

Abstract
There is increasing evidence that neuron death in neurode-generative diseases, such as Parkinson's disease, is due to the activation of programmed cell death. However, the upstream mediators of cell death remain largely unknown. One approach to the identification of upstream mediators is to perform gene expression analysis in disease models. Such analyses, performed in tissue culture models induced by neurotoxins, have identified up-regulation of CHOP/GADD153, a transcription factor implicated in apoptosis due to endoplasmic reticulum stress or oxidative injury. To evaluate the disease-related significance of these findings, we have examined the expression of CHOP/GADD153 in neurotoxin models of parkinsonism in living animals. Nuclear expression of CHOP protein is observed in developmental and adult models of dopamine neuron death induced by intrastriatal injection of 6-hydroxydopamine (6OHDA) and in models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). CHOP is a mediator of neuron death in the adult 60HDA model because a null mutation results in a reduction in apoptosis. In the chronic MPTP model, however, while CHOP is robustly expressed, the null mutation does not protect from the loss of neurons. We conclude that the role of CHOP depends on the nature of the toxic stimulus. For 6OHDA, an oxidative metabolite of dopamine, it is a mediator of apoptotic death.
Keywords: apoptosis, endoplasmic reticulum stress, oxidative stress, Parkinson's disease, programmed cell death, substantia nigra
There is an emerging consensus that programmed cell death (PCD) is likely to play a role in neuron death in neurode-generative disease (Mattson 2000; Yuan and Yankner 2000). For Parkinson's disease (PD), this consensus is based on studies in animal models and human post-mortem material demonstrating either apoptotic morphology or immunohistochemical evidence for activation of caspases (reviewed in Vila and Przedborski 2003). One of the hallmarks of PCD is that in many contexts, it requires the transcription of genes that mediate cell death (Martin et al. 1988; Oppenheim et al. 1990). Therefore, a useful strategy to attempt to identify genes that mediate neuronal degeneration is to screen gene expression in models of disease. Such a strategy has been implemented for PD by performing serial analysis of gene expression in PC12 cells, a catecholaminergic cell line (Greene and Tischler 1976), treated with 6-hydroxydopamine (6OHDA), a neurotoxin which is an oxidative metabolite of endogenous dopamine (Senoh and Witkop 1959; Kostrzewa and Jacobowitz 1974). Among the up-regulated transcripts identified by this analysis, and of particular potential relevance to neuronal death, was a striking induction of the transcription factor CHOP/GADD153 (Ryu et al. 2002). CHOP has been implicated as a mediator of apoptosis in the contexts of both endoplasmic reticulum (ER) stress (Matsumoto et al. 1996; Zinszner et al. 1998; Kawahara et al. 2001; Maytin et al. 2001; Gotoh et al. 2002; Oyadomari and Mori 2004) and oxidative stress (Guyton et al. 1996; Mengesdorf et al. 2002). In keeping with a possible role of either of these forms of cellular stress in mediating CHOP induction and neuron death, the analysis of gene expression also identified the induction of many other genes involved in ER and oxidative stress (Ryu et al. 2002, 2005).
A similar induction of CHOP was also observed by Holtz and O'Malley in a gene expression screen of neurotoxin models of parkinsonism (Holtz and O'Malley 2003). These investigators used Affymetrix gene arrays to screen dopaminergic MN9D cells following exposure to either 6OHDA or MPP+, and noted that the most highly expressed transcript, for both neurotoxins, was that for CHOP (Holtz and O'Malley 2003).
These findings in gene expression screens performed in vitro are potentially relevant to human PD because the classes of transcripts induced, those related to oxidative stress and ER stress, relate to important current hypotheses for pathogenesis. The possibility that the oxidative metabolism of dopamine may be injurious to dopaminergic neurons is one of the longest-standing hypotheses (Fahn and Cohen 1992). More recently, ER stress has been postulated to play a role. An important genetic cause of PD is loss of function mutations in parkin (Ishikawa and Tsuji 1996; Kitada et al. 1998). These mutations have been implicated in abnormal protein processing because parkin is an E3 ubiquitin-ligase (Shimura et al. 2000) and, as such, it plays a role in targeting cellular proteins for destruction by the proteasome (Ciechanover 1998). One putative protein target of parkin, Pael-R, is a difficult-to-fold protein, and it has been postulated that its accumulation may result in dopaminergic neuron death due to ER stress (Imai et al. 2000, 2001).
The possible implications of these in vitro observations for the pathogenesis of PD depend on whether they generalize to the in vivo context. We have therefore investigated the expression of CHOP in several neurotoxin models of parkinsonism in living animals: substantia nigra (SN) dopamine neuron degeneration induced by intrastriatal injection of 6OHDA in both developing (Marti et al. 1997) and adult rodents (Sauer and Oertel 1994), and by both the acute (Heikkila et al. 1984) and chronic (Tatton and Kish 1997) systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). In addition, we have sought to determine whether CHOP plays a functional role as an essential mediator of dopamine neuron death by examining the vulnerability of homozygous CHOP null mice.
Materials and methods
Animals
For the study of postnatal rats, timed pregnant females were obtained from Charles River Laboratories (Wilmington, MA, USA). The date of delivery was defined as postnatal day (PND) 1. For adult mouse studies utilizing the 6OHDA and MPTP models, C57BL/6 mice were obtained from Charles River. CHOP null mice were produced by homologous recombination to replace all of the CHOP coding sequence (except for the final 34 C-terminal residues) with the coding sequence for β-galactosidase containing a nuclear localization signal. The neomycin selection cassette was then removed by Cre recombinase. There was no detectable CHOP protein in cells and tissues derived from these animals (Fig. 1). These mice were back-crossed into the C57BL/6 strain five times before breeding for experiments. The CHOP null mice were genotyped by PCR analysis of tail DNA using three-primer PCR analysis as previously described (Zinszner et al. 1998), with the modification that the primer to detect the mutant allele was based on the β-galactosidase sequence and produced a 300 bp product.
Fig. 1.

Absence of CHOP protein expression in CHOP null mice. Immunoblot of nuclear extract of untreated and tunicamycin-treated (2 μg/mL, 6 h) wild-type and CHOP−/− cells blotted with antisera reactive with CHOP, ATF4 (a positive control) and p75, a ubiquitously-expressed nuclear protein that serves as a loading marker. No protein CHOP expression is observed in CHOP null cells after tunicamycin treatment. The antibody to ATF4 was raised against a full-length bacterially-expressed fusion protein and is characterized in Ron and Habener (1992). The p75 band was detected by an antiserum to Drosophila protein, described in Immanuel et al. (1995).
Animal models
The models used in this investigation are summarized in Table 1. The 6OHDA model in postnatal rats was performed as previously described (Marti et al. 1997). Briefly, rat pups at PND7 were pre-treated with 25 mg/kg desmethylimipramine, anesthetized by hypothermia and placed prone on an ice pack. 6-OHDA hydrobromide (Regis, Morton Grove, IL, USA) was prepared at 15 μg (total weight)/1.0 μL in 0.9% NaCl/0.02% ascorbic acid, and infused by pump (Harvard Apparatus, Holliston, MA, USA) at a rate of 0.25 μL/min for 4 min (total dose 15 μg). Postnatal mice were injected in a similar fashion except that the solution was prepared at a concentration of 20 μg/μL and infused for 2 min, for a total dose of 10 μg. For experiments in postnatal mice, littermate wild-type and heterozygote animals were examined in comparison with nulls. Adult mice were infused with a concentration of 5 μg/μL at a rate of 0.5 lL/min for 8 min for a total dose of 20 μg. For experiments in adult mice, C57BL/6 adults were used as controls.
Table 1.
Models used to assess the role of CHOP/GADD153 in apoptosis in SN dopamine neurons
| Treatment | Species | Age | Route | Morphology of cell death |
|---|---|---|---|---|
| None (natural cell death) |
Rat | Developmental | N.A. | Apoptosis |
| Axotomy | Rat | Developmental | N.A. | Apoptosis |
| 6OHDA | Rat or Mouse | Developmental | Intrastriatal | Apoptosis |
| 6OHDA | Mouse | Adult | Intrastriatal | Apoptotic and non-apoptotic |
| MPTP | Mouse | Adult | I.P., acute | Non-apoptotic |
| MPTP | Mouse | Adult | I.P., chronic | Apoptotic and non-apoptotic |
Abbreviations: N.A., not applicable; I.P., intraperitoneal.
The medial forebrain bundle (MFB) axotomy model in postnatal rats was performed as previously described (El-Khodor and Burke 2002). Briefly, rat pups were anesthetized by hypothermia. Animals were positioned in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA, USA) to conform with the neonatal brain atlas of Heller et al. (1979). The MFB was transected by lowering a retractable wire knife (Kopf Instruments) through a skull burr hole 1.4 mm posterior and 2.5 mm lateral to bregma to a ventral position of 6.5 mm below bregma.
For the acute MPTP lesion model, mice received four i.p. injections of MPTP-HCl (20 mg/kg free base; Sigma, St Louis, MO, USA) dissolved in saline, 2 h apart in 1 day as previously described (Teismann et al. 2003). Control mice received saline only. MPTP handling and safety measures were in accordance with our published guidelines (Przedborski et al. 2001). For the chronic MPTP model, mice received one i.p. injection of MPTP-HCl per day (30 mg/kg per day of free base) for 5 consecutive days as described (Tatton and Kish 1997).
All procedures were approved by the Institutional Animal Care and Use Committee of Columbia University.
Immunohistochemistry
For CHOP immunoperoxidase histochemistry, animals were perfused intracardially first with 0.9% NaCl and then with 4% paraformaldehyde and 0.1 M phosphate buffer (PB). The brains were then removed and post-fixed in the same fixative for 3 h. Each brain was then cryoprotected in 20% sucrose for 24 h. The brains were then rapidly frozen in isopentane on dry ice, and sections were cut in a cryostat at 30 μm. Sections were processed free-floating. After a phosphate-buffered saline (PBS) wash and treatment with PBS, 0.5% bovine serum albumen and 0.1% Triton X-100, sections were incubated with rabbit anti-CHOP at 1 : 500 for 48 h at 4°C. After a wash, sections were then incubated with biotinylated protein A (prepared in this laboratory) at 1 : 100 for 1 h at ambient room temperature. Sections were further incubated with avidin-biotinylated-horseradish peroxidase complexes (ABC; Vector Laboratories, Burlingame, CA, USA) at 1 : 600 for 1 h. After incubation with diaminobenzidine, sections were mounted onto subbed slides and counterstained with thionin. The primary antibody had been previously characterized and used for immunohistochemistry (Ron and Habener 1992; Zinszner et al. 1998). For immunofluorescence double-labeling for CHOP and tyrosine hydroxylase (TH), sections were collected into Tris-buffered saline (TBS) and then treated with TBS/0.2% Triton/2% goat serum/2% horse serum. They were then incubated in the same solution with anti-CHOP (1 : 500) and mouse anti-TH (1 : 40) (Chemicon, Temecula, CA, USA) for 48 h at 4°C. The sections were next treated with Texas red horse anti-mouse (Vector) at 1 : 75 and biotinylated goat anti-rabbit (Vector) at 1 : 75 for 1 h at ambient room temperature, followed by treatment with Fluor-avidin (Vector) at 1 : 100 for 1 h. Sections were then mounted onto gelatin-coated glass slides and coverslipped with Dako anti-fade medium (Carpinteria, CA, USA). The sections were examined by epifluorescence with a Nikon Eclipse 800 microscope.
For TH immunoperoxidase histochemistry, animals were per-fused, as described above, and then post-fixed in the same fixative for 1 week. Each brain was cryoprotected in 20% sucrose for 24–48 h and then rapidly frozen. A complete set of serial sections through the SN was cut at 30 μm. Sections were saved individually in serial order at 4°C, and individual sections at regular intervals were then selected for TH immunostaining, in conformity with the fractionator method of sampling (Coggeshall and Lekan 1996) (see below). Sections were processed free-floating, as described above for CHOP. The primary antibody was a rabbit anti-TH (Calbiochem, La Jolla, CA, USA) at 1 : 1000. After treatment with biotinylated protein A and ABC, sections were mounted on subbed slides in serial order and thionin-counterstained.
Quantitative morphology
For the analysis of the time course of appearance of CHOP-positive nuclear profiles and apoptosis in the postnatal 6OHDA model in rats, counts were performed as previously described (Oo et al. 2003; Ganguly et al. 2004). CHOP-positive nuclear profiles were counted in identical fashion on the same sections.
The number of SN dopaminergic neurons in the lesion experiments with CHOP null and C57BL/6 control mice was determined by stereological analysis. A complete set of TH-immunostained serial sections, sampled as every fourth section through the SN, was analyzed by a stereological method for each animal. Each analysis was performed under blinded conditions on coded slides. For each animal, the SN on each side of the brain was analyzed. For each section, the entire SN was identified as the region of interest. Using StereoInvestigator software (Micro Bright Field, Inc., Williston, VT, USA) a fractionator probe was established for each section. The number of TH-positive neurons in each counting frame was then determined by focusing down through the section, using a 100× objective under oil, as required by the optical dissector method (Coggeshall and Lekan 1996). Our criterion for counting an individual TH-positive neuron was the presence of its nucleus either within the counting frame, or touching the right or top frame lines (green) but not touching the left or bottom lines (red). The total number of TH-positive neurons for each SN on one side was then determined by the StereoInvestigator program. The total volume of the SN was also determined by the StereoInvestigator program for each brain on the basis of the sum of volumes derived from the area of each individual serial section and the tissue height represented by that section.
Northern analysis and non-radioactive in situ hybridization analysis (NRISH) of BiP
Rat BiP cDNA was subcloned into pCMS-EGFP (BD Biosciences, San Jose, CA, USA) as described (Ryu et al. 2002) and used for creation of an antisense RNA probe. Northern analysis was performed as previously described (El-Khodor et al. 2001). Briefly, RNA was isolated from microdissected SN using the Qiagen RNAeasy Mini kit (Valencia, CA, USA). The RNA concentration of each sample was determined by measuring absorption at 260 nm on a GenQuant spectro-photometer (Amersham Pharmacia Biotech, Piscataway, NJ, USA). A 20 μg aliquot of each RNA was electrophoresed in 1.4% agarose-formaldehyde gel and transferred onto an Immobilon (+) membrane (Millipore, Bedford, MA, USA). The hybridization was performed overnight at 68°C in Ultrahyb buffer from Ambion (Austin, TX, USA). The membrane was then exposed to phosphorimager cassettes, scanned and analyzed by Image Quant software (Molecular Dynamics, Indianapolis, IN, USA).
For NRISH, brains were rapidly removed from 6OHDA-injected adult mice at 48 h post-injection, and rapidly frozen in embedding medium on dry ice. Sections (14 μm) were thaw-mounted on glass slides (Superfrost Plus, Fisher, Hampton, NH, USA). For hybridization, sections were warmed on a slide warmer at 37°C for 20 min, and then fixed by immersion in 4% paraformaldehyde in 0.1 m PBS. After washing, sections were acetylated by treatment with acetic anhydride in triethanolamine. After another wash, sections were treated with a pre-hybridization solution, as previously described (Burke et al. 1994), for 2 h at ambient room temperature. Sections were then covered with hybridization solution and incubated overnight at 68°C in a humidified chamber. Hybridization solution contained the BiP riboprobe labeled with digoxigenin-UTP (1 μL/slide) (200–400 ng/mL), prepared as per the manufacturer's instructions (Roche Diagnostics, Penzberg, Germany). The size and integrity of labeled probe were confirmed by gel electrophoresis. The same probe used for northern analysis was used for the in situ hybridization. After a wash in 0.5× saline sodium citrate (SSC) for 10 min, followed by a wash in 0.2× SSC at 68°C for 30 min, sections were incubated with an anti-digoxigenin antibody (Roche) at 1 : 5000 overnight at 4°C. After additional washes, sections were incubated with a solution containing nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate (Promega Corporation, Madison, WI, USA) in a darkened humidified chamber overnight. Sections were then washed and coverslipped with Dako aqueous mounting medium.
RT-PCR/Southern blot analysis of the XBP-1 splice variant
To perform Southern analysis of the x-box binding protein-1 (XBP-1) splice variant, we first generated a DNA probe. We performed reverse transcription using RNA isolated from mouse kidney after treatment with tunicamycin. We then performed PCR of the 422 bp region of mouse XBP-1 containing the site of the unconventional splice, using primers based on nucleotide number 363 (Accession no. BC029197) (5′- CCTTGTGGTTGAGAACCAGG-3′) (forward) and nucleotide number 810 (5′-GAGGCTTGGTGTATACATGG-3′) (reverse). The band containing the spliced DNA fragment of XBP-1 was isolated from an agarose gel, subcloned in the pGEM-T vector (Promega) and sequenced. The DNA fragment containing the site of the XBP-1 unconventional splice was isolated from this clone using SalI and NcoI restriction enzymes (Promega). This fragment was then used to generate a 32P-labeled DNA probe with the Rediprime II Kit, random prime labeling system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). For XBP-1 splice variant Southern blot analysis, RNA was isolated from tissues using the Qiagen RNAeasy Mini Kit, as described above. First strand cDNA was then synthesized from isolated RNA by the RT system (Promega). PCR was performed individually with each cDNA sample using the above primers with Taq polymerase from Roche. A 10 μg aliquot of each DNA sample was electrophoresed in a 2% agarose gel. The DNA was then transferred onto a Hybond-N membrane (Amersham Pharmacia Biotech), hybridized with the XBP-1 DNA probe in Ultrahyb solution (Ambion) overnight at 42°C, washed as recommended, then exposed to phosphorimager cassettes, scanned and analyzed by Image Quant software (Molecular Dynamics).
Statistical analysis
The time course of appearance of apoptotic and CHOP-positive profiles in the postnatal 6OHDA model was analyzed by anova with a Tukey post hoc analysis. Stereological determination of the number of SN dopaminergic neurons in the 6OHDA and MPTP lesion experiments was analyzed by anova with a Tukey post hoc analysis. The number of apoptotic profiles in wild-type and CHOP null adult mice in the 6OHDA model was analyzed by the t-test. All statistical analyses were performed using SigmaStat software (SPSS Science, Chicago, IL, USA).
Results
CHOP protein expression is induced in a developmental neurotoxin model of parkinsonism
We initially performed in vivo experiments in a rat developmental model in which the intrastriatal injection of 6OHDA results in the induction of death in dopamine neurons of the SN, exclusively with the morphology of apoptosis (Marti et al. 1997). In this model, the unilateral intrastriatal injection of 6OHDA resulted in the unilateral induction of CHOP protein expression, demonstrated by immunohistochemistry (Fig. 2a). On the side of injection, CHOP expression was observed only in the SNpc, the exclusive site of neuron death in this model (Marti et al. 1997). CHOP expression was characterized at a cellular level by performing double-label immunofluorescence for CHOP and TH, to identify dopaminergic neurons of the SN. This analysis revealed that CHOP was expressed predominantly in the nucleus (Figs 2b,b′). To determine the cellular sites of CHOP expression within the SNpc, we examined 50 representative CHOP-positive nuclear profiles among six sections derived from two animals. This analysis showed that all CHOP-positive nuclei were within TH-positive, dopaminergic neurons of the SNpc. Thus, there was a precise correlation at the cellular level between the neuronal population that undergoes death in this model, and CHOP expression (Fig. 2b). All of the CHOP- and TH-positive neuronal profiles identified by the double-labeling procedure had a normal neuronal morphology: abundant cytoplasm, with a polygonal shape, and tapered proximal dendrites. We know from previous studies of this model that the vast majority of dopamine neurons die (Marti et al. 1997) and therefore, CHOP-positive profiles (all of which were TH-positive) are exceedingly likely to be destined to die. We therefore interpret the normal-appearing morphology to mean that if CHOP is to be implicated as a death mediator, it is expressed early in the death process, before any morphological change at the cellular level.
Fig. 2.

Localization and time course of CHOP expression following developmental 6OHDA lesion in postnatal rats. (a) Low power photomicrographs at PLD6 of the substantia nigra contralateral (control: Con) and ipsilateral (experimental: Exp) to an intrastriatal injection of 6OHDA in a PND7 rat. CHOP protein expression is demonstrated by immunoperoxidase staining without a counterstain. CHOP-positive nuclei therefore appear as punctate brown profiles at this power. On the contralateral control side (a′), there is an absence of staining. On the ipsilateral experimental side, numerous CHOP-positive profiles are observed within the SNpc (b′). No positive profiles were observed in the SNpr or in the midbrain dorsal to the SNpc. (b) Double-immunofluorescence labeling for CHOP and TH in the SNpc at PLD4 following intrastriatal injection of 6OHDA in a PND7 rat. TH immunostaining is demonstrated by Texas Red (a′), CHOP by fluorescein (b′), and the merged image is shown in c′. CHOP immunostaining was predominantly nuclear. Following 6OHDA injection, CHOP staining was observed strictly within TH-positive, dopaminergic profiles of the SNpc. Note that CHOP-positive profiles appear normal morphologically; there is no apparent change in neuronal shape or proximal dendrites in comparison with adjacent, CHOP-negative, TH-positive neurons. Bar in c′ = 10 μm. (c) Time course for the appearance of apoptotic and CHOP-positive profiles in SN following intrastriatal injection of 6OHDA in PND7 rats. A total of 24 rats was studied: n = 4 at PLD0 and 2; n = 5 at PLD4 and 6; n = 6 at PLD8. CHOP-positive and apoptotic profiles were counted in the same sections from each animal, as described in Methods. The number of CHOP-positive profiles reached a peak at PLD4 (**p < 0.02 vs. PLD0, 2 and 8; anova, Tukey post hoc). The number of apoptotic profiles also reached a peak at PLD4 (*p < 0.05 vs. PLD0 and 8; anova, Tukey post hoc). However, the time of induction for the two types of profile differed at PLD2; for apoptotic profiles, the number at PLD2 was induced and not significantly different from the number at peak, whereas for CHOP profiles, there was no induction at PLD2. As discussed in the text, this difference may suggest that there are non-CHOP-dependent, as well as CHOP-dependent mechanisms of cell death in this model.
We investigated the time course of CHOP expression at the population level in this model. We recognize that since apoptosis occurs rapidly (Oppenheim 1991), and since at any given time of killing of the animal there will be a heterogeneous population of dying cells in varying stages of the death process, this population analysis will not resolve the cellular sequence of events. Nevertheless, it is informative to determine whether, at the population level, the appearance of CHOP-positive profiles correlates with the appearance of apoptotic profiles. CHOP expression at the population level in this model correlated at most times with the induction of apoptotic death (Fig. 2c). The occurrence of the peak number of CHOP-positive nuclear profiles corresponded precisely with the occurrence of the peak number of apoptotic profiles at post-lesion day (PLD) 4. However, one exception to this correlation was that apoptosis was induced as early as PLD2, in the absence of any induction of CHOP, suggesting that an early component of apoptosis in this model is not associated with CHOP induction, as discussed further below.
Having demonstrated a co-localization between CHOP expression and the dopaminergic neuronal phenotype, and a temporal correlation between CHOP expression and apoptosis in the SN, we next examined the generality of the relationship in other developmental models in which apoptosis occurs. During the postnatal development of SN dopamine neurons, there is naturally-occurring cell death, exclusively with the morphology of apoptosis (Janec and Burke 1993; Oo and Burke 1997). Immunostaining for CHOP was performed on SN sections obtained from PND 14 rats (during the second phase of naturally-occurring cell death). We examined 36 SN sections among n = 4 rats and no instance of CHOP positivity was identified. Among these sections, 124 apoptotic profiles were identified, due to natural cell death (Fig. 3a). This naturally-occurring cell death can be augmented by an axotomy lesion of the medial forebrain bundle during the postnatal period (El-Khodor and Burke 2002). Examination of 18 SN sections from three PND6 rats at 24 h post-axotomy failed to reveal any CHOP-positive profiles (Fig. 3b). Among these sections, numerous apoptotic profiles were identified in SN, as described (El-Khodor and Burke 2002), and sections from 6OHDA-treated animals processed in parallel were positive for CHOP (Fig. 3c). Thus, we conclude that in the postnatal developmental period, CHOP protein expression is induced by the neurotoxin 6OHDA, but not by naturally-occurring cell death or a physical lesion that augments it.
Fig. 3.

CHOP is expressed in neurotoxin models of induced death in SN dopamine neurons. CHOP immunoperoxidase histochemistry was performed on free-floating sections, as described in Methods, with rabbit anti-CHOP (Zinszner et al. 1998) at 1 : 500 for 48 h, followed by thionin counterstain. (a) CHOP expression does not occur in SN during the apoptotic postnatal natural cell death event. A representative field showing a single apoptotic profile (arrow) in a PND14 rat is negative for CHOP immunostaining. (b) The naturally-occurring cell death event in SN can be augmented by postnatal axotomy of the dopaminergic axonal projection (El-Khodor and Burke 2002), as it is for many other developing neural projections (Oppenheim 1991). As for natural cell death, CHOP expression does not occur in this context, as shown by a representative field in a PND6 rat at 1 day post-lesion. An apoptotic profile is shown (arrow). (c) Unlike naturally-occurring cell death and axotomy, cell death induced by 6OHDA in PND7 rat results in the expression of CHOP in many neuronal profiles in the SNpc (broad arrowheads). In this model, CHOP-positive profiles rarely show basophilic apoptotic chromatin clumps (narrow arrow) (2% of instances). As discussed in the text, this rare association between CHOP expression and apoptotic nuclear morphology suggests that if CHOP is implicated in mediating death, it is likely to be an early participant, typically before morphological change. Bar = 20 μm for a, b and c. (d) A representative neuronal profile with a CHOP-positive nucleus (broad arrow) is shown at PLD6 following intrastriatal injection of 6OHDA in an adult mouse. (e, f) CHOP nuclear staining is also observed in SNpc neurons following MPTP injection in adult mice by either the chronic (C) or acute (A) regimens. Bar = 10 μm for d, e, f.
CHOP protein expression in adult neurotoxin models of parkinsonism
To investigate the expression of CHOP in adult neurotoxin models, we exclusively studied mice to permit comparison between the 6OHDA model and the widely used MPTP mouse model of parkinsonism (Heikkila et al. 1984; Przedborski and Vila 2003). Adult mice injected into the striatum with 6OHDA demonstrated numerous CHOP-positive nuclei within neurons of the SNpc (Fig. 3d). For the study of MPTP effects on CHOP expression, we evaluated two dose regimens in common current use. Most widely used is an acute set of injections, 20 mg/kg for four doses, 2 h apart on a single day. This dosing regimen induces SN dopamine neuron death in the absence of apoptotic morphology (Jackson-Lewis et al. 1995). A second regimen utilizes a chronic set of injections, 30 mg/kg daily for 5 days (Tatton and Kish 1997), and results in neuron death with the morphological characteristics of apoptosis. In both of these MPTP models, numerous CHOP-positive neuronal profiles were identified within the SN (Figs 3e and f). In all of these adult contexts, positive nuclear CHOP expression was identified in neurons which otherwise appeared normal, suggesting, as previously discussed, that if CHOP is to be implicated as a death mediator in these models, then it is expressed prior to degenerative morphological changes. We conclude from these studies that CHOP is generally expressed in the SNpc in neurotoxin models of parkinsonism.
CHOP mediates neuron death in the adult 6OHDA model
Having demonstrated close relationships between CHOP expression and the death of SN dopamine neurons in these neurotoxin models, we next sought to determine whether CHOP plays a critical functional role in mediating this death, as it has been shown to do in non-neuronal models of cell death due to ER stress (Zinszner et al. 1998) and oxidative stress. For this assessment, we compared the sensitivity of homozygous CHOP null mice with wild-type controls in their degree of sensitivity to neurotoxin-induced neuron death. In the postnatal 6OHDA model, we found that there was no difference between homozygous CHOP nulls and either heterozygous mice or wild-type controls in the degree of apoptosis among SN dopaminergic neurons induced by intrastriatal 6OHDA (Fig. 4). However, we recognized that in this model, death is known to be mediated not only by the direct effect of the neurotoxin but also, in the developmental period, by an ‘axotomy’ effect due to destruction of dopaminergic terminals during a period of target dependence (Marti et al. 1997). Since we had shown directly that axotomy does not induce CHOP expression, we considered the possibility that this admixture of death mechanisms may obscure a role played by CHOP in death due to the neurotoxin. Such a possibility was also suggested by the time course analysis in Fig. 2(c), which showed an early apoptotic component in the absence of CHOP induction. We therefore examined the sensitivity of adult CHOP null mice to intrastriatal injection of 6OHDA, as adult dopamine neurons do not have target dependence (Kelly and Burke 1996).
Fig. 4.
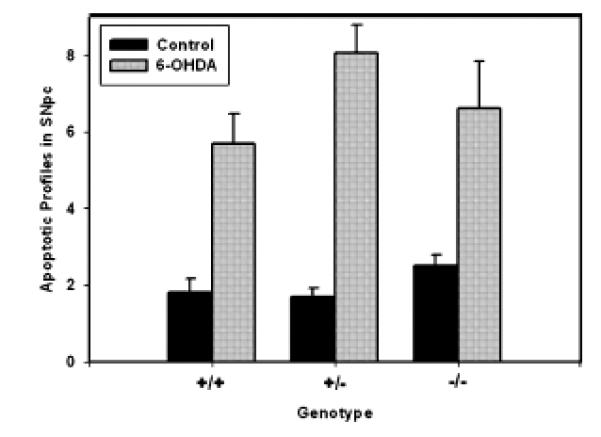
The CHOP null mutation does not protect from induction of apoptosis in the developmental 6OHDA model. In total, 20 PND6 mice (wild-type n = 5; heterozygous n = 10; null n = 5) received a unilateral intrastriatal 6OHDA injection and were killed at PLD4 for the determination of apoptotic profiles within the TH-immunostained substantia nigra, as described in Methods. In all three genotypes, there was a robust induction of apoptosis, as previously described for rats (Marti et al. 1997) (anova p < 0.001 for the 6OHDA effect). There were, however, no differences among the genotypes for this effect.
In adult mice, there was a clear protective effect of the homozygous CHOP null mutation (Fig. 5). CHOP null animals demonstrated a 65% reduction in the number of apoptotic profiles in the SNpc at the sixth post-lesion day. To determine whether this reduction in the magnitude of neuron death resulted in a lasting protection from the neurotoxin, we examined the number of surviving TH-positive neurons in the SN at 28 days post-lesion. This analysis revealed that the null mutation did provide a substantial, lasting protective effect; there was a 79% increase in the number of surviving TH-positive neurons in comparison with wild-type controls (control: 857 ± 131; null: 1531 ± 173 neurons per SN) (Fig. 5b). Nevertheless, the absolute magnitude of the protective effect in the nulls, expressed as 31% survival, while significantly greater than that in the wild-type (19%, p < 0.02), was considerably less than anticipated based on a 65% suppression of apoptotic death in the acute period. In addition, at 28 days post-lesion, there was no evidence for sparing of dopaminergic innervation of the striatum in the nulls. In the nulls, there was a 28.0 ± 3.2 sparing of the optical density of TH-positive fibers within the striatum, as there was in wild-type controls (28.3 ± 3.6).
Fig. 5.
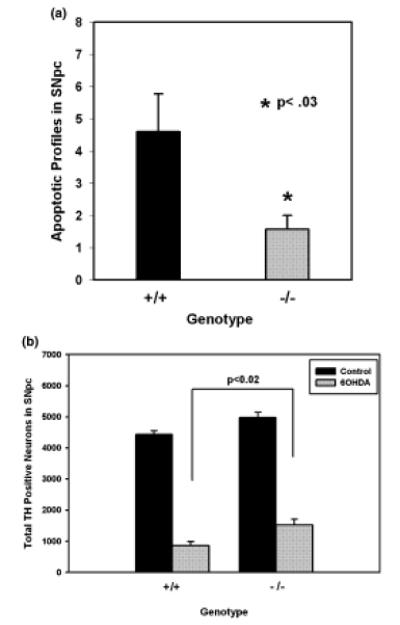
The CHOP null mutation protects from apoptotic cell death in the adult 6OHDA model. (a) Wild-type (n = 5) and CHOP homozygous null (n = 6) adult mice were injected into the striatum with 6OHDA. They were killed 6 days later for TH immunostaining of the SN and counting of apoptotic profiles within the SNpc. The CHOP null animals demonstrated a 65% reduction in the level of apoptosis (p < 0.03, t-test). (b) Wild-type (n = 7) and CHOP null (n = 8) adult mice were injected with 6OHDA and killed 28 days later for TH immunostaining of serial sections for stereologic determination of the number of surviving dopaminergic neurons. In both genotypes, the 6OHDA injection led to a significant reduction in the number of SN dopamine neurons (p < 0.001, anova; Tukey post-hoc). In the CHOP null animals, there was a 79% increase in the number of surviving neurons (p < 0.02, Tukey post hoc). Nevertheless, the absolute magnitude of preservation of neurons (31%) was less than anticipated, based on a much greater level of suppression of death in the acute phase.
CHOP mediates a cellular response to injury, but not neuron death, in the chronic MPTP model
Given that CHOP expression is induced in both the acute and chronic MPTP models, we sought to determine whether it plays a role as a death mediator, as it does in the adult 6OHDA model. Since the role of CHOP as a death mediator has previously been identified in non-neuronal cells in the context of apoptosis (Zinszner et al. 1998), we examined its role in the chronic MPTP model in which apoptosis has been identified (Tatton and Kish 1997). Based on our results in the adult 6OHDA model demonstrating a disparity between the ability of the CHOP null mutation to protect from death in the acute period following the lesion as compared with the chronic period, we conducted separate assessments of both of these post-lesion periods. We found that the CHOP null mutation provided a protective effect in the acute (PLD4) period following the chronic administration of MPTP. The CHOP null animals demonstrated only a non-significant trend for a decrease in the number of TH-positive SN neurons at this time, whereas wild-type controls demonstrated a 65% decrease (Figs 6a and b). However, this difference could not be attributed to a difference in the magnitude of apoptotic death between the two genotypes. While there was a trend towards fewer apoptotic profiles in these sections among the CHOP null mice (2.7 ± 0.8 profiles/SN), this did not achieve significance in comparison with the wild-type (5.2 ± 1.1, p > 0.1, Tukey post hoc). We therefore attribute the marked difference in number of TH-positive neurons between the two genotypes to the well described suppression of TH phenotype following MPTP treatment (Jackson-Lewis et al. 1995). In keeping with this interpretation, in the chronic setting at 21 days post-lesion, there was only a 36% decrease in TH neuron number following MPTP in the wild-type animals. This apparent increase in the number of TH-positive neurons between the acute and chronic lesion periods has previously been shown to be due to a recovery of phenotype (Jackson-Lewis et al. 1995). In the chronic period, in the MPTP-treated mice, unlike the 6OHDA-treated mice, there was no protective effect of the null mutation on the number of surviving TH-positive neurons (Fig. 6c). This difference between the two models is in keeping with the lack of an effect of the null mutation on the magnitude of cell death in the acute period of the MPTP model, whereas there was a pronounced effect in the 6OHDA model. As would be expected from the lack of a protective effect of the null mutation on TH-positive neuron number, there was also no protective effect on striatal TH-positive fiber density (data not shown). We therefore conclude that in the chronic MPTP model, CHOP appears primarily to play a role in the loss of phenotype response that accompanies cellular injury, rather than in cell death, as it does in the 6OHDA model.
Fig. 6.
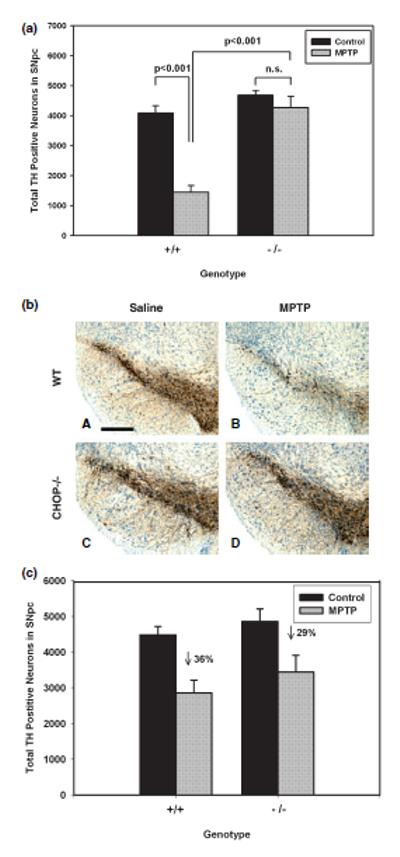
The CHOP null mutation provides early protection from loss of phenotype, but not from neuron death, in the chronic MPTP model. (a) Wild-type and CHOP null adult mice were injected with saline or MPTP (30 mg/kg/day) for 5 days (n = 4 each group except wild-type saline, n = 3) and killed 4 days after the last dose for immunostaining and stereologic determination of TH-positive neuron number. Remarkably, there was minimal apparent effect in the CHOP nulls treated with MPTP. The wild-type mice showed a 65% decrease in number of TH-positive profiles. This difference could not be attributed to a change in the magnitude of apoptosis, as discussed in the text. (b) Representative low power photomicrographs demonstrating the resistance of SN dopamine neurons in CHOP null mice to the early effect (4 days post-lesion) of MPTP in the chronic injection model. These sections are derived from mice studied by stereologic analysis of TH-positive neuron number, shown in (a). Bar 300 μm. (c) Wild-type and CHOP null mice were injected with saline=or MPTP (n = 4–5 each group) and killed 21 days following the final injection for TH immunostaining and stereology. At this late post-lesion day, when the acute suppression of phenotype has recovered, it is apparent that there has been only a 36% loss of SN dopamine neurons in wild-type mice. While there was a trend for a reduction in the number of neurons lost in the CHOP null mice (29% loss), this did not achieve significance (p > 0.5, Tukey post-hoc).
CHOP induction in neurotoxin models is not accompanied by changes in BiP mRNA expression, or the appearance of the XBP-1 splice variant
The induction of CHOP alone cannot be taken as compelling evidence for the occurrence of the ER stress response because CHOP can be induced by other cell stressors, such as oxidative stress, arsenite exposure and amino acid limitation (Bruhat et al. 1997; Jousse et al. 1999; Entingh et al. 2001; Mengesdorf et al. 2002). Therefore, to determine whether the induction of CHOP observed in these models was indicative of the broader ER stress response, we examined the mRNA expression of an ER-resident chaperone, BiP (also know as Grp78) (Gething 1999; Kaufman 1999). Induction of BiP mRNA has previously been shown to occur in vitro in conjunction with CHOP induction upon exposure of neuronal cells to 6OHDA (Ryu et al. 2002; Holtz and O'Malley 2003). In addition, we assessed nigral tissue by PCR for the presence of a splice variant of the transcription factor x-box binding protein-1 (XBP-1) (Yoshida et al. 2001; Calfon et al. 2002), a specific marker for the unfolded protein response.
Northern analysis of SN tissue from mice treated according to the chronic MPTP regimen on the last day of injection (PLD0) or 2 days after the final injection failed to demonstrate any change in BiP mRNA in comparison with saline-treated controls (not shown). To conduct an analysis of BiP mRNA expression at the SNpc regional and cellular levels in the adult 6OHDA model, we performed NRISH. As previously reported by others for normal rat (Little et al. 1996), we observed widespread constitutive expression of BiP mRNA in brain (not shown). However, we did not observe any induction in SNpc, at the regional or cellular level, following unilateral intrastriatal 6OHDA injection at PLD2. A similar analysis of MPTP-treated mice failed to show any difference in BiP mRNA expression in SNpc in comparison with saline-treated controls (not shown).
Southern analysis of PCR reaction products for the XBP-1 unspliced and spliced variants was performed with the inclusion of a positive control derived from renal tissue of tunicamycin-treated mice, in which the ER stress response has previously been demonstrated (Zinszner et al. 1998). This analysis was performed for SN tissues derived from 6OHDA-treated mice at 1 and 3 days post-lesion, and for tissues derived from mice treated with MPTP according to both the acute and chronic regimens. In no instance was the XBP-1 splice variant identified in SN tissues, in spite of its clear presence in tunicamycin-treatment renal tissue. We conclude that in spite of the induction of CHOP protein in these models, there is no additional biochemical evidence of an unfolded protein response using these methods at the tissue level.
Discussion
The hypothesis that PCD plays a role in neural degeneration in PD rests principally on two forms of evidence. First, in rodent neurotoxin models, there is histological and biochemical evidence for activation of PCD mediators, such as the caspases, and functional evidence from genetic and pharmacological studies (reviewed in Vila and Przedborski 2003). Second, while traditional morphological assessments of human PD post-mortem brains for apoptosis have been controversial, there has been growing evidence for activation of caspases (Hartmann et al. 2000, 2001; Viswanath et al. 2001). While this evidence validates PCD as a target for the development of neuroprotective therapeutics, much remains unknown, particularly about upstream mediators that would make attractive therapeutic targets (Yuan and Yankner 2000).
The identification of CHOP as a markedly up-regulated transcript following the treatment of catecholaminergic cell lines with dopaminergic neurotoxins (Ryu et al. 2002; Holtz and O'Malley 2003) and with rotenone, a mitochondrial Complex 1 inhibitor (Ryu et al. 2002), is of particular interest because as a transcription factor, it would be likely to play an upstream regulatory role. In keeping with that possibility, a gene activated by CHOP, DOC6, is homologous to gelsolin, a mediator of cytoskeletal collapse during apoptosis (Wang et al. 1998). CHOP is also of particular interest in relation to PD because it has been implicated as an apoptotic mediator in the setting of oxidative stress (Guyton et al. 1996; Mengesdorf et al. 2002), which has been long postulated to play a role in PD (reviewed in Fahn and Cohen 1992), and ER stress (Matsumoto et al. 1996; Zinszner et al. 1998; Kawahara et al. 2001; Maytin et al. 2001; Gotoh et al. 2002; Oyadomari and Mori 2004), which has likewise recently been implicated in this disease (Imai et al. 2000, 2001).
We have determined that CHOP is expressed in neurotoxin animal models of parkinsonism. In a developmental model of apoptosis induced in dopamine neurons of the SN by the intrastriatal injection of 6OHDA (Marti et al. 1997), there was robust induction of CHOP protein expression exclusively within the SNpc. At a cellular level, CHOP expression was nuclear, as expected for a transcription factor, and exclusively within dopaminergic neurons. CHOP expression was also observed in neurotoxin models in the adult setting following intrastriatal 6OHDA, and either acute or chronic systemic MPTP exposure. In these adult models, as in the developmental 6OHDA model, CHOP expression was strictly within the SNpc at a regional level, and within the nucleus of otherwise normal-appearing neurons at a cellular level. CHOP expression, however, is not a universal feature of apoptosis in dopamine neurons; in the developmental setting, it is observed neither during naturally-occurring cell death (Janec and Burke 1993; Oo and Burke 1997), nor with augmentation of this death by axotomy (El-Khodor and Burke 2002). On the basis of classic neurotrophic theory (Clarke 1985), the naturally-occurring cell death event and its augmentation by axotomy would be considered to be regulated by the availability of neurotrophic support. Our observations that CHOP is not induced in these conditions, but it is by neurotoxic insults, are comparable with the in vitro observations of Ryu et al. (2002), who noted that CHOP is induced by neurotoxins, but not by neurotrophic withdrawal.
The principal finding of these investigations was that adult CHOP null mice were resistant to apoptotic death in SN dopamine neurons induced by the intrastriatal injection of 6OHDA. We considered the possibilities that this reduction may be due to a change in the time course of apoptosis, or to the rate of clearance of apoptotic profiles in the null mice, rather than an actual reduction in the eventual magnitude of the death event. We therefore assessed the final surviving number of SN DA neurons at PLD28 and found that they were increased, indicating that the null mutation did in fact reduce the magnitude of death. We therefore conclude that CHOP is an important functional mediator of apoptosis in the 6OHDA model. Given that CHOP is highly expressed prior to any morphologic change in dopamine neurons destined to die in this model, we postulate that CHOP is likely to be an early mediator in the death process. Although the CHOP null mutation was protective in this model, the degree of preservation of SN dopamine neurons in absolute terms, 31%, was less than anticipated based on a 65% suppression of apoptotic death in the early post-lesion period. This discrepancy suggests that some of the death which ultimately occurs in the CHOP nulls is delayed. There are two possible explanations for this delay. First, death mediators other than CHOP may eventually come into play (Ryu et al. 2005) and bring about the loss of the majority of dopamine neurons. Second, in these non-temporally-regulated nulls, compensatory changes may have taken place to provide alternate death pathways. These two possibilities are not mutually exclusive.
In view of the ability of the CHOP null mutation to provide neuroprotection in the adult 6OHDA model, the question arises as to why it did not also provide protection in the postnatal model, in which CHOP expression is clearly induced. Our interpretation of this difference rests on our previous demonstrations that during the first two postnatal weeks, SN dopamine neurons are dependent on interactions with their target, the striatum, as envisioned by classic neurotrophic theory (Clarke 1985), whereas in adults they are not (Macaya et al. 1994; Kelly and Burke 1996; Stefanis and Burke 1996). Therefore, during this postnatal period, the death of SN dopamine neurons following the destruction of their nerve terminals with 6OHDA is likely to be mediated by an ‘axotomy’ effect as well as a direct neurotoxic effect. This interpretation is supported not only by the aforementioned studies of the developmental time course of striatal target dependence, but also by our demonstrations that the postnatal 6OHDA model is characterized by two cellular patterns of caspase activation: a perinuclear pattern, as observed in naturally-occurring cell death (Jeon et al. 1999; El-Khodor and Burke 2002; Oo et al. 2002), and a cytoplasmic pattern, observed in direct neurotoxic injury (Jeon et al. 1999; Oo et al. 2002). Given this likelihood of an axotomy effect in the postnatal 6OHDA model, and based on our demonstration herein that CHOP is not expressed following developmental axotomy, we would anticipate that a functional role for CHOP would be difficult to discern in the postnatal 6OHDA lesion.
MPP+, the toxic metabolite of MPTP, induced CHOP expression in in vitro models (Ryu et al. 2002; Holtz and O'Malley 2003). MPTP treatment in vivo likewise induced the expression of CHOP protein, but in the chronic MPTP model, unlike the 6OHDA model, the CHOP null mutation did not significantly diminish the level of apoptosis or increase the number of surviving neurons. The null mutation did, however, prevent the loss of TH immunoreactivity in the period early after the MPTP injections. We interpret this relative preservation of TH immunoreactivity in the absence of protection from cell death to be attributable to protection from the loss of phenotype, which is well documented in this (Jackson-Lewis et al. 1995) and other neuronal injury models (Wooten et al. 1978). We conclude that while CHOP plays a role in regulating cellular phenotype in the MPTP model, it is not likely to play a role as an important death mediator. This difference in the role of CHOP between the 6OHDA and MPTP models in living animals is consistent with the observations made in vitro by Holtz and O'Malley (2003). Following treatment with 6OHDA, they observed a greater induction of CHOP and a more general induction of other ER stress markers than with MPTP treatment.
To determine whether the CHOP induction observed in these neurotoxin models was specifically due to ER stress, we assayed mRNA expression of the ER-resident chaperone BiP (Gething 1999; Kaufman 1999) and the splice variant of XBP-1 (Yoshida et al. 2001; Calfon et al. 2002). In none of the models was there a change observed in BiP mRNA expression or the appearance of the XBP-1 splice variant. These results were not unexpected for the MPTP model in view of in vitro results that showed no induction of BiP or XBP-1 by MPP+ (Holtz and O'Malley 2003). However, the results were unexpected for the 6OHDA model as both prior in vitro studies had demonstrated clear evidence for a full ER stress response induced by 6OHDA (Ryu et al. 2002; Holtz and O'Malley 2003). There are two principal interpretations of these negative results. First, it is possible that CHOP induction in the 6OHDA model in living animals is not part of a full ER stress response, the in vitro results notwithstanding. It is well established that 6OHDA produces oxidative stress (Heikkila and Cohen 1973; Cohen and Heikkila 1974). It is therefore possible that its induction of CHOP in living animals is mediated principally by cellular oxidative stress (Guyton et al. 1996; Mengesdorf et al. 2002). Alternatively, it is possible that the studies of BiP and the XBP-1 splice variant that were performed at the tissue level lacked the sensitivity to detect changes, which, for CHOP, were detected at the cellular level by immunohistochemistry. Thus, our inability to detect other markers for ER stress in the 6OHDA model does not permit us to definitively conclude that it is not present.
We conclude that these investigations performed in living animals are largely supportive of the in vitro results suggesting the possibility of a role for CHOP in the neurodegeneration associated with PD. We find, as predicted from these gene expression screens, that CHOP is expressed in diverse neurotoxin models of dopamine neuron death. These observations support the validity of the in vitro screens for genes of potential relevance to disease. In addition, we find that CHOP can play a role as a mediator of cell death, depending on the context; in the 6OHDA model, CHOP is a necessary death mediator. The context specificity of CHOP is an important feature, because it suggests that it may be possible in designing neuroprotection strategies to target disease-related death pathways without interfering with other apoptotic pathways that may be important for survival of the organism.
Acknowledgements
This work was supported by NS26836, NS38370, DAMD17-03-1-0492, ES08681, NS43628, The Parkinson's Disease Foundation and the Michael J. Fox Foundation. We gratefully acknowledge the quantitative morphological analysis performed by Ms Rebecca Greene.
Abbreviations used
- ABC
avidin-biotinylated-horseradish peroxidase complexes
- ER
endoplasmic reticulum
- MFB
medial forebrain bundle
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NRISH
non-radioactive in situ hybridization
- 6OHDA
6-hydroxydopamine
- PB
phosphate buffer
- PBS
phosphate-buffered saline
- PCD
programmed cell death
- PD
Parkinson's disease
- PLD
post-lesion day
- PND
post-natal day
- SSC
saline sodium citrate
- SN
substantia nigra
- TBS
Tris-buffered saline
- TH
tyrosine hydroxylase
References
- Bruhat A, Jousse C, Wang XZ, Ron D, Ferrara M, Fafournoux P. AminoacidlimitationinducesexpressionofCHOP,aCCAAT/enhancer binding protein-related gene, at both transcriptional and post-transcriptional levels. J. Biol. Chem. 1997;272:17 588–17 593. doi: 10.1074/jbc.272.28.17588. [DOI] [PubMed] [Google Scholar]
- Burke RE, Franklin SO, Inturrisi CE. Acute and persistent suppression of preproenkephalin mRNA expression in the striatum following developmental hypoxic-ischemic injury. J. Neurochem. 1994;62:1878–1886. doi: 10.1046/j.1471-4159.1994.62051878.x. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PGH. Neuronal death in the development of the vertebrate nervous system. Trends Neurosci. 1985;8:345–349. [Google Scholar]
- Coggeshall RE, Lekan HA. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J. Comp. Neurol. 1996;364:6–15. doi: 10.1002/(SICI)1096-9861(19960101)364:1<6::AID-CNE2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Cohen G, Heikkila RE. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J. Biol. Chem. 1974;249:2447–2452. [PubMed] [Google Scholar]
- El-Khodor BF, Burke RE. Medial forebrain bundle axotomy during development induces apoptosis in dopamine neurons of the substantia nigra and activation of caspases in their degenerating axons. J. Comp. Neurol. 2002;452:65–79. doi: 10.1002/cne.10367. [DOI] [PubMed] [Google Scholar]
- El-Khodor BF, Kholodilov NG, Yarygina O, Burke RE. The expression of mRNAs for the proteasome complex is developmentally regulated in the rat mesencephalon. Brain Res. Dev. Brain Res. 2001;129:47–56. doi: 10.1016/s0165-3806(01)00181-x. [DOI] [PubMed] [Google Scholar]
- Entingh AJ, Law BK, Moses HL. Induction of the C/EBP homologous protein (CHOP) by amino acid deprivation requires insulin-like growth factor I, phosphatidylinositol 3-kinase, and mammalian target of rapamycin signaling. Endocrinology. 2001;142:221–228. doi: 10.1210/endo.142.1.7906. [DOI] [PubMed] [Google Scholar]
- Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson's disease: Evidence supporting it. Ann. Neurol. 1992;32:804–812. doi: 10.1002/ana.410320616. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Oo TF, Rzhetskaya M, Pratt R, Yarygina O, Momoi T, Kholodilov N, Burke RE. CEP11004, a novel inhibitor of the mixed lineage kinases, suppresses apoptotic death in dopamine neurons of the substantia nigra induced by 6-hydroxydopamine. J. Neurochem. 2004;88:469–480. doi: 10.1046/j.1471-4159.2003.02176.x. [DOI] [PubMed] [Google Scholar]
- Gething MJ. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Gotoh T, Oyadomari S, Mori K, Mori M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J. Biol. Chem. 2002;277:12 343–12 350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl Acad. Sci. USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Xu Q, Holbrook NJ. Induction of the mammalian stress response gene GADD153 by oxidative stress: role of AP-1 element. Biochem. J. 1996;314:547–554. doi: 10.1042/bj3140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Hunot S, Michel PP, et al. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc. Natl Acad. Sci. USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis. J. Neurosci. 2001;21:2247–2255. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila RE, Cohen G. 6-Hydroxydopamine: evidence for superoxide radical as an oxidative intermediate. Science. 1973;181:456–457. doi: 10.1126/science.181.4098.456. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Hess A, Duvoisin RC. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science. 1984;224:1451–1453. doi: 10.1126/science.6610213. [DOI] [PubMed] [Google Scholar]
- Heller A, Hutchens JO, Kirby ML, Karapas F, Fernandez C. Stereotaxic electrode placement in the neonatal rat. J. Neurosci. Meth. 1979;1:41–76. doi: 10.1016/0165-0270(79)90006-2. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J. Biol. Chem. 2003;278:19 367–19 377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000;275:35 661–35 664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Immanuel D, Zinszner H, Ron D. Association of SARFH (sarcoma-associated RNA-binding fly homolog) with regions of chromatin transcribed by RNA polymerase II. Mol. Cell Biol. 1995;15:4562–4571. doi: 10.1128/mcb.15.8.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Tsuji S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology. 1996;47:160–166. doi: 10.1212/wnl.47.1.160. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Janec E, Burke RE. Naturally occurring cell death during postnatal development of the substantia nigra of the rat. Mol. Cell Neurosci. 1993;4:30–35. doi: 10.1006/mcne.1993.1004. [DOI] [PubMed] [Google Scholar]
- Jeon BS, Kholodilov NG, Oo TF, Kim S, Tomaselli KJ, Srinivasan A, Stefanis L, Burke RE. Activation of caspase-3 in developmental models of programmed cell death in neurons of the substantia nigra. J. Neurochem. 1999;73:322–333. doi: 10.1046/j.1471-4159.1999.0730322.x. [DOI] [PubMed] [Google Scholar]
- Jousse C, Bruhat A, Harding HP, Ferrara M, Ron D, Fafournoux P. Amino acid limitation regulates CHOP expression through a specific pathway independent of the unfolded protein response. FEBS Lett. 1999;448:211–216. doi: 10.1016/s0014-5793(99)00373-7. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Oyadomari S, Gotoh T, Kohsaka S, Nakayama H, Mori M. Induction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cells. FEBS Lett. 2001;506:135–139. doi: 10.1016/s0014-5793(01)02898-8. [DOI] [PubMed] [Google Scholar]
- Kelly WJ, Burke RE. Apoptotic neuron death in rat substantia nigra induced by striatal excitotoxic injury is developmentally dependent. Neurosci. Lett. 1996;220:85–88. doi: 10.1016/s0304-3940(96)13216-x. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM, Jacobowitz DM. Pharmacological actions of 6-hydroxydopamine. Pharmacol. Rev. 1974;26:199–288. [PubMed] [Google Scholar]
- Little E, Tocco G, Baudry M, Lee AS, Schreiber SS. Induction of glucose-regulated protein (glucose-regulated protein 78/BiP and glucose-regulated protein 94) and heat shock protein 70 transcripts in the immature rat brain following status epilepticus. Neuroscience. 1996;75:209–219. doi: 10.1016/0306-4522(96)00267-9. [DOI] [PubMed] [Google Scholar]
- Macaya A, Munell F, Gubits RM, Burke RE. Apoptosis in substantia nigra following developmental striatal excitotoxic injury. Proc. Natl Acad. Sci. USA. 1994;91:8117–8121. doi: 10.1073/pnas.91.17.8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti MJ, James CJ, Oo TF, Kelly WJ, Burke RE. Early developmental destruction of terminals in the striatal target induces apoptosis in dopamine neurons of the substantia nigra. J. Neurosci. 1997;17:2030–2039. doi: 10.1523/JNEUROSCI.17-06-02030.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano P, Lowry O, Carter J, Johnson E. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 1996;395:143–147. doi: 10.1016/0014-5793(96)01016-2. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001;267:193–204. doi: 10.1006/excr.2001.5248. [DOI] [PubMed] [Google Scholar]
- Mengesdorf T, Althausen S, Paschen W. Genes associated with pro-apoptotic and protective mechanisms are affected differently on exposure of neuronal cell cultures to arsenite. No indication for endoplasmic reticulum stress despite activation of grp78 and gadd153 expression. Brain Res. Mol. Brain Res. 2002;104:227–239. doi: 10.1016/s0169-328x(02)00384-4. [DOI] [PubMed] [Google Scholar]
- Oo TF, Burke RE. The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Dev. Brain Res. 1997;98:191–196. doi: 10.1016/s0165-3806(96)00173-3. [DOI] [PubMed] [Google Scholar]
- Oo TF, Siman R, Burke RE. Distinct nuclear and cytoplasmic localization of caspase cleavage products in two models of induced apoptotic death in dopamine neurons of the substantia nigra. Exp. Neurol. 2002;175:1–9. doi: 10.1006/exnr.2002.7881. [DOI] [PubMed] [Google Scholar]
- Oo TF, Kholodilov N, Burke RE. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal GDNF in vivo. J. Neurosci. 2003;23:5141–5148. doi: 10.1523/JNEUROSCI.23-12-05141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Ann. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: Evidence for the role of cell death genes. Dev. Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Vila M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson's disease. Ann. N Y Acad. Sci. 2003;991:189–198. [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J. Neurochem. 2001;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J. Neurosci. 2002;22:10 690–10 698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol. Dis. 2005;18:54–74. doi: 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6 hydroxydopamine a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Senoh S, Witkop B. Formation and rearrangements of aminochromes from a new metabolite of dopamine and some of its derivatives. J. Am. Chem. Soc. 1959;81:6231–6235. [Google Scholar]
- Shimura H, Hattori N, Kubo S, et al. Familial parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Burke RE. Transneuronal degeneration in substantia nigra pars reticulata following striatal excitotoxic injury in adult rat: Time course, distribution, and morphology of cell death. Neuroscience. 1996;74:997–1008. doi: 10.1016/0306-4522(96)00175-3. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Natl Acad. Sci. USA. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003;4:365–375. doi: 10.1038/nrn1100. [DOI] [PubMed] [Google Scholar]
- Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J. Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. EMBO J. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten GF, Park DH, Joh TH, Reis DJ. Immunochemical demonstration of reversible reduction in choline acetyltransferase concentration in rat hypoglossal nucleus after hypoglossal nerve transection. Nature. 1978;275:324–325. doi: 10.1038/275324a0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]