

Abstract
Previous studies demonstrated that targeted deletion of the Ron receptor tyrosine kinase (TK) domain in mice leads to marked hepatocyte protection in a well-characterized model of lipopolysaccharide (LPS)-induced acute liver failure in D-galactosamine (GalN)-sensitized mice. Hepatocyte protection in TK−/− mice was observed despite paradoxically elevated serum levels of tumor necrosis factor alpha (TNFα). To understand the role of Ron in the liver, purified populations of Kupffer cells and hepatocytes from wild-type (TK+/+) and TK−/− mice were studied. Utilizing quantitative RT-PCR, we demonstrated that Ron is expressed in these cell-types. Moreover, we also recapitulated the protected hepatocyte phenotype and exaggerated cytokine production observed in the TK−/− mice in vivo through the use of purified cultured cells ex vivo. We show that isolated TK−/− Kupffer cells produce increased levels of TNFα and select cytokines compared to TK+/+ cells following LPS stimulation. We also show that conditioned media from LPS-treated TK−/− Kupffer cells was more toxic to hepatocytes than control media, suggesting the exaggerated levels of cytokines produced from the TK−/− Kupffer cells are detrimental to wild type hepatocytes. In addition, we observed that TK−/− hepatocytes were more resistant to cell death compared to TK+/+ hepatocytes, suggesting that Ron functions in both the epithelial and inflammatory cell compartments to regulate acute liver injury. These findings were confirmed in vivo in mice with hepatocyte and macrophage cell-type-specific conditional Ron deletions. Mice with Ron loss selectively in hepatocytes exhibited less liver damage and increased survival compared to mice with Ron loss in macrophages. In conclusion, we have dissected cell-type-specific roles for Ron such that this receptor modulates cytokine production from Kupffer cells and inhibits hepatocyte survival in response to injury.
Keywords: Met Receptor, Acute Liver Injury, Hepatocyte Growth Factor-Like Protein, Receptor Tyrosine Kinase, Nuclear Factor Kappa B
Acute liver failure (ALF) is an often fatal condition resulting in hepatocellular apoptosis and hemorrhagic necrosis. The most frequent cause of ALF in adults is due to drug toxicity, with a wide spectrum of etiologies responsible for the remaining cases (1). The cascade of events that leads to ALF is complex and not well understood. An established model for studying acute hepatocellular injury in mice is by the co-administration of the hepatocyte-specific transcriptional inhibitor galactosamine (GalN) and the bacterial endotoxin, lipopolysaccharide (LPS) (2). This model is principally a macrophage/monocyte-mediated model of shock and liver injury with secreted tumor necrosis factor alpha (TNFα) required for hepatic injury (3–4). In this model, LPS stimulates the release of TNFα, a pleiotropic cytokine that is capable of inducing proliferation or apoptosis in hepatocytes and other cell types (5), depending on the physiologic conditions, and numerous other cytokines and chemokines present in the microenvironment secreted from Kupffer cells, the resident tissue macrophage in the liver. After partial hepatectomy, TNFα is crucial for tissue regeneration, while in the setting of a toxic insult, TNFα induces cell death. The transcription factor NF-κB is reported to play an important role in determining which way the TNFα balance will tilt (6).
Ron is a cell surface receptor tyrosine kinase that participates in divergent processes, including modulation of inflammatory responses (7). Ron is expressed in a variety of cells but is most abundant in epithelial cells and macrophages (8). Ron is activated by its ligand, hepatocyte growth factor-like protein (HGFL), also known as macrophage-stimulating protein (9). HGFL is an 85 kDa circulating protein produced and secreted primarily by hepatocytes (10). Activation of Ron in peritoneal macrophages has been shown to stimulate macrophage shape changes, chemotaxis, adhesion, and phagocytosis (x11). Ron has also been shown in alveolar and peritoneal macrophages to limit select cytokine responses in inflammatory cells through attenuation of NF-κB by a mechanism that has yet to be identified (12–13).
Previous studies from our laboratory showed increased inflammatory responses and shortened survival times in mice with a deleted Ron tyrosine kinase domain (TK−/−) compared to wild-type control mice during the induction of bacterial peritonitis and in a lung injury model (14–15). Paradoxically, utilizing the well-characterized model of LPS/GalN induced ALF in mice, though serum levels of TNFα were elevated, livers from TK−/− mice exhibited marked hepatocyte protection compared with controls (16). To investigate the function of Ron in regulating hepatocyte survival, purified populations of Kupffer cells and hepatocytes from wild-type and TK−/− mice were isolated. Utilizing purified cells, we recapitulated ex vivo the protected hepatocyte phenotype and exaggerated cytokine production observed in the TK−/− mice in vivo. Furthermore, by using mice with targeted deletions of Ron in hepatocytes and macrophages, we were able to substantiate our findings ex vivo. In total, our data suggests that Ron loss selectively in hepatocytes provides a survival benefit during ALF despite increased cytokine production by deregulated Kupffer cell activation.
Experimental Procedures
Mice
Ron tyrosine kinase deficient mice (TK−/−) and floxed Ron mice (TKfl/fl) were generated as described and were backcrossed into a C57BL/6 background (7). Age matched male mice between 14 and 24 weeks old were used for all experiments. C57BL/6 albumin-Cre and lysozyme-Cre mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Cre expressing mice were crossed with floxed Ron (TKfl/fl) mice to create the targeted knockouts. Deletion of the Ron TK domain was determined by semi-quantative competitive PCR as described (7, 17). All mice were maintained under specific pathogen-free conditions and were treated in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Cellular Isolation and Immunocytochemistry
Mouse Kupffer cells and hepatocytes were isolated using the technique described by Kuboki et al. (18). Cell staining was performed with antibodies against F4/80 (ab6640, Abcam, Cambridge, MA), Albumin (Bethyl Laboratories, Inc., Montgomery, TX), Ron (AF431, R&D Systems, Minneapolis, MN) or isotype control antibodies. Mounting media contained DAPI for nuclear staining. THP-1 cells were purchased from ATCC (Manassas, VA) and were differentiated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA).
RNA and Quantitative Real-Time PCR
RNA was isolated using TriZol (Invitrogen, Carlsbad, CA). One μg of RNA was converted to cDNA with the high capacity RNA to cDNA kit according to manufacturer’s instructions (Applied Biosystems, Foster City, CA). Real time PCR was performed using FastStart SYBR Green (F. Hoffmann-La Roche Ltd, Nutley, NJ). The following genes and corresponding sequences were chosen: Ron (5′-TCCCATTGCAGGTCTGTGTAGA-3′; 5′-CGGAAGCTGTATCGTTGATGTC-3′), β-glucuronidase (GusB) (5′-TTGAGAACTGGTATAAGACGCATCAG-3′; 5′-TCTGGTACTCCTCACTGAACATGC-3′). TNFα (5′-CATCTTCTCAAAATTCGAGTGACAA-3′; 5′-TGGGAGTAGACAAGGTACAACCC-3′), KC (5′-TGCACCCAAACCGAAGTCAT-3′; 5′-TTGTCAGAAGCCAGCGTTCAC-3′), HGFL ( 5 ′-TGGTACAGTGTTCAAGGGCTCTT-3′; 5′-GCATGGCTGCTCATG-3′) and EGR1 (5′-TCTTGGTGCCTTTTGTGTGAC-3′; 5′-CTCTTCCTCGTTTTTGCTCTC-3′). Expression levels were normalized to GusB as internal control. Relative gene expression results are reported. Real-time analyses were repeated twice with similar results using samples from 3 independent isolations.
Cytokine Production
Kupffer cells were plated in Williams E media supplemented with 5% FBS. Conditioned media was generated by replacing the Kupffer cell media with fresh media plus 500μg/ml LPS (E. coli serotype 0111:B4; Sigma, St Louis, MO) and collected at the time points indicated. For the cytokine array, conditioned media was collected and incubated with the mouse cytokine antibody array (Panel A) from R&D Systems (Minneapolis, MN). Detection of replicate spots is by horseradish peroxidase-based chemiluminescence and film. Film was scanned and spots were quantitated using ImageJ from the National Institute of Health. TNFα levels were measured by ELISA (R&D Systems, Minneapolis, MN). Recombinant HGFL was supplied by R&D systems.
Luciferase Reporter Assays
Twenty-four hours before LPS exposure, Kupffer cells or primary hepatocytes were transfected with a NF-κB reporter (pNF-κB luc) plasmid or an empty vector (pTAL luc), and a control plasmid expressing Renilla (pRL-TK) utilizing Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Kupffer cells were treated with LPS (1μg/ml) in complete media for 2 hours. Hepatocytes were treated with 10 ng/ml of TNFα for 6 hours. Cell lysates were collected and luciferase activity was determined using the Dual-Luciferase Assay System (Promega, Madison, WI). Samples were run in duplicate and averaged.
Conditioned Kupffer Media on Hepatocytes
Conditioned media was generated by placing fresh media, with or without 500μg/ml LPS, onto Kupffer cells. This media was subsequently collected after 1 or 2 hours. AML12 mouse hepatocytes (American Type Culture Collection, Manassas, VA) were grown in DMEM/F12 media supplemented with 10% FBS and ITS (Invitrogen, Carlsbad, CA). Cells were pretreated with 200ng/ml of actinomycin D (ActD) for 30 minutes. Media was then changed to 150μl of conditioned Kupffer cell media plus 200ng/ml ActD. After 18 hours, media was removed and cells were fixed. Cells were quantitated by crystal violet assays and hepatocyte number was calculated based on a standard curve (19). TNFα antibody (AB-410-NA) was from R&D systems.
TNFα Hepatocyte Assay
Primary hepatocytes were pretreated with 200ng/ml of ActD in Williams E media + 5% FBS for 30 minutes. Media was then changed to include increasing amounts of recombinant mouse TNFα (Peprotech, Inc., Rocky Hill, NJ) plus ActD. After 18 hours, media was removed and cells were fixed and quantitated by crystal violet assays.
Western Analyses
Cells were collected and homogenized in 2X Laemmli Buffer. For HGFL determination, cells were cultured in serum free media and the media was collected after 36 hours. Media was concentrated with Amicon Ultra-4 centrifugal filters (Millipore, Billerica, MA). Protein concentrations were determined using the Micro BCA Kit (Pierce Biotechnology, Rockford, IL). Primary antibodies used were anti-NF-κB p65, anti-pNF-κB p65 Ser536, anti-pIKKα/β, anti-IKKβ anti-Caspase-3 (Cell Signaling, Boston, MA), anti-HGFL (T-19, Santa Cruz Biotechnology, Santa Cruz, CA).
LPS and GalN Injection of Mice
Mice were injected with 0.8 μg LPS and 30 mg GalN in saline and normalized to 30 g body weight (500μl total volume) or GalN alone. This low dose of LPS does not alter mortality in the Ron TK deficient mice, but when combined with GalN induces significant liver injury (7, 16). Blood was collected and plasma alanine aminotransferase levels were determined at Shriners Hospital.
Terminal Deoxynucleotidyl Transferase–Mediated Deoxyuridine Triphosphate Nick-End Labeling (TUNEL) Analysis
Paraffin-embedded sections of liver tissue were analyzed by TUNEL staining (16). For each liver tissue section per mouse, the number of TUNEL-positive cells in 3 random high-powered fields was counted by an investigator blinded to treatment group.
Statistical Analyses
Statistical significance for all analyses was determined by Student’s t-test, Logrank or one-way ANOVA using GraphPad Prism 3.03 software (GraphPad Software, Inc. La Jolla, CA). Error bars represent SEM.
Results
The Ron receptor is expressed in Kupffer cells and hepatocytes
Quantitative data directly comparing Ron expression in liver macrophages (Kupffer cells) and in liver parenchymal cells in the mouse is lacking. To examine Ron expression in mouse Kupffer cells and hepatocytes, populations of murine Kupffer cells and hepatocytes were collected. The isolated Kupffer cells ranged in purity from 90–95% following centrifugal elutriation based on F4/80 immunostaining (Figure 1A) and the ability of the cells to ingest fluorescent beads (data not shown). Hepatocyte identity was confirmed with albumin immunostaining and purity was over 99% (Figure 1A). Ron protein expression was observed in isolated Kupffer cells and hepatocytes by immunocytochemistry (Figure 1A). Quantitative real-time PCR was performed to compare Ron RNA expression levels in Kupffer cells, hepatocytes, AML12 cells and peritoneal macrophages as shown in Figure 1B. The expression of Ron in TK−/− Kupffer cells and hepatocytes was undetectable. AML12 cells and primary hepatocytes express and secrete HGFL (Figure 1C).
Figure 1. Ron is expressed in Kupffer cells and hepatocytes.

(A) Representative pictures of isolated Kupffer cells and hepatocytes. Top, Immunocytochemical staining of Kupffer cells by immunofluorescence for the macrophage specific surface marker F4/80 or for Ron compared to control staining with an isotype-matched primary antibody. Nuclei were stained with DAPI. Bottom, Representative pictures depicting Albumin and Ron staining of hepatocytes with DAPI stained nuclei. Control staining with an isotype-matched primary antibody on hepatocytes is shown. (B) Quantitative real-time PCR analysis of Ron mRNA expression from Kupffer cells, hepatocytes, AML12 cells and peritoneal macrophages. Normalized relative Ron expression levels are expressed as the mean ± the standard error and are representative of two independent experiments. Ron expression levels are relative to β-glucuronidase as an internal control. TK−/− Kupffer cells and hepatocytes serve as a negative control. (C) HGFL is expressed at the mRNA level and is secreted by AML12 cells and primary mouse hepatocytes.
Ron regulates Kupffer cell responses to LPS
To investigate the role of Ron as a potential mediator of Kupffer cell inflammation, we examined cytokine production from wild type (TK+/+) and TK−/− Kupffer cells in response to LPS ex vivo. Figure 2A demonstrates that Ron loss leads to increases in TNFα production from Kupffer cells in response to LPS. To examine the extent of cytokine changes regulated by Ron, an antibody array was utilized to simultaneously compare 40 cytokines in conditioned media from LPS stimulated wild type and Ron TK−/− Kupffer cells. Figure 2B displays relative values of select cytokines whereby MCP-1, MIP-2, interleukin-1 receptor antagonist (IL-1ra), and IL-6 levels were elevated approximately two-fold in the media from the Ron TK−/− cells compared to controls. Keratinocyte chemoattractant (KC) and tissue inhibitor of metalloproteinase (TIMP-1) showed moderate increases in the TK−/− conditioned media while no changes between groups were observed in the levels of IL-1α or IL-13. The results for all cytokines are listed in Supplemental Table S1. No differences were observed in the basal conditioned media between the TK+/+ and TK−/− Kupffer cells in the absence of LPS treatment, with the exception of IL-1ra and TIMP-1 expression, which were higher basally in the TK−/− Kupffer cells and did not respond to LPS treatment (data not shown). To examine the role of HGFL in suppressing cytokine production, TK+/+ Kupffer cells were treated with HGFL and stimulated with LPS. As shown in Figure 2C, HGFL treatment suppresses the release of TNFα and reduces the expression of TNFα, KC and Early Growth Response 1 (EGR1), well-documented NF-κB responsive genes.
Figure 2. Ron regulates cytokine production in Kupffer cells.

(A) TNFα ELISA of Kupffer cell media after stimulation with LPS for the indicated times. TNFα production increases in TK+/+ and TK−/− Kupffer cells following LPS stimulation with significantly higher levels in the TK−/− media by 2 hours. *P<0.05. (B) Densitometric analysis of an antibody-based cytokine array of select cytokines from TK+/+ and TK−/− Kupffer cells following LPS stimulation for 2h. Error bars are the range of two spots of each cytokine and is representative of a least two experiments. (C) TNFα ELISA and quantitative real-time PCR of TNFα, KC, and EGR1 showing reduced expression of NF-κB target genes in LPS-stimulated Kupffer cells after overnight treatment with 100 ng/ml HGFL. *P<0.05.
Loss of Ron signaling leads to increased NF-κB activity
As demonstrated in alveolar and peritoneal macrophages, Ron functions to inhibit cytokine signaling by limiting potentially damaging hyper-signaling through the NF-κB pathway (12, 20–21). To investigate whether the increased cytokine expression observed in Ron TK−/− Kupffer cells is associated with increased NF-κB signaling, NF-κB activation in TK+/+ and TK−/− Kupffer cells was examined by luciferase reporter assays. Kupffer cells from TK+/+ and TK−/− mice were transfected with a vector containing a NF-κB response element upstream of luciferase or an empty vector control. As shown in Figure 3A, TK−/− Kupffer cells had significantly higher reporter activity compared to TK+/+ cells in response to LPS treatment (2.5 fold versus 1.5 fold over the basal level). Basal levels of reporter activity were similar between genotypes. EMSA analysis confirmed an increased NF-κB response in the TK−/− cells (data not shown). To support this data and further define the mechanism by which Ron may regulate cytokine production, the human macrophage cell line THP-1 was utilized. Differentiated THP-1 cells express abundant levels of Ron (data not shown). As shown in Figure 3B, LPS stimulation induces the phosphorylation of NF-κB at Ser536, a verified marker of NF-κB transcriptional activity (22), in THP-1 cells. Treatment with HGFL decreases this phosphorylation. In addition, the stimulation of IκB kinase (IKK) phosphorylation, an upstream kinase which regulates NF-κB activation, is reduced by HGFL treatment, similar to recently reported findings (21). Similar results are also observed with Kupffer cells which exhibit less NF-κB and IKK phosphorylation in response to HGFL treatment than that observed with LPS alone (Figure 3C and data not shown).
Figure 3. TK−/− Kupffer cells exhibit exaggerated NF-κB signaling in response to LPS.

(A) Kupffer cells from TK+/+ and TK−/− mice were transfected with a vector containing an NF-κB response element upstream of firefly luciferase and a control plasmid. After LPS stimulation for 4h, TK−/− Kupffer cells had dramatically higher reporter activity than TK+/+ cells. Bars represent the average of 3 independent experiments with at least two mice per genotype per experiment. *P<0.002. (B) Differentiated THP-1 cells were stimulated with 500 ng/ml of LPS with or without overnight HGFL treatment. Western analysis of cell lysates depicts diminished NF-κB and IKKα/β phosphorylation in HGFL treated cells. Samples from two independent experiments are shown. Total NF-κB, IKKβ, and Tubulin serve as loading controls. (C) Western analysis of LPS-stimulated Kupffer cell lysates with or without HGFL pretreatment overnight (O/N) or for 30 minutes (30′). HGFL treatment before LPS stimulation leads to reduced NF-κB Ser536 phosphorylation (p-p65) compared to controls. Total NF-κB and Tubulin serve as loading controls.
Conditioned media from LPS-treated Ron TK−/− Kupffer cells is detrimental to hepatocytes
Given the significant decrease in hepatocyte apoptosis observed in TK−/− mice in response to LPS/GalN treatment in vivo (16) and the alteration of cytokine signaling observed in the TK−/− Kupffer cells ex vivo, we initially hypothesized the altered cytokine milieu produced by the TK−/− Kupffer cells was capable of affording protection to hepatocytes during the induction of acute liver failure. To test this hypothesis, we isolated Kupffer cells from TK+/+ and TK−/− mice and stimulated them ex vivo with LPS or vehicle. Equal amounts of conditioned media were collected after 2 hours, and were applied to the AML12 hepatocyte cell line, in the presence of the transcriptional inhibitor actinomycin D (ActD) to emulate conditions in vitro of hepatocyte death observed in previous in vivo experiments. As shown in Figure 4, while the viability of hepatocytes exposed to control media or conditioned media from either untreated TK+/+ or TK−/− Kupffer cells did not significantly differ from one another, the percent hepatocyte death was significantly increased in the presence of conditioned media from LPS-treated TK−/− Kupffer cells compared to similarly treated TK+/+ Kupffer cells. These results demonstrate that the LPS-induced cytokine milieu from the TK−/− Kupffer cells is capable of inducing increased hepatocyte death compared to control cells under these conditions. In Figure 4C, neutralization of TNFα in the TK−/− Kupffer cell conditioned media restored hepatocyte viability to near 100%, suggesting that TNFα has a prominent role in inducing hepatocyte death in ActD treated hepatocytes.
Figure 4. Conditioned media from LPS-stimulated TK−/− Kupffer cells is detrimental to hepatocytes.

(A) Kupffer cells from TK+/+ or TK−/− mice were treated in the presence or absence of LPS for 2 hours. Conditioned media was collected and applied to AML12 hepatocytes with ActD. After 18h, the hepatocytes were fixed and quantified by crystal violet staining. (B) Media from the TK−/− Kupffer cells increased hepatocyte death by 35% compared to control cells. The data is expressed as the mean ± standard error and is representative of three independent experiments. *P<0.05. (C) Treatment of TK−/− Kupffer cell conditioned media with an anti-TNFα antibody for 30 minutes leads to a significant reduction in hepatocyte death to less than 5%. Goat IgG was used as a control. Bar graph is the average of 3 experiments. *P<0.05.
Ron TK−/− hepatocytes are afforded protection from TNFα-induced apoptosis
Given that our prior studies in TK−/− mice demonstrated lessened hepatocyte apoptosis in vivo, despite elevated serum levels of TNFα, following the induction of acute liver failure by LPS/GalN treatment, we next sought to determine if Ron signaling regulates hepatocyte survival. Using a well-established method to induce apoptosis in hepatocytes by the co-administration of ActD and TNFα (23), hepatocytes from TK+/+ and TK−/− mice were treated with increasing amounts of TNFα for 18 hours and hepatocyte viability was subsequently assessed. As shown in Figure 5, hepatocytes derived from TK−/− mice were significantly protected from TNFα induced apoptosis compared to TK+/+ hepatocytes at a TNFα concentration range from 0.5 to 5.0ng/ml. At a TNFα concentration of 1ng/ml, 90% of the TK−/− hepatocytes were viable compared to 75% viability observed in hepatocytes from wild-type mice. This data suggests that Ron signaling in hepatocytes may be an important mediator of hepatocyte survival following liver injury.
Figure 5. TK−/− hepatocytes are protected from TNFα-induced cell death.
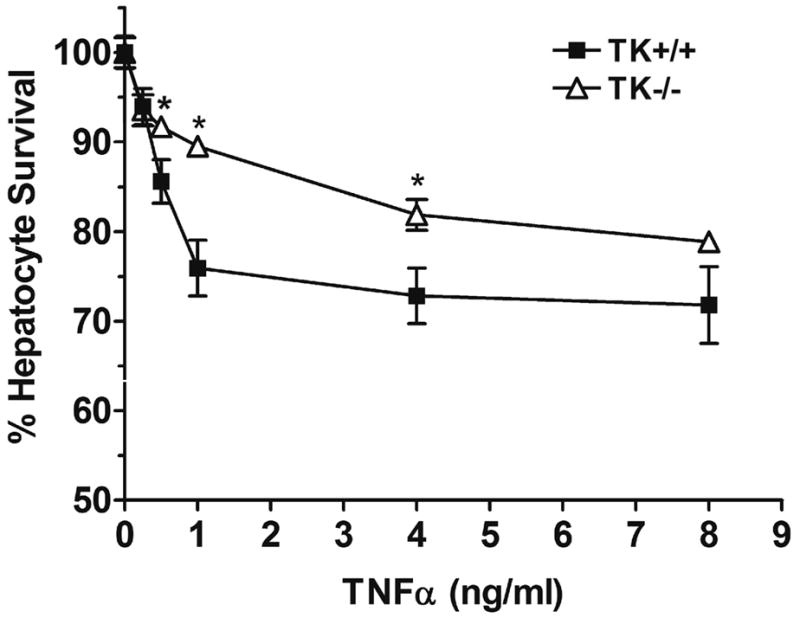
Primary hepatocytes from TK+/+ or TK−/− mice were treated for 30 minutes with ActD followed by the addition of increasing amounts of TNFα. After 18 hours, the hepatocytes were fixed and cell survival was determined by crystal violet staining with cell number quantitated based on a standard curve of hepatocytes. The data is expressed as the mean ± standard error and is representative of three independent experiments. *P<0.01 compared to the corresponding control treated sample.
Inhibition of NF-κB signaling attenuates the survival of Ron TK−/− hepatocytes
While we and others have shown that Ron regulates NF-κB in macrophages, including Kupffer cells (Figure 3), nothing is known about Ron signaling in hepatocytes (12, 20). To test whether the differential hepatocyte survival observed in TK−/− hepatocytes may be due to differential NF-κB activation, we examined survival of TK+/+ and TK−/− hepatocytes in response to constant levels of TNFα and ActD with the inclusion of increasing concentrations of the irreversible NF-κB activation inhibitor BAY 11-7085 (24). As depicted in Figure 6A, TK+/+ and TK−/− hepatocyte survival converged with increasing concentrations of Bay 11-7085. This data suggests that blunting NF-κB activation in TK−/− hepatocytes negates the survival advantage in these cells. Indeed, as shown in Figure 6B, TK−/− hepatocytes have elevated phosphorylated NF-κB after TNFα treatment compared to wild-type hepatocytes. Basal levels of pNF-κB did not differ between genotypes and are similar to that observed at the 6 hour time point (data not shown). To confirm exaggerated NF-κB signaling in TK−/− hepatocytes, NF-κB luciferase reporter assays were performed. TK+/+ and TK−/− hepatocytes were stimulated with TNFα and reporter activity was determined after 6 hours. As shown in Figure 6C, TK−/− hepatocytes exhibited 1.5X more luciferase activity than TK+/+ hepatocytes.
Figure 6. NF-κB inhibition abrogates survival of TK−/− hepatocytes in response to TNFα-induced death.

(A) Hepatocytes from TK+/+ or TK−/− mice were treated for 30 minutes with ActD plus increasing amounts of the NF-κB activation inhibitor, Bay 11-7085 followed by 5ng/ml TNFα. After 18 hours, the hepatocytes were fixed and cell survival was determined by crystal violet staining. The data is expressed as the mean ± standard error. *P<0.01 (B) Hepatocytes from TK+/+ or TK−/− mice were treated for 30 minutes with ActD followed by 5 ng/ml TNFα. Cells from 3 independent experiments of each cell type were examined after 10 minutes and 6 hours by Western analysis. pNF-κB (phosphorylated p65 subunit at Ser536) band density was normalized to Actin. *P<0.05. (C) TK+/+ and TK−/− hepatocytes were transfected with a vector containing an NF-κB response element upstream of firefly luciferase and a control plasmid. After 10 ng/ml TNFα stimulation for 6h, TK−/− hepatocytes had 1.5 fold higher reporter activity than TK+/+ cells. Values represent the average of two independent experiments relative to untreated cells. *P<0.05.
Targeted deletion of Ron TK in hepatocytes leads to a protected phenotype in vivo
Our ex vivo data suggests that despite an elevated cytokine profile, including increased TNFα, TK−/− hepatocytes are protected from damage compared to wild type hepatocytes. In order to test our ex vivo findings in vivo, we employed Cre-LoxP technology to generate mice with cell type-specific deletion of Ron from hepatocytes [i.e. albumin-Cre (Alb-Cre or AC) Ron TKfl/fl mice] or myeloid lineage cells [i.e. lysozyme-Cre (Lys-Cre or LC) Ron TKfl/fl mice]. By semi-quantitative competitive PCR, the TK region of Ron appeared completely ablated in Alb-Cre Ron TKfl/fl hepatocytes (Supplemental Figure S1). Ron TK ablation was observed in the majority of Lys-Cre Ron TKfl/fl Kupffer cells (Supplemental Figure S1), ranging from approximately 60–80%.
Mice were injected with LPS/GalN, and then liver tissue was analyzed for histopathology and blood was analyzed for ALT levels. In Figure 7A–C, hematoxylin-eosin staining of representative liver sections shows the greatest hemorraghic necrosis and pyknotic nuclei in the Lys-Cre Ron TKfl/fl liver. Alb-Cre Ron TKfl/fl liver sections displayed the least damage of the three groups. ALT levels at 5 hours in Lys-Cre Ron TKfl/fl plasma displayed a more rapid rise than the other two groups (Figure 7G). Unfortunately, by 6 hours a majority of Lys-Cre Ron TKfl/fl mice were moribund or dead and adequate blood could not be collected for ALT analysis. Importantly, however, at 6 hours, ALT levels in Ron TKfl/fl control plasma were significantly higher than Alb-Cre Ron TKfl/fl plasma, suggesting protection of the TK−/− hepatocytes.
Figure 7. Deletion of the Ron TK domain in hepatocytes is protective whereas deletion in myeloid derived cells is detrimental to mice treated with LPS/GalN.

Histopathology of LPS/GalN mice after 5 hours: (A) Ron TKfl/fl, (B) Alb-Cre Ron TKfl/fl, (C) Lys-Cre Ron TKfl/fl. (D, E, F) Representative TUNEL stained slides (bar=100 μm), positive nuclei stained red. (D) Ron TKfl/fl, (E) Alb-Cre Ron TKfl/fl, (F) Lys-Cre Ron TKfl/fl. (G) Plasma ALT levels of LPS/GalN treated mice at 4.5, 5, and 6 hours. At 6 hours, Ron TKfl/fl control mice had significantly higher ALT than Alb-Cre Ron TKfl/fl mice (*P<0.05). ND, not determined as Lys-Cre mice were moribund or dead at 6 hours. (H) Positive TUNEL counts per 100X field. *P<0.05
TUNEL staining was performed on liver sections at 5h to assess the level of apoptosis (Figure 7D–F). Alb-Cre Ron TKfl/fl liver had statistically lower levels of TUNEL positive cells followed by control Ron TKfl/fl and Lys-Cre Ron TKfl/fl livers (Figure 7H). This result is consistent with Western analyses for active (cleaved) Caspase-3 observed in liver lyates with the lowest levels of active Caspase-3 detected in the Alb-Cre Ron TKfl/fl livers (Supplemental Figure S2).
To test for survival differences, mice from each group were allowed to proceed until death from ALF. Kaplan-Meier survival curves (Figure 8A) clearly show that Lys-Cre Ron TKfl/fl mice have a significantly shorter survival time. The mean survival time of each genotype is depicted in Figure 8B. While Figure 7 demonstrates protection from hepatocyte apoptosis and liver injury in the Alb-Cre Ron TKfl/fl mice compared to the other genotypes, overt survival of the Alb-Cre Ron TKfl/fl mice was longer than the Ron TKfl/fl mice but was not statistically different (P=0.07).
Figure 8. Alb-Cre Ron TKfl/fl and Ron TKfl/fl mice have a survival advantage over Lys-Cre Ron TKfl/fl mice treated with LPS/GalN.

(A) Kaplan-Meier survival graph of mice injected with LPS/GalN. Lys-Cre Ron TKfl/fl n=5, Ron TKfl/fl n=8, Alb-Cre Ron TKfl/fl n=6. Survival of the Lys-Cre Ron TKfl/fl mice was significantly less than the other two groups. Logrank test P=0.005. (B) Comparison of survival times in hours. *P<0.05 between Alb-Cre and Lys-Cre mice.
Discussion
Herein, we dissected the role of Ron receptor TK in an endotoxin-induced model of acute liver failure to further studies demonstrating that mice with a global deletion of Ron TK had a protected liver injury phenotype (16). Examination of hepatocytes and Kupffer cells showed expression of Ron in both, and importantly, ex vivo experiments showed Ron signaling is critical for both suppressing hepatotoxic Kupffer cell cytokine production and for sensitizing hepatocytes to Kupffer cell-derived products such as TNFα. Together with in vivo experiments using mice with either hepatocyte- or Kupffer cell-specific deletion of Ron TK, we determined that Ron TK signaling in both cell types has important effects on hepatocyte survival following exposure to endotoxin in this model of acute liver failure. However, the cell type likely responsible for Ron-dependent hepatic protection in this liver injury model is the hepatocyte, given that lack of Ron signaling in hepatocytes ex vivo and in vivo is sufficient to afford a hepatocyte survival benefit.
Following stimulation with LPS, TK−/− Kupffer cells have altered cytokine production ex vivo compared to TK+/+ Kupffer cells, with the most significantly elevated cytokines being IL-1ra (2.5 fold), IL-6 (2 fold), TIMP-1 (1.5 fold), MCP-1 (3.5 fold) and MIP-2 (1.9 fold). A potential mechanism for these perturbations is increased NF-κB signaling in the Ron-deficient state. These perturbations in cytokine production in response to LPS are consistent with earlier in vivo observations in this liver injury model (16), as well as in models of endotoxic shock or nickel-induced acute lung injury (12, 15, 20, 25). Interestingly, the effect of the altered cytokine milieu generated by LPS-treated TK−/− Kupffer cells was more toxic to mouse hepatocytes in vitro compared to TK+/+ Kupffer cells. This may be due to higher TNFα levels, the composition of cytokines, or the presence of an untested cytokine in the conditioned media. Moreover, it is possible that the kinetics of cytokine expression is as important as the composition of cytokines produced.
TK−/− hepatocytes are protected compared to TK+/+ hepatocytes from TNFα/ActD initiated cell death over the range of 0.5 to 5ng/ml of TNFα, which are similar to the serum TNFα levels observed in the previously reported in vivo experiment (16). Whether the observed differences in hepatocyte viability are sufficient to explain the in vivo observations by Leonis et al. is not discernable by this assay. Hepatocytes plated ex vivo are without feedback mechanisms to Kupffer cells and other hepatic cell types, and thus, the magnitude of the effect of TNFα ex vivo may not completely mimic what is observed in vivo. However, the results from the conditional deletion of Ron selectively in hepatocytes support the significance of our ex vivo culture conditions and prior in vivo experiments.
Previously, we demonstrated that the protected liver injury response to treatment with LPS/GalN in Ron TK−/− mice was associated with a 1–2 hour delay in the progression to death based on survival analyses (16). The lack of a more significant discrepancy in the time to mortality may be multifactorial and confounded by the sensitivity of the Ron TK−/− mice to LPS alone and by the severe necrosis and endothelial damage that is observed in the model. Despite the modest overall survival benefit of the TK−/− mice compared to controls, less liver injury was observed in the TK−/− mice as judged by liver histopathology, ALT levels, hepatocyte TUNEL staining and the extent of hepatic apoptosis. Similar protected phenotypes were observed in the Alb-Cre Ron TKfl/fl mice as the Ron TK−/− mice compared to control mice. This is based on a reduction in liver histopathology and significantly decreased ALT levels and TUNEL staining in the Alb-Cre Ron TKfl/fl mice compared to controls. Interestingly, a 1- to 2-hour increase in survival time in the Alb-Cre Ron TKfl/fl mice was also observed compared to controls. Therefore, the data is consistent with a protected liver phenotype in the Alb-Cre Ron TKfl/fl mice compared to controls. Our data also show a significant decrease in survival of the LysCre mice and associated worsened liver phenotypes in these mice compared to Alb-Cre Ron TKfl/fl and wild type mice and supports the premise that Ron functions in both cellular compartments in vivo.
Our results show increased NF-κB activation in hepatocytes that lack Ron signaling. NF-κB inhibition has been shown to tilt the balance of TNFα signaling toward apoptosis in hepatocytes; the converse is found as well, that increased NF-κB signaling provides a survival benefit (6, 26–30). In addition, Ron has been shown to regulate NF-κB (12, 20–21). Pretreatment of primary hepatocytes with the NF-κB inhibitor Bay-11-7085 abrogated the survival advantage of Ron deficient cells, suggesting that the elevated NF-κB levels observed in the early time points in these cells may at least be partly responsible for the protective phenotype. Although the exact mechanism for how NF-κB activity protects cells from apoptosis is not clear, up-regulation of anti-apoptotic proteins is one important effect of NF-κB signaling. Several anti-apoptotic proteins can be up-regulated by TNFα through NF-κB, however, we have seen no difference in two such proteins, C-IAP-2 or XIAP (31), between TK+/+ and TK−/− hepatocytes ex vivo following exposure to TNFα when examined by western blotting (data not shown).
Thus, we have demonstrated that Ron signaling is detrimental to hepatocyte survival when challenged with TNFα and that Ron is a regulator of hepatotoxic cytokine signaling in Kupffer cells. While the exact mechanism has not been elucidated, our ex vivo and in vivo studies suggest that Ron signaling appears to limit NF-κB signaling in both hepatocytes and Kupffer cells, leading to an overall sensitization of hepatocytes to Kupffer cell-derived products. Further research on the cell-type specific effects of Ron, and on how Ron regulates NF-κB is important in order to understand the mechanisms underlying this receptor’s effects on hepatocyte survival and before positing strategies that may lead to therapies for ALF or other liver pathologies, such as obesity related steatohepatitis and alcohol induced liver disease, that may, in part, have liver injury mediated by endotoxin.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by the Public Health Services Grants DK-73552 (S.E.W.), CA-111819 (M.A.L.) and the Digestive Diseases Research Development Center grant DK-064403 (S.E.W.) from the National Institutes of Health, as well as by grant project #8950 (S.E.W.) from Shriners Hospital for Children.
The authors would like to acknowledge the technical assistance provided by William Niehaus.
List of Abbreviations
- TK
tyrosine kinase
- LPS
lipopolysaccharide
- ALF
acute liver failure
- GalN
D(+)-galactosamine hydrochloride
- TNFα
tumor necrosis factor α
- NF-κB
nuclear factor-kB
- IL
interleukin
- GusB
β-glucuronidase
- MCP-1
macrophage chemoattractant protein-1
- IFN-γ
interferon γ
- HGFL
hepatocyte growth factor-like protein
- ActD
actinomycin D
- KC
keratinocyte chemoattractant
- TIMP-1
tissue inhibitor of metalloproteinase
- ELISA
enzyme-linked immunosorbent assay
- MIP-2
macrophage inflammatory protein-2
- IL-1ra
interleukin-1 receptor antagonist
- TUNEL
Terminal Deoxynucleotidyl Transferase–Mediated Deoxyuridine Triphosphate Nick-End Labeling
- ALT
alanine aminotransferase
References
- 1.Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, McCashland TM, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 2.Bohlinger I, Leist M, Gantner F, Angermuller S, Tiegs G, Wendel A. DNA fragmentation in mouse organs during endotoxic shock. Am J Pathol. 1996;149:1381–1393. [PMC free article] [PubMed] [Google Scholar]
- 3.Galanos C, Freudenberg MA. Mechanisms of endotoxin shock and endotoxin hypersensitivity. Immunobiology. 1993;187:346–356. doi: 10.1016/S0171-2985(11)80349-9. [DOI] [PubMed] [Google Scholar]
- 4.Josephs MD, Bahjat FR, Fukuzuka K, Ksontini R, Solorzano CC, Edwards CK, 3rd, Tannahill CL, et al. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-alpha and not by Fas ligand. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1196–1201. doi: 10.1152/ajpregu.2000.278.5.R1196. [DOI] [PubMed] [Google Scholar]
- 5.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, et al. NF-kappaB inactivation converts a hepatocyte cell line TNF-alpha response from proliferation to apoptosis. Am J Physiol. 1998;275:C1058–1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 7.Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, Wang MH, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J Clin Invest. 2001;108:567–576. doi: 10.1172/JCI11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunelleschi S, Penengo L, Lavagno L, Santoro C, Colangelo D, Viano I, Gaudino G. Macrophage stimulating protein (MSP) evokes superoxide anion production by human macrophages of different origin. Br J Pharmacol. 2001;134:1285–1295. doi: 10.1038/sj.bjp.0704356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene. 1993;8:1195–1202. [PubMed] [Google Scholar]
- 10.Skeel A, Yoshimura T, Showalter SD, Tanaka S, Appella E, Leonard EJ. Macrophage stimulating protein: purification, partial amino acid sequence, and cellular activity. J Exp Med. 1991;173:1227–1234. doi: 10.1084/jem.173.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard EJ, Skeel AH. Enhancement of spreading, phagocytosis and chemotaxis by macrophage stimulating protein (MSP) Adv Exp Med Biol. 1979;121B:181–194. doi: 10.1007/978-1-4684-8914-9_16. [DOI] [PubMed] [Google Scholar]
- 12.Lentsch AB, Pathrose P, Kader S, Kuboki S, Collins MH, Waltz SE. The Ron receptor tyrosine kinase regulates acute lung injury and suppresses nuclear factor kappaB activation. Shock. 2007;27:274–280. doi: 10.1097/01.shk.0000239755.82711.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu QP, Fruit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN-gamma and lipopolysaccharide by the STK/RON receptor tyrosine kinase. J Immunol. 1999;163:6606–6613. [PubMed] [Google Scholar]
- 14.Caldwell CC, Martignoni A, Leonis MA, Ondiveeran HK, Fox-Robichaud AE, Waltz SE. Ron receptor tyrosine kinase-dependent hepatic neutrophil recruitment and survival benefit in a murine model of bacterial peritonitis. Crit Care Med. 2008;36:1585–1593. doi: 10.1097/CCM.0b013e318170a8c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDowell SA, Mallakin A, Bachurski CJ, Toney-Earley K, Prows DR, Bruno T, Kaestner KH, et al. The role of the receptor tyrosine kinase Ron in nickel-induced acute lung injury. Am J Respir Cell Mol Biol. 2002;26:99–104. doi: 10.1165/ajrcmb.26.1.4621. [DOI] [PubMed] [Google Scholar]
- 16.Leonis MA, Toney-Earley K, Degen SJ, Waltz SE. Deletion of the Ron receptor tyrosine kinase domain in mice provides protection from endotoxin-induced acute liver failure. Hepatology. 2002;36:1053–1060. doi: 10.1053/jhep.2002.36822. [DOI] [PubMed] [Google Scholar]
- 17.Nikolaidis NM, Kulkarni RM, Gray JK, Collins MH, Waltz SE. Ron receptor deficient alveolar myeloid cells exacerbate LPS-induced acute lung injury in the muring lung. Innate Immunity. 2010 doi: 10.1177/1753425910383725. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuboki S, Okaya T, Schuster R, Blanchard J, Denenberg A, Wong HR, Lentsch AB. Hepatocyte NF-kappaB activation is hepatoprotective during ischemia-reperfusion injury and is augmented by ischemic hypothermia. Am J Physiol Gastrointest Liver Physiol. 2007;292:G201–207. doi: 10.1152/ajpgi.00186.2006. [DOI] [PubMed] [Google Scholar]
- 19.Gillies RJ, Didier N, Denton M. Determination of cell number in monolayer cultures. Anal Biochem. 1986;159:109–113. doi: 10.1016/0003-2697(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 20.Nikolaidis NM, Gray JK, Gurusamy D, Fox W, Stuart WD, Huber N, Waltz SE. Ron receptor tyrosine kinase negatively regulates TNFalpha production in alveolar macrophages by inhibiting NF-kappaB activity and Adam17 production. Shock. 2010;33:197–204. doi: 10.1097/SHK.0b013e3181ae8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray M, Yu S, Sharda DR, Wilson CB, Liu Q, Kaushal N, Prabhu KS, et al. Inhibition of TLR4-induced IkappaB kinase activity by the RON receptor tyrosine kinase and its ligand, macrophage-stimulating protein. J Immunol. 2010;185:7309–7316. doi: 10.4049/jimmunol.1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luedde T, Heinrichsdorff J, de Lorenzi R, De Vos R, Roskams T, Pasparakis M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc Natl Acad Sci U S A. 2008;105:9733–9738. doi: 10.1073/pnas.0800198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leist M, Gantner F, Kunstle G, Bohlinger I, Tiegs G, Bluethmann H, Wendel A. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med. 1996;2:109–124. [PMC free article] [PubMed] [Google Scholar]
- 24.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 25.Mallakin A, Kutcher LW, McDowell SA, Kong S, Schuster R, Lentsch AB, Aronow BJ, et al. Gene expression profiles of Mst1r-deficient mice during nickel-induced acute lung injury. Am J Respir Cell Mol Biol. 2006;34:15–27. doi: 10.1165/rcmb.2005-0093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: effects on liver homeostasis and beyond. Endocr Rev. 2007;28:365–386. doi: 10.1210/er.2006-0031. [DOI] [PubMed] [Google Scholar]
- 27.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heyninck K, Wullaert A, Beyaert R. Nuclear factor-kappa B plays a central role in tumour necrosis factor-mediated liver disease. Biochem Pharmacol. 2003;66:1409–1415. doi: 10.1016/s0006-2952(03)00491-x. [DOI] [PubMed] [Google Scholar]
- 29.Libert C, Van Bladel S, Brouckaert P, Shaw A, Fiers W. Involvement of the liver, but not of IL-6, in IL-1-induced desensitization to the lethal effects of tumor necrosis factor. J Immunol. 1991;146:2625–2632. [PubMed] [Google Scholar]
- 30.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha- induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.