5 min. After cooling to 4°C, the precipitated protein was collected as sediment after centrifugation. The supernatant was used to determine the cAMP and cGMP contents by EIA kits following acetylation of the samples as described by the manufacturer.
Measurement of intracellular Ca2+mobilization
After centrifuging platelet-rich plasma at 500×g, 25°C for 8 min, isolated platelets were resuspended in Ca2+-free Tyrode's solution. Platelet suspension was protected from light and incubated with Fura 2-AM (5 µM) at 37°C for 30 min. Human platelets were then prepared as previously described. The platelet count was adjusted to 3 × 108 mL−1 platelets. Just before [Ca2+]i measurements were performed, Ca2+ was added back to platelets to a final concentration of 1.0 mM, then NP-313, collagen, thrombin, AA, A23187, thapsigargin or OAG was added. The rise in [Ca2+]i was measured using a F4500 Fluorometer (Hitachi, Japan) at excitation wavelengths of 340 and 380 nm, and an emission wavelength of 500 nm. Fluorescence was calibrated with lysed platelets (0.1% Triton X-100) in the absence and presence of 10 mM EGTA in each run to obtain maximum and minimum fluorescence values, and [Ca2+]i was calculated from the fluorescence, using 224 nM as the Ca2+–Fura 2 dissociation constant.
LDH assay
The LDH activity released was measured spectrophotometrically by recording the decrease in the optical density of beta-nicotinamide adenine dinucleotide at 340 nm, as previously described (Jin et al., 2004). NP-313 (80 µM) was incubated with platelet suspension for 60 min and then centrifuged for 4 min at 1312×g. The LDH activity in the platelet suspension and cellular LDH activity from platelets, which was lysed with 1% Triton-X 100, were determined. Total LDH activity was the summation of both released and cellular LDH activities. The released LDH activity was expressed as a percentage of total LDH activity.
Fluorescein sodium–induced platelet thrombus formation in mesenteric venules of mice
Platelet plug formation in mesenteric microvessels was performed according to a previously described method with modifications (Chang et al., 1998). In brief, male ICR mice (12–14 g) were anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.), and then the fluorescein sodium (12.5 mg·kg−1) was i.v. injected into the lateral tail vein of the mouse. Venules with diameters of 30–40 µm were selected to be irradiated to produce a microthrombus. In the epi-illumination system, the area of irradiation (wavelength above 520 nm) was approximately 50 µm in diameter on the focal plane. After the operation, the mouse was i.v. injected with phosphate-buffered saline (vehicle), aspirin (100 or 200 mg·kg−1) or various doses of NP-313 through the other lateral tail vein. Five minutes after administration of these drugs, the irradiation by filtered light was started, and the time to occlusion (TTO, upon cessation of blood flow) was recorded.
Tail bleeding time in mice
The bleeding time was measured by use of transection of the tail in a mouse model (Dejana et al., 1982). Various doses of NP-313 or vehicle were given i.v. 5 min prior to tail cutting.
Ex vivo mouse platelet aggregation
Male ICR mice (20–30 g) were treated i.v. with various doses of NP-313 or vehicle before being anaesthetizied with sodium pentobarbital (50 mg·kg−1 i.p.). Blood samples were collected at 5 min by intracardiac puncture after drug treatment. PRP was obtained by centrifuging the blood sample at 180×g for 4 min. PPP was obtained by centrifugation of remaining blood at 2000×g for 4 min. PRP was adjusted to 3.75 × 108 platelets·mL−1 with PPP. Platelet aggregation was measured as described above.
Statistical analysis
The experimental results are expressed as the means ± SEM. The statistical comparisons were made by anova (following a paired Students't-test), and differences were considered to be significant at P-value < 0.05.
Results
Effect of NP-313 on platelet aggregation
As shown in Figure 2, NP-313 inhibited collagen (10 µg·mL−1), AA (200 µM), thapsigargin (0.1 µM), thrombin(0.1 U·mL−1) and A23187 (8 µM) induced platelet aggregation of washed human platelets in a concentration-dependent manner, with the following IC50 values: 1.7 ± 0.1, 2.4 ± 0.2, 3.8 ± 0.2, 7.7 ± 0.2 and 20.0 ± 0.3 µM respectively. However, in response to thrombin and A23187-induced platelet aggregation, the maximal degree of inhibition with NP-313 reached around 80% and 60%, respectively.
Figure 2.
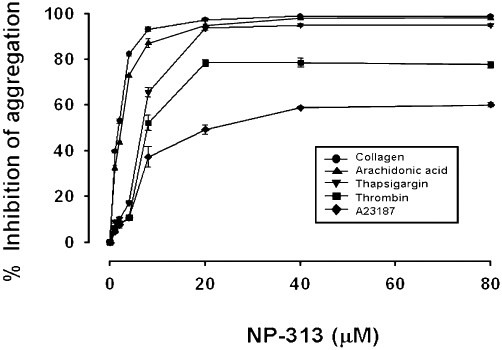
Effect of NP- 313 on aggregation of washed human platelets. Washed human platelets were incubated with DMSO (vehicle control) or NP-313 at 37°C for 3 min, and then collagen (10 µg·mL−1), arachidonic acid (AA; 200 µM), thapsigargin (0.1 µM), thrombin (0.1 U·mL−1) or A23187 (8 µM) was added to trigger platelet aggregation. Data are presented as the mean ± SEM (n = 5).
Effect of NP-313 on collagen-, thrombin- and AA-induced granule secretion and TXB2 formation in platelets
We examined whether NP-313 could inhibit the processes of platelet activation, such as granule secretion, and TXB2 formation. NP-313 concentration-dependently inhibited P-selectin expression (Figure 3A), and also TXB2 generation induced by collagen or thrombin (Figure 3B).
Figure 3.
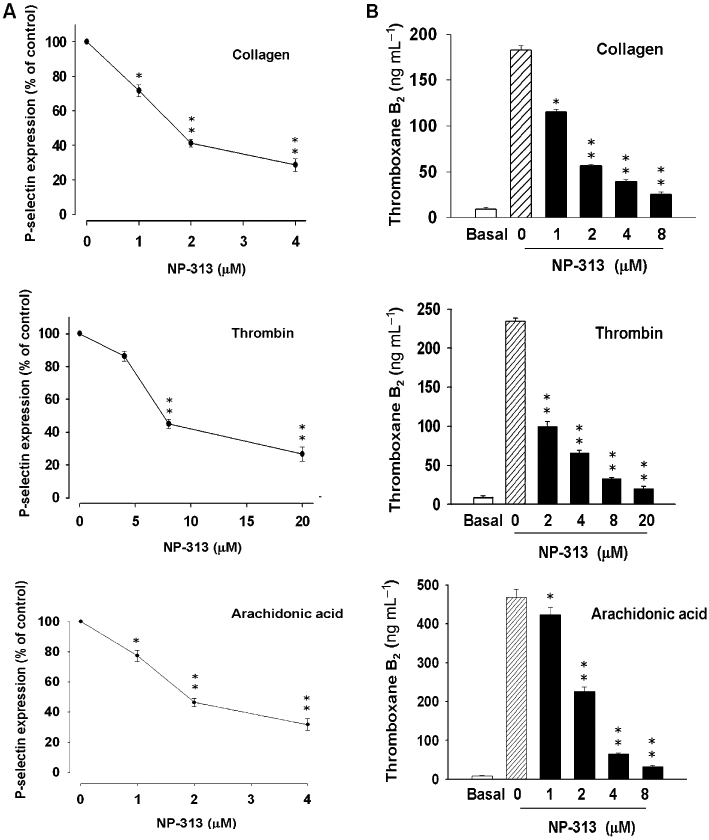
Effect of NP-313 on collagen-, thrombin- and arachidonic acid (AA)-induced P-selectin expression and thromboxane B2 (TXB2) formation in human platelets. (A) Washed human platelets were preincubated with DMSO (vehicle control) or NP-313 for 3 min and then treated with collagen (10 µg·mL−1), thrombin (0.1 U·mL−1) or AA (200 µM) in the presence of FITC-conjugated anti-CD62P for 15 min at room temperature. Data are presented as the means ± SEM (n = 3). *P < 0.05 and **P < 0.01 as compared with the corresponding stimulus control, one-way anova (Dunnett's post hoc test). (B) Platelet suspensions were preincubated with DMSO (vehicle control) or NP-313 for 3 min at 37°C, and then collagen (10 µg·mL−1), thrombin (0.1 U·mL−1) or AA (200 µM) was added to trigger TXB2 formation. TXB2 formation was terminated by the addition of EDTA (2 mM) and indomethacin (50 µM). *P < 0.05 and **P < 0.01 as compared with the corresponding stimulus control, one-way anova (Dunnett's post hoc test).
Effect of NP-313 on COX-1, TXA2 synthase, phospholipase PLA2 and PLC activity
It has been reported that some 1,4-naphthoquinone derivatives inhibit TXA2 synthase (Jin et al., 2005a,b;). NP-313 inhibited COX-1 and TXA2 synthase concentration-dependently with IC50 values of 1.5 ± 0.4 and 3.9 ± 0.3 µM respectively (Figure 4A,B). Consistent with these data, NP-313 concentration-dependently inhibited TXB2 formation in platelets stimulated by collagen, thrombin and AA (Figure 3B). In contrast, NP-313 at 10 µM had little inhibitory effect on cPLA2 and PLC activities (data not shown).
Figure 4.
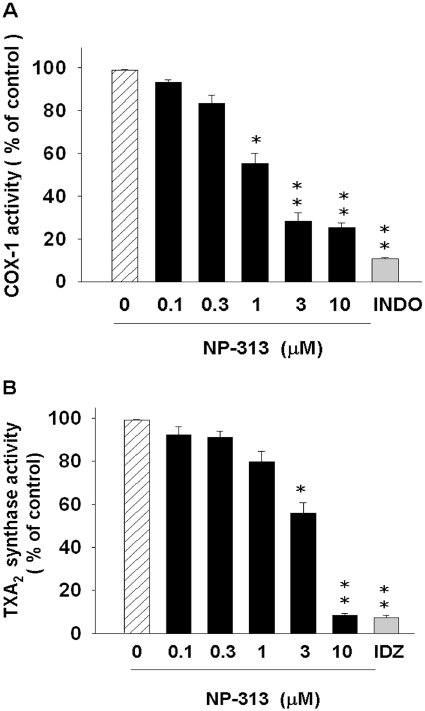
Effect of NP-313 on cyclooxygenase-1 (COX-1) and thromboxane A2 (TXA2) synthase activities. (A) The human platelet COX-1 in a reaction buffer was treated with NP-313, indomethacin (INDO; 0.3 µM) or DMSO (vehicle control) at 37°C for 15 min, and then 100 µM AA was added for 15 min. COX enzyme activities were reflected by the amount of PGE2 produced. Data are expressed as % of PGE2 formed in the presence of assay buffer, and data represent the mean ± SEM (n = 3). **P < 0.01 as compared with the vehicle control, one-way anova (Dunnett's post hoc test). (B) After preincubation with indomethacin (50 µM) at 37°C for 2 min, platelet suspensions containing NP-313, imidazole (IDZ; 50 mM) or DMSO (vehicle control) were further incubated for 3 min, and then 5 µM PGH2 was added for 5 min. Data are expressed as % of the TXB2 formed in the presence of assay buffer, and data represent the mean ± SEM (n = 3) *P < 0.05 and **P < 0.01 as compared with the vehicle control, one-way anova (Dunnett's post hoc test).
Effect of NP-313 on cyclic nucleotide levels in human platelets
1,4-Naphthoquinone derivatives also have been shown to inhibit platelet aggregation by suppression of intracellular calcium mobilization; this is mediated by the inhibition of IP3 production or elevation of cAMP level (Jin et al., 2004; 2005b;). To assess whether the action of NP-313 was due to elevation of intracellular levels of cAMP and/or cGMP, two major inhibitory messengers in regulating platelet aggregation (Schwarz et al., 2001), the effect of NP-313 on cyclic nucleotide levels in platelets was examined. In washed human platelets, the addition of NTG and PGE1 significantly increased cGMP formation and cAMP respectively; however, NP-313 affected neither cAMP levels nor cGMP levels as compared with that of resting platelets (Figure 5).
Figure 5.
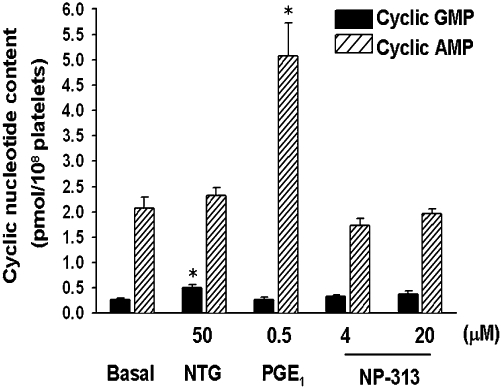
Effects of NP- 313 on platelet cGMP and cAMP levels. Washed platelets were incubated at 37°C for 2 min with NP-313, PGE1 or NTG. The reaction was stopped, platelets were then pelleted, and the supernatants were assayed for cGMP and cAMP by enzyme immunoassay (EIA). Values are presented as mean ± SEM (n = 4). *P < 0.05 as compared with the basal control, one-way anova (Dunnett's post hoc test).
Effect of NP-313 on collagen-, thrombin-, AA- and A23187 –induced intracellular Ca2+ mobilization in human platelets
Treatment of the Fura-2-loaded platelet suspension with NP-313 inhibited collagen-, thrombin-, AA- and A23187-evoked increase in [Ca2+]i in a concentration-dependent manner (Figure 6). NP-313 almost completely inhibited the elevation of the [Ca2+]i in response to collagen and AA. Nevertheless, in response to thrombin- and A23187-evoked increase of [Ca2 +]i, the residual 20% and 40% of [Ca2+]i mobilization, respectively, were unaffected by NP-313, even used at 80 µM.
Figure 6.
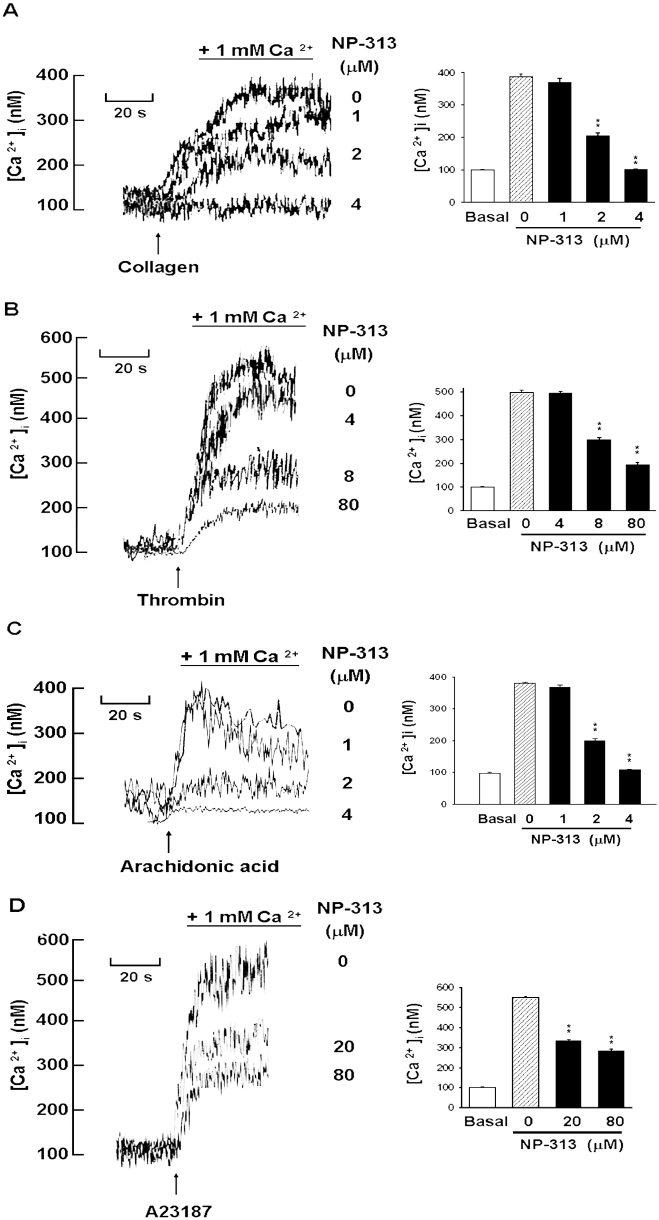
Effect of NP-313 on collagen-, thrombin- and A23187–induced intracellular [Ca2+]i mobilization in human platelets. Calcium (1 mM) was added to the platelet suspension 30 s before data collection started (zero time). Various concentrations of NP-313 were added to the platelets at 10 s, and collagen (10 µg·mL−1), thrombin (0.1 U·mL−1) or A23187 (8 µM) was added at 30 s. The traces shown in left panel are from a representative experiment; similar results were obtained from three separate experiments, and average data are presented in the right panel (A, B and C). *P < 0.05 as compared with the corresponding stimulus control, one-way anova (Dunnett's post hoc test).
Effect of NP-313 on thrombin- and A23187-induced Ca2+ release from internal stores and influx of extracellular Ca2+ in human platelets
Cytosolic Ca2+ elevation occurs as a consequence of release of Ca2+ from intracellular stores and influx from the extracellular medium. In the following experiments, Fura-2-loaded platelets were preincubated in the buffer containing CP (10 mM), CPK (1 U·mL−1) and indomethacin (100 µM), to exclude the effects of ADP and TXA2 on [Ca2+]i elevation during agonist-induced stimulation. The data in Figure 6A showed the effect of NP-313 on intracellular Ca2+ mobilization and Ca2+ influx mediated by thrombin. As platelets were incubated in Ca2+-free medium containing 1 mM EGTA, and followed by the addition of thrombin, the increase in [Ca2+]i was substantially smaller compared with Ca2+ influx (Figure 6, left). NP-313, even used at 80 µM, failed to inhibit thrombin-induced release of intracellular stored Ca2+. Next, after agonist-mediated Ca2+ release from intracellular store, the effect of NP-313 on Ca2+ influx was assessed by the addition of 2 mM Ca2+. In contrast, the increase in [Ca2+]i induced by thrombin in the presence of extracellular Ca2+ was concentration-dependently inhibited by NP-313 (Figure 6A, right).These results indicate NP-313 mainly prevents the entry of Ca2+ into the cytoplasm but has little influence on Ca2+ mobilization from the dense tubular system. In addition, the influence of NP-313 on A23187-evoked Ca2+ mobilization was similar to that observed with thrombin (Figure 6B). It is known that A23187, a Ca2+ ionophore, mobilizes Ca2+ across membranes and directly increases [Ca2+]i. Furthermore, the residual [Ca2+]i mobilization observed both in thrombin- and A23187-induced Ca2+ influx were unaffected by NP-313 even at 80 µM.
Effect of NP-313 on thrombin- and A23187-induced Ca2+ release from internal stores and influx of extracellular Ca2+ in human platelets
Cytosolic Ca2+ elevation occurs as a consequence of release of Ca2+ from intracellular stores and influx from the extracellular medium. In the following experiments, Fura-2-loaded platelets were preincubated in the buffer containing CP (10 mM), CPK (1 U·mL−1) and indomethacin (100 µM), to exclude the effects of ADP and TXA2 on [Ca2+]i elevation during agonist-induced stimulation. The data in Figure 7A show the effect of NP-313 on intracellular Ca2+ mobilization and Ca2+ influx mediated by thrombin. As platelets were incubated in Ca2+-free medium containing 1 mM EGTA, and followed by the addition of thrombin, the increase of [Ca2+]i was substantially smaller compared with Ca2+ influx (Figure 7A, left). NP-313, even used at 80 µM, failed to inhibit thrombin-induced release of intracellular stored Ca2+. Next, after agonist-mediated Ca2+ release from intracellular store, the effect of NP-313 on Ca2+ influx was assessed by the addition of 2 mM Ca2+. In contrast, the increase in [Ca2+]i by thrombin in the presence of extracellular Ca2+ was concentration-dependently inhibited by NP-313 (Figure 7A, right). These results indicate NP-313 mainly prevents the entry of Ca2+ into the cytoplasm but has little influence on Ca2+ mobilization from the dense tubular system. Furthermore, the influence of NP-313 on A23187-evoked Ca2+ mobilization was similar to that observed with thrombin (Figure 7B). Nevertheless, A23187 can diffuse through membranes and directly increase [Ca2+]i from intracellular stores without increasing IP3 and through extracellular Ca2+-influx, which is also partly due to SOCE activation after Ca2+ depletion of ER (Fuse et al., 2001; Kunzelmann-Marche et al., 2001).
Figure 7.
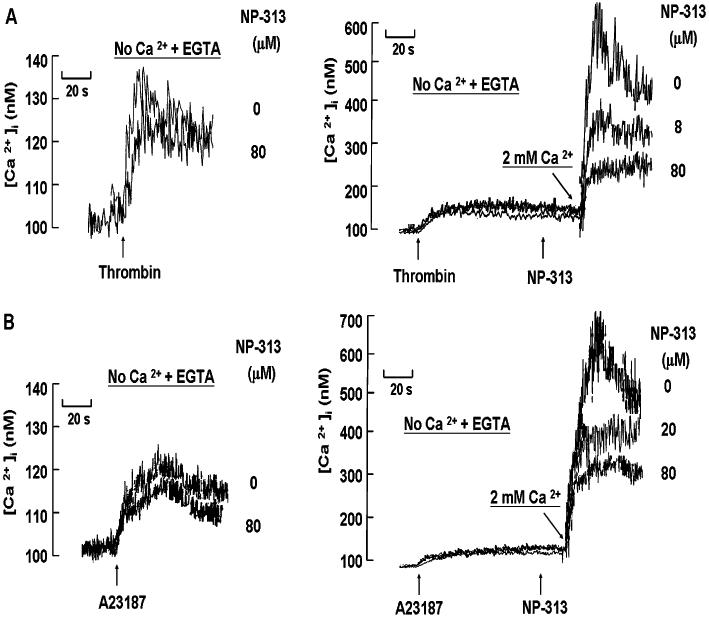
Effect of NP-313 on thrombin- and A23187-induced Ca2+ release from internal stores and influx of extracellular Ca2+ in human platelets. Fura-2-loaded platelets were suspended in Tyrode's buffer containing creatining phosphate (CP; 10 mM), creatining phosphatase (CPK; 1 U·mL−1) and indomethacin (100 µM). Effect of NP-313 on the release of the internally stored calcium in thrombin (0.1 U·mL−1; A, left) and A23187 (8 µM; B, left) stimulated platelets. External calcium was not added to the platelet suspension; 1 mM EGTA was added 30 s prior to data collection (zero time). After 10 s, various concentrations of NP-313 were added; 20 s later, thrombin or A23187 was added. Effect of NP-313 on calcium influx initiated after mobilization of intracellular Ca2+ by thrombin (0.1 U·mL−1; A, right) and A23187 (8 µM; B, right). Platelets were preincubated in the absence of extracellular Ca2+ and in the presence of 1 mM EGTA. Thrombin or A23187 was added at 10 s, and various concentrations of NP-313 were added to the platelets at 90 s. At 110 s, when [Ca2+]i was declining because of depletion of intracellular stores, 2 mM Ca2+ was added to the platelets. A representative of three experiments is shown. EGTA, ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetra acetic acid.
Effect of NP-313 on thapsigargin- and OAG-induced Ca2+ entry
There are mainly two Ca2+ entry channels on platelet plasma membrane, that is SOCC and SOCC-independent channel. Studies were carried out to determine if NP-313 had an impact on Ca2+ entry mediated by these two channels. It is well known that thapsigargin mediates Ca2+ influx through SOCC by inhibiting the sarcoplasmic/ endoplasmic reticulum Ca2+-ATPase (SERCA) pump without increasing the level of IP3, thus promoting a loss of Ca2+ via a ‘leak’ process in the ER (Pozzan et al., 1994; Sage, 1997). This Ca2+-depleted condition of the ER then causes an increase in Ca2+ influx through SOCC. Figure 8A shows that NP-313 markedly elicited a concentration-dependent inhibition of thapsigargin-mediated Ca2+ mobilization. However, because NP-313 had no effect on the thapsigargin-evoked release of Ca2+ from the intracellular stores in the absence of extracellular Ca2+ (data not shown), the effect of NP-313 to inhibit the Ca2+ influx was most probably caused by a more direct inhibitory effect on SOCC itself and not indirectly by preventing the loss of Ca2+ from the ER. In contrast, NP-313, at concentrations up to 80 µM, had no effect on the moderate and slow [Ca2+]i increase evoked by a DAG analogue, OAG (Figure 8B), which was reported to activate store-independent Ca2+ entry (Hassock et al., 2002). Thus, NP-313 is considered to selectively inhibit Ca2+ influx mediated by SOCC.
Figure 8.
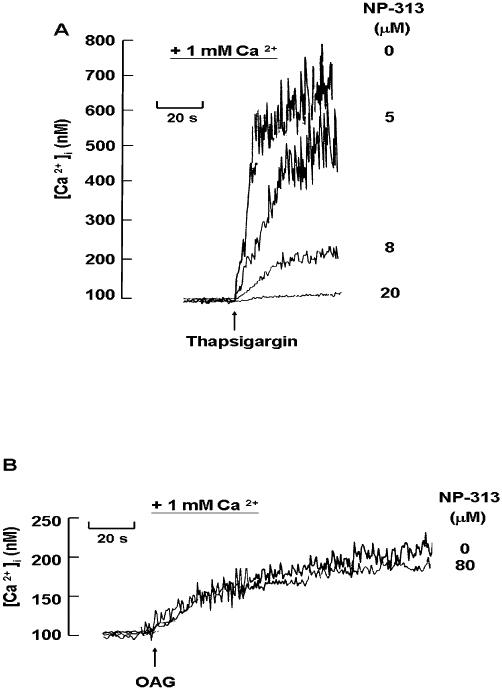
Effect of NP-313 on thapsigargin- and OAG- induced Ca2+ entry. Fura-2-loaded platelets were suspended in Tyrode's buffer containing creatine phosphate (CP; 10 mM), creatine phosphokinase (CPK; 1 U·mL−1) and indomethacin (100 µM). Calcium (1 mM) was added to the platelets 30 s before data collection was started plus various concentrations of NP-313 were added at 10 s, and 0.1 µM thapsigargin (A) or 60 µM OAG (B) was added at 30 s. A representative of three experiments is shown. OAG, 1-oleoyl-2-acetyl-sn-glycerol.
Lack of cytotoxic effect of NP-313 on human platelets
No significant increase in LDH release was observed with NP-313 (80 µM) and vehicle-treated platelets, even when the incubation time of NP-313 with platelets was prolonged to 30 min (about 2.7% vs. 2.6% release), suggesting that it neither affects platelet permeabilization nor induces platelet cytolysis at the concentration used.
Effect of NP-313 on in vivo thrombosis and bleeding time in a mouse model
It has been previously demonstrated that administration of fluorescein sodium and the subsequent irradiation of mesenteric venules induces the formation of marked mixed thrombi, composed of activated platelets and fibrin clots (Chang et al., 1998). Figure 8A shows that the effects of NP-313 on average TTO. In the vehicle-treated group, i.v. application of 12.5 µg·g−1 fluorescein sodium induced an average TTO of 95.0 ± 7.5 s (n = 6). However, after i.v. administration at 4, 8 and 16 µg·g−1, NP-313 significantly prolonged the average TTO to 127.0 ± 12.1, 188.3 ± 25.7 and 242.5 ± 16.4 s respectively (n = 6; P < 0.01 for each, one-way anova, Dunnett's post hoc test).
As shown in Figure 9B, NP-313 did not affect the bleeding time in mice compared with vehicle-treated mice after i.v. administration at effective antithrombotic dose of 4 and 8 µg·g−1. In contrast, aspirin caused a marked prolongation of tail bleeding time when an effective antithrombotic dose of 200 µg·g−1 was administered i.v. However, NP-313 slightly prolonged bleeding time when administered at 16 µg·g−1. Furthermore, we evaluated the ex vivo antiplatelet action of NP-313 on PRP. As shown in Figure 9C, upon i.v. administration of NP-313 at 4, 8 and 16 µg·g−1 for mice, NP-313 inhibited ex vivo platelet aggregation of PRP caused by collagen (10 µg·mL−1) 5 min after drug administration.
Figure 9.
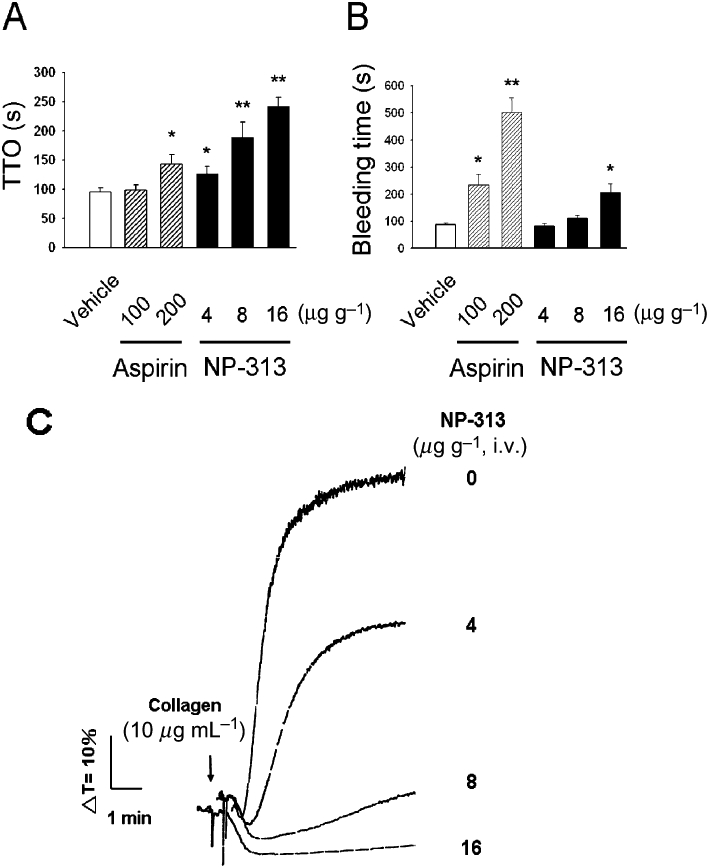
Effect of NP-313 on in vivo thrombosis and bleeding time in a mouse model. (A) Effect of NP-313 on the time to occlusion (TTO) measured 5 min after its i.v. administration upon light irradiation of mesenteric venules of mice pretreated with fluorescein sodium. Data are presented as the mean ± SEM (n = 6). (B) Effect of NP-313 on tail bleeding time of mice measured 5 min after i.v. administration. Data are presented as the mean ± SEM (n = 6). *P < 0.05 and **P < 0.01 as compared with the vehicle control, one-way anova (Dunnett's post hoc test). (C) Effect of NP-313 on ex vivo mouse platelet aggregation of PRP induced by collagen (10 µg·mL−1). Aggregation traces shown are representative of three independent experiments.
Discussion
The present study has shown that NP-313 inhibits collagen- and thrombin-induced platelet aggregation and platelet activation, such as α-granule secretion, TXA2 formation and intracellular Ca2+ mobilization in a concentration-dependent manner. However, NP-313 preferentially inhibited collagen-induced platelet aggregation as compared that induced by thrombin (IC50, 1.7 vs. 7.7 µM, respectively). On being stimulated by collagen or thrombin, platelets synthesize and release TXA2, which amplifies platelet aggregation (Jackson et al., 2003). It is known that collagen and AA depend on the formation of TXA2 in triggering platelet aggregation (Parise et al., 1984; Atkinson et al., 2003). On the other hand, collagen activates platelets through a tyrosine kinase-based signalling pathway and sequential activation of PLCγ2, leading to intracellular Ca2+ mobilization (Jackson et al., 2003), and these processes are exclusively dependent on glycoprotein VI activation. However, GPVI alone mediates only a part of platelet aggregation, and full-cell response and aggregation secondary mediators (like TXA2) are needed (Atkinson et al., 2003). Enzymatic assays showed that NP-313 exhibits a potent inhibitory effect on COX-1 and TXA2 synthase activities, while it has little effect on PLA2 or PLC activity. NP-313 also concentration-dependently inhibited platelet aggregation and TXB2 generation induced by AA. These results suggest that NP-313 produces its effect on TXA2 synthesis mainly by inhibiting both COX-1 and TXA2 synthase activities, which may contribute to the inhibition of platelet aggregation with collagen. Nevertheless, unlike the TXA2 dependency of aggregation with collagen (Nieswandt and Watson, 2003), thrombin induces intracellular Ca2+ mobilization and platelet aggregation independent of the TXA2 pathway (Freedman, 2005), in line with our results that the majority of the platelet responses were unaffected even in the presence of ADP scavenger and COX inhibitors (Figure 7A). Furthermore, NP-313, below or at a concentration of 4 µM, abolished thrombin-mediated TXB2 formation while not inhibiting thrombin-induced PLC-dependent intracellular Ca2+ mobilization (Figure 3B, right panel and Figure 6B).
The mechanism of calcium release from the IP3-sensitive internal stores is well characterized. NP-313 has no effect on intracellular Ca2+ mobilization by thrombin in the absence of extracellular Ca2+, indicating that it does not interfere with the intracellular IP3R–Ca2+ channel activity (Figure 7A, left panel). Ca2+ entry is thought to occur predominantly as a consequence of stored Ca2+ depletion and has been referred to as SOCE or capacitative Ca2+ entry through SOCC (Putney et al., 2001). The identity of the SOCC in platelets remains elusive. Among them, Orai 1 has been found to be expressed in human platelets, and its function has been linked to thrombus formation in vitro as well as in vivo (Braun et al., 2009). Orai 1 belongs to a family of channels that have four putative transmembrane domains and contain the pore-forming subunits of SOCE channels. Orai 1–deficient platelets showed an almost absence of Ca2+ entry after store depletion by thapsigargin and much reduced Ca2+ entry when stimulated by agonists. On the other hand, activation of the SOCE-independent channel, transient receptor potential canonical (TRPC)6 with high expression in human platelets, by DAG and some of its metabolites also have been shown to contribute to the Ca2+ entry via G-protein–linked receptors (such as thrombin receptor) (Authi, 2009). NP-313 was found to inhibit the platelet aggregation caused by thapsigargin that bypasses receptor-mediated processes. Thapsigargin, a tool used to study SOCC, effectively elevates intracellular [Ca2+]i by inhibiting the calcium ATPase pump of the dense tubular system without increasing the level of IP3 (Pozzan et al., 1994). On the other hand, the DAG analogue OAG has been used to investigate SOCE-independent Ca2+ entry (TRPC)6 channel, attributable to direct activation of this channel independent of PKC (Hofmann et al., 1999). In the present study, thapsigargin-induced cytosolic Ca2+ mobilization was almost completely inhibited by NP-313 at a concentration of 20 µM (Figure 5), which has no influence on OAG-mediated intracellular Ca2+ mobilization in platelets (Figure 8A,B), suggesting that NP-313 selectively inhibits SOCE rather than blocking SOCE-independent channels. This may also explain why NP-313 inhibited thrombin-induced Ca2+ influx but did not affect the residual 30% of [Ca2+]i influx (Figure 7A, right panel). NP-313 preferentially inhibits collagen-induced platelet aggregation and cytosolic Ca2+-mobilization compared with these effects induced by thrombin, consistent with the recent observation that Orai 1–mediated Ca2+ entry is particularly essential for collagen receptor-, a GPVI-ITAM-, mediated cell activation (Authi, 2009). In addition, the inhibitory action of NP-313 on TXA2 synthesis was not only mediated by inhibiting both COX-1 and TXA2 synthase activities but also possibly by the inhibition of cytosolic free Ca2+. It is well known that upon stimulation of platelets, AA is liberated from the sn-2-position of membrane phospholipids through the action of cPLA2, activated by an increase in cytosolic free Ca2+ (McNicol and Shibou, 1998). However, the question remains open regarding whether NP-313 acts primarily on this SOCC or indirectly on some molecule that regulates SOCC. Furthermore, we found that NP-313 has an inhibitory action on PKCα, since NP-313 at 10 µM inhibited PKCα (90% inhibition), and PMA (300 nM)-induced platelet aggregation in a concentration-dependent manner. A complete inhibition was observed at 15 µM (data not shown). The inhibitory effect on PKCα may contribute to the observation that NP-313 also could partly inhibit thrombin-induced aggregation (Konopatskaya et al., 2009).
We further evaluated the antithrombotic efficacy/ side effect profile of NP-313 in a mouse model. Intravenous administration of NP-313 (4–8 µg·g−1) dose-dependently prevented thrombus formation caused by the irradiation of mesenteric vessel of the fluorescein sodium–pretreated mice. However, it did not significantly prolong bleeding time, implying that NP-313 preferentially inhibits thrombus formation with little effect on haemostasis. In addition, it was found that the antithrombotic activity of NP-313 was in parallel with ex vivo antiplatelet activity. Notably, it was shown that NP-313 is more effective at inducing an antithrombotic action than the antiplatelet agent, aspirin, which mainly affects only the TXA2 pathway of platelet activation. The promising antithrombotic efficacy of NP-313 may result from a dual inhibition of TXA2 synthesis and a suppression of Ca2+ influx through SOCC. It has been shown that mice deficient in Orai 1 are markedly protected systemically and arterially from ischaemia-induced thrombosis but have only a slightly prolonged bleeding time (Braun et al., 2009). However, further investigation is needed to address the precise mechanism of the antithrombotic effect of NP-313.
In conclusion, this study demonstrated that NP-313, a 1,4-naphthoquinone derivative, possesses the ability to inhibit both TXA2 synthesis and SOCE. This compound does not affect cyclic nucleotide levels or induce LDH release. With its distinct action on the Ca2+ channels involved in modulating intracellular Ca2+ mobilization, NP-313 may become a new pharmacological tool for investigating the signal transduction pathways involved in regulating [Ca2+]i through platelet SOCC. NP-313 also shows in vivo protection against thrombous formation with little effect on haemostasis, suggesting that it may be a potential candidate for development as an antithrombotic agent.
Acknowledgments
This work was supported by grants from the National Science Council, Taiwan and China Medical University (CMU93-PC-02) of Taiwan.
Glossary
Abbreviations
- AA
arachidonic acid
- CP
creatine phosphate
- CPK
creatine phosphokinase
- cPLA2
cytosolic phospholipase A2
- DAG
diacylglycerol
- EDTA
ethylene diamine tetraacetic acid
- EGTA
ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- Fura 2-AM
Fura-2 acetoxymethyl ester
- IP3
inositol 1,4,5-trisphosphate
- IP3R
inositol 1,4,5-triphosphate receptors
- LDH
lactate dehydrogenase
- NP-313
2-acetylamino-3-chloro-1,4-naphthoquinone
- NTG
nitroglycerin
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- PIP2
phospholipid phosphatidylinositol 4,5- bisphosphate
- PLA2
phospholipase A2
- PLC
phospholipase C
- PMA
phorbol-12-myristate-13-acetate
- PRP
platelet-rich plasma
- SOCC
store-operated Ca2+ channels
- SOCE
store-operated Ca2+ entry
- TXA2
thromboxane A2
Conflicts of interest
N/A.
Supporting Information
Teaching Materials; Figs 1–9 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita H, Nakano T, Hanasaki K. Thromboxane A2: its generation and role in platelet activation. Prog Lipid Res. 1989;28:273–301. doi: 10.1016/0163-7827(89)90002-7. [DOI] [PubMed] [Google Scholar]
- Atkinson BT, Jarvis GE, Watson SP. Activation of GPVI by collagen is regulated by alpha2beta1 and secondary mediators. J Thromb Haemost. 2003;1:1278–1287. doi: 10.1046/j.1538-7836.2003.00245.x. [DOI] [PubMed] [Google Scholar]
- Authi KS. Orai1: a channel to safer antithrombotic therapy. Blood. 2009;113:1872–1873. doi: 10.1182/blood-2008-11-185959. [DOI] [PubMed] [Google Scholar]
- Berridge M. Conformational coupling: a physiological calcium entry mechanism. Sci STKE. 2004;243:pe33. doi: 10.1126/stke.2432004pe33. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- Born GV, Cross MJ. The aggregation of blood platelets. J Physiol. 1963;168:178–195. doi: 10.1113/jphysiol.1963.sp007185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113:2056–2063. doi: 10.1182/blood-2008-07-171611. [DOI] [PubMed] [Google Scholar]
- Chang MC, Lin HK, Peng HC, Huang TF. Antithrombotic effect of crotalin, a platelet membrane glycoprotein Ib antagonist from venom of Crotalus atrox. Blood. 1998;91:1582–1589. [PubMed] [Google Scholar]
- Dejana E, Villa S, de Gaetano G. Bleeding time in rats: a comparison of different experimental conditions. Thromb Haemost. 1982;48:108–111. [PubMed] [Google Scholar]
- Dogne JM, de Leval X, Hanson J, Frederich M, Lambermont B, Ghuysen A, et al. New developments on thromboxane and prostacyclin modulators part I: thromboxane modulators. Curr Med Chem. 2004;11:1223–1241. doi: 10.2174/0929867043365260. [DOI] [PubMed] [Google Scholar]
- Dogne JM, Hanson J, de Leval X, Pratico D, Pace-Asciak CR, Drion P, et al. From the design to the clinical application of thromboxane modulators. Curr Pharm Des. 2006;12:903–923. doi: 10.2174/138161206776055921. [DOI] [PubMed] [Google Scholar]
- Freedman JE. Molecular regulation of platelet-dependent thrombosis. Circulation. 2005;112:2725–2734. doi: 10.1161/CIRCULATIONAHA.104.494468. [DOI] [PubMed] [Google Scholar]
- Fuse I, Higuchi W, Uesugi Y, Aizawa Y. Pathogenetic analysis of three cases with a bleeding disorder characterized by defective platelet aggregation induced by Ca2+ ionophores. Br J Haematol. 2001;112:603–608. doi: 10.1046/j.1365-2141.2001.02637.x. [DOI] [PubMed] [Google Scholar]
- Hassock SR, Zhu MX, Trost C, Flockerzi V, Authi KS. Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood. 2002;100:2801–2811. doi: 10.1182/blood-2002-03-0723. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM. Antiplatelet therapy: in search of the ‘magic bullet’. Nat Rev Drug Discov. 2003;2:775–789. doi: 10.1038/nrd1198. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Nesbitt WS, Kulkarni S. Signaling events underlying thrombus formation. J Thromb Haemost. 2003;1:1602–1612. doi: 10.1046/j.1538-7836.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- Jin YR, Hwang KA, Cho MR, Kim SY, Kim JH, Ryu CK, et al. Antiplatelet and antithrombotic activities of CP201, a newly synthesized 1,4-naphthoquinone derivative. Vascul Pharmacol. 2004;41:35–41. doi: 10.1016/j.vph.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Jin YR, Cho MR, Lee KS, Lee JJ, Lim Y, Han XH, et al. An antithrombotic agent, NQ301, inhibits thromboxane A2 receptor and synthase activity in rabbit platelets. Basic Clin Pharmacol Toxicol. 2005a;97:162–167. doi: 10.1111/j.1742-7843.2005.pto_973123.x. [DOI] [PubMed] [Google Scholar]
- Jin YR, Cho MR, Ryu CK, Chung JH, Yuk DY, Hong JT, et al. Antiplatelet activity of J78 (2-Chloro-3-[2′-bromo, 4′-fluoro-phenyl]-amino-8-hydroxy-1,4-naphthoquinone), an antithrombotic agent, is mediated by thromboxane (TX) A2 receptor blockade with TXA2 synthase inhibition and suppression of cytosolic Ca2+ mobilization. J Pharmacol Exp Ther. 2005b;312:214–219. doi: 10.1124/jpet.104.073718. [DOI] [PubMed] [Google Scholar]
- Kang WS, Chung KH, Chung JH, Lee JY, Park JB, Zhang YH, et al. Antiplatelet activity of green tea catechins is mediated by inhibition of cytoplasmic calcium increase. J Cardiovasc Pharmacol. 2001;38:875–884. doi: 10.1097/00005344-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans JM, Karim ZA, et al. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann-Marche C, Freyssinet JM, Martinez MC. Regulation of phosphatidylserine transbilayer redistribution by store-operated Ca2+ entry: role of actin cytoskeleton. J Biol Chem. 2001;276:5134–5139. doi: 10.1074/jbc.M007924200. [DOI] [PubMed] [Google Scholar]
- Lee GY, Chang TS, Lee KS, Khil LY, Kim D, Chung JH, et al. Antiplatelet activity of BRX-018, (6aS,cis)-malonic acid 3-acetoxy-6a9-bis-(2-methoxycarbonyl-acetoxy)-6,6a,7,11b-tetrahydro-indeno [2,1-c]chromen-10-yl ester methylester. Thromb Res. 2005;115:309–318. doi: 10.1016/j.thromres.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Lien JC, Huang LJ, Teng CM, Wang JP, Kuo SC. Synthesis of 2-alkoxy 1,4-naphthoquinone derivatives as antiplatelet, antiinflammatory, and antiallergic agents. Chem Pharm Bull (Tokyo) 2002;50:672–674. doi: 10.1248/cpb.50.672. [DOI] [PubMed] [Google Scholar]
- McNicol A, Shibou TS. Translocation and phosphorylation of cytosolic phospholipase A2 in activated platelets. Thromb Res. 1998;92:19–26. doi: 10.1016/s0049-3848(98)00097-8. [DOI] [PubMed] [Google Scholar]
- Mustard JF, Perry DW, Ardlie NG, Packham MA. Preparation of suspensions of washed platelets from humans. Br J Haematol. 1972;22:193–204. doi: 10.1111/j.1365-2141.1972.tb08800.x. [DOI] [PubMed] [Google Scholar]
- Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- Parise LV, Venton DL, Le Breton GC. Arachidonic acid-induced platelet aggregation is mediated by a thromboxane A2/prostaglandin H2 receptor interaction. J Pharmacol Exp Ther. 1984;228:240–244. [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Putney JW, Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. Mechanisms of capacitative calcium entry. J Cell Sci. 2001;114(Pt 12):2223–2229. doi: 10.1242/jcs.114.12.2223. [DOI] [PubMed] [Google Scholar]
- Rink TJ, Sage SO. Calcium signaling in human platelets. Annu Rev Physiol. 1990;52:431–449. doi: 10.1146/annurev.ph.52.030190.002243. [DOI] [PubMed] [Google Scholar]
- Rodriguez S, Wolfender JL, Hakizamungu E, Hostettmann K. An antifungal naphthoquinone, xanthones and secoiridoids from Swertia calycina. Planta Med. 1995;61:362–364. doi: 10.1055/s-2006-958102. [DOI] [PubMed] [Google Scholar]
- Sage SO. The Wellcome Prize Lecture. Calcium entry mechanisms in human platelets. Exp Physiol. 1997;82:807–823. doi: 10.1113/expphysiol.1997.sp004066. [DOI] [PubMed] [Google Scholar]
- Schwarz UR, Walter U, Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153–1161. doi: 10.1016/s0006-2952(01)00760-2. [DOI] [PubMed] [Google Scholar]
- Son DJ, Cho MR, Jin YR, Kim SY, Park YH, Lee SH, et al. Antiplatelet effect of green tea catechins: a possible mechanism through arachidonic acid pathway. Prostaglandins Leukot Essent Fatty Acids. 2004;71:25–31. doi: 10.1016/j.plefa.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Szczeklik A, Musial J, Undas A, Sanak M. Aspirin resistance. J Thromb Haemost. 2005;3:1655–1662. doi: 10.1111/j.1538-7836.2005.01372.x. [DOI] [PubMed] [Google Scholar]
- Verma RP. Anti-cancer activities of 1,4-naphthoquinones: a QSAR study. Anticancer Agents Med Chem. 2006;6:489–499. doi: 10.2174/187152006778226512. [DOI] [PubMed] [Google Scholar]
- Walsh PN. Platelet coagulation-protein interactions. Semin Thromb Hemost. 2004;30:461–471. doi: 10.1055/s-2004-833481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.