


Abstract
Tiling microarrays have proven to be a valuable tool for gaining insights into the transcriptomes of microbial organisms grown under various nutritional or stress conditions. Here, we describe the use of such an array, constructed at the level of 20 nt resolution for the Escherichia coli MG1655 genome, to observe genome-wide changes in the steady-state RNA levels in mutants defective in either RNase E or RNase III. The array data were validated by comparison to previously published results for a variety of specific transcripts as well as independent northern analysis of additional mRNAs and sRNAs. In the absence of RNase E, 60% of the annotated coding sequences showed either increases or decreases in their steady-state levels. In contrast, only 12% of the coding sequences were affected in the absence of RNase III. Unexpectedly, many coding sequences showed decreased abundance in the RNase E mutant, while more than half of the annotated sRNAs showed changes in abundance. Furthermore, the steady-state levels of many transcripts showed overlapping effects of both ribonucleases. Data are also presented demonstrating how the arrays were used to identify potential new genes, RNase III cleavage sites and the direct or indirect control of specific biological pathways.
INTRODUCTION
The analysis of the post-transcriptional processing, maturation and decay of RNA molecules in prokaryotes such as Escherichia coli has historically focused on individual classes of molecules such as rRNAs, tRNAs, mRNAs and small RNAs (sRNAs). Thus, considerable effort has been invested in understanding the maturation of 30S rRNA precursors into mature 16S, 23S and 5S rRNA species (1), the processing of tRNA precursors (2–7), the mechanisms of mRNA decay (8) and the processing and degradation of sRNAs (9–11) (http://exosal.org).
Although it was originally thought that different ribonucleases might be involved in the processing, maturation and decay of particular classes of RNA molecules, work over the past 15 years has clearly demonstrated that a limited set of ribonucleases mediate all aspects of RNA metabolism in E. coli (7,8,12). For example, the essential endoribonuclease RNase E, encoded by the rne gene, is involved in many aspects of RNA metabolism, including mRNA decay (13–17), sRNA processing and decay (10,18), tRNA processing (3,5,19) and rRNA maturation (20,21). In contrast, RNase G, a paralog of RNase E, appears to have a much more limited range of substrates, including some mRNAs and 16S rRNA precursors (21–25). On the other hand, RNase III is primarily known for its role in rRNA maturation (26), and also has been shown to be involved, to a limited extent, in mRNA degradation and sRNA processing (27–32). In addition, recent studies have demonstrated that some E. coli sRNAs regulate the stability and translation initiation efficiency of specific mRNAs through RNase III-dependent cleavages (33,34).
Historically, the analysis of RNA transcripts has relied on either northern blots or, to a lesser extent, qRT-PCR. Northern analysis is a particularly powerful method for studying RNA processing and decay, since it permits the visualization of both a full-length or mature transcript and its degradation or processing intermediates. However, both methods have significant limitations in that they are time consuming and cannot easily discern interactions among ribonucleases or provide an overview of general pathways of RNA processing and decay. Thus, despite years of research, many questions remain unanswered regarding the overall in vivo roles of ribonucleases such as RNase E and RNase III in E. coli RNA metabolism.
The development of DNA macro- and microarrays led to studies in E. coli that explored gene regulation in response to various stresses and growth conditions (35,36). A further application of macro- and microarrays has been to study the effect of nuclease mutations on overall mRNA abundance. For example, Mohanty and Kushner (37) used macroarrays to determine how the deletion of either polynucleotide phosphorylase (PNPase) or RNase II (both 3′ → 5′ exonucleases) affected the steady-state levels of all the E. coli open reading frames. In addition, Lee et al. (23) demonstrated that the steady-state levels of 40% of the coding sequences (CDSs) in a mutant containing an RNase E deletion, kept viable by a 174-fold increase in RNase G levels (25), changed in abundance compared with a wild type control. However, these studies relied upon relatively low-resolution gene expression arrays, which only included information about mRNA abundances.
In contrast, tiling DNA microarrays provide the ability to study RNA processing on a transcriptome-wide scale. It is therefore now possible to simultaneously examine the role of any ribonuclease on all coding and non-coding RNAs in the transcriptome. Other potential advantages of the increased resolution associated with tiling microarrays are the ability to predict the approximate locations of RNase cleavage sites, the identification of potentially novel genes and small RNAs and the examination of the effects of a particular ribonuclease on specific biological pathways.
In the work presented here, we have compared the transcriptomes, at 20 nt resolution, of wild type E. coli to both an RNase E deletion mutant (rneΔ1018), kept viable by an altered RNase G protein (rng-219) (25) and an RNase III null mutant (rnc-14::ΔTn10) (38). The RNase E deletion mutant was particularly useful because it contained only about a 12-fold increase in a mutant RNase G protein containing a single amino acid substitution in its RNase H domain (25) as compared with the strain employed by Lee et al. (23), which contained a 174-fold increase in the level of an extended form of RNase G (25).
Our analysis of the tiling microarray data for the rne deletion strain showed that 1520 CDSs (35% of the annotated CDSs) contained one or more regions (at least 100 nt or two contiguous probes) that were increased in abundance compared with the wild type control by at least 1.5-fold. In addition, 1096 CDS (25% of CDSs) had at least one region of decreased abundance when compared with the wild type control. We also determined that at least 47 annotated non-coding RNAs (ncRNAs) were affected directly or indirectly by the absence of RNase E.
A second experiment employing total RNA isolated from an RNase III null mutant revealed that 391 CDSs (9%) contained one or more regions that were increased in abundance as compared with wild type, while 120 CDSs (3%) exhibited decreased abundance. This is the first reported array study in E. coli using an RNase III null mutant and the data indicate that RNase III has a more widespread role in cellular RNA processing than previously envisioned. Furthermore, there appears to be a high level of redundancy and/or possible cooperation between RNase III/RNase E cleavage events for a number of transcripts. Of considerable interest was the number of changes in the abundance of small regulatory RNAs in the two mutants versus the wild type control, which in and of itself could be responsible for a number of the observed changes throughout the transcriptome. The data have also permitted the identification of RNase III cleavage sites, including one within an mRNA coding sequence, and a large number of potentially novel transcripts.
MATERIALS AND METHODS
Construction of the E. coli tiling microarray
Both strands of the MG1655 genome (version U00096.2, GI:48994873) were tiled using 60 nt length probes containing 20 bp overlaps at each end. Sixty nucleotide long, negative control probes were created by generating random sequences with a 50% GC content and selecting those with low cross-reactivity against E. coli probes using OligoPicker (39). The final array design contained 231 984 E. coli probes, 6415 duplicate E. coli probes and 3000 random probes. Probe sequences were submitted to Agilent Technologies (Palo Alto, CA, USA) for array manufacture (AMADID 015365). Probes were spotted randomly on the microarray slide to reduce any potential effects of background non-specific hybridization.
Bacterial strains and plasmids
All E. coli strains used in this study were derived from MG1693 (thyA715 rph-1) provided by the E. coli Genetic Stock Center, Yale University. SK3564 [rneΔ1018::bla thyA715 rph-1 recA56 srlD::Tn10 (Tcr) /pDHK30(rng-219 Smr/Spr)/pWSK129 (Kmr)] is an RNase E deletion strain in which cell viability is supported by a mutant RNase G (rng-219) protein synthesized from a single copy plasmid (25). SK4455 (rnc-14::ΔTn10 thyA715 rph-1) was constructed by moving the rnc-14::ΔTn10 allele from HT115 (38) via P1 transduction into MG1693 and subsequent selection for Tcr. The presence of the rnc-14::ΔTn10 allele in SK4455 was confirmed by analysis of genomic DNA using PCR (data not shown) and by western blot analysis with monoclonal anti-RNase III antibodies, which were a gift from D. Court (data not shown).
Growth of bacterial strains and isolation of total RNA
Strains were grown with shaking at 37°C in Luria broth supplemented with thymine (50 µg/ml) until a cell density of approximately 2.5 x 108 cfu/ml (60 Klett units above background, No. 42 green filter) was reached. RNA was extracted using the method described by O’Hara et al. (40), with the exception that 10% trimethyl(tetra-decyl)ammonium bromide (Sigma) was used in place of Catrimox-14 (41). RNA was quantified on a NanoDropTM (Thermo Scientific) apparatus. Five hundred nanogram of each RNA sample were run on a 1% Agarose–Tris-acetate-EDTA gel and visualized with ethidium bromide to ensure accurate quantities and sufficient quality for further analysis. RNA to be used for microarray hybridization was further treated to remove any contaminating DNA with the DNA-free kitTM (Ambion) and was further analyzed for quality on a Bioanalyzer (Agilent Technologies).
Microarray analysis
Array hybridization, image processing, data normalization and visualization were performed as described in Hiley et al. (42). RNA samples were directly labeled using the Ulysses system from Invitrogen. The genome annotations used in this study were obtained from E. coli strain MG1655, genome version NC_000913.2, GI:49175990. Small RNA annotation information contained in Supplementary Table S2 was obtained from the EcoCyc database (43). Fold change numbers derived from the array data were calculated by averaging the normalized log2 ratios for all probes which either map within the genomic feature or are within 100 nt of the feature to account for spacer regions. Experimental artifacts due to non-specific hybridization background were minimized in the averages by excluding the highest and lowest 10% of individual signal ratios as outliers. All fold change ratios reported in this study have been converted from log2 ratios to linear ratios. Greatest change fold ratios from the array data were obtained from PeakFinder (44) average binding ratios representing the greatest change within a genomic feature. Values represented as negative fold changes in this study were derived by taking the negative inverse of the linear binding ratio to better describe the fold RNA abundance change between mutant and wild type.
Gene Ontology analysis for changes with P ≤ 0.1 for SK4455 was performed using the Wallenius distribution method, described in Young et al. (45) and the gene ontology database (46). P-values were corrected for multiple testing using the Benjamini and Hochberg method (47).
Due to the large amounts of data generated in this study, all array data and electronic files necessary for viewing and evaluation of the data are available online on the Kushner laboratory web site (http://www.genetics.uga.edu/kushner/).
Northern analysis
Northern analysis was performed as described in O’Hara et al. (40). In the case of RNA species less than 500 nt in length, 10 µg of total RNA were run on 6% polyacrylamide–8.3 M urea gels. For RNA species larger than 500 nt, 10 µg of total RNA was run on 1.2% agarose/3-(N-morpholino)propanesulfonic acid-acetate-EDTA gels using the protocol of Vincze and Bowra (48). Northern blots were probed with 32P-5′-end-labeled oligonucleotides (41) and were scanned with a PhosphorImager (StormTM 840, GE Healthcare). The data were quantified using ImageQuant TL software (GE Healthcare). The relative quantities (RQs) reported were normalized based on the loading controls (either 5S or 23S rRNA) determined for each northern blot.
Primer extension
Primer extension analysis for the nirB transcript was performed as described previously (4) with the exception of primers that were specifically designed for nirB.
Microarray data and oligonucleotide sequences
All microarray data and oligonucleotide sequences used in this study are available upon request. Oligonucleotide sequences used for the northern blots in Figure 4 were as follows: cspE (5′-GACGTATCTTACAGAGCGAT-3′); dnaK (5′-TCATGTGTTTCGGACCGGTCGCGTCTGCAGTGATGTAT-3′); ompF (5′-TCACCGTTACCCTTGGAAAAATAATGCAGACCAACAGCTTT-3′); ryhB [probe used was the same sequence as probe EM1 from Massé and Gottesman (49) 5′-AAGTAATACTGGAAGCAATGTGAGCAATGTCGTGCTTTCAGGTTCTC-3′]; 6S RNA (ssrS) (5′-ATATCGGCTCAGGGGACT-3′); 5S rRNA (5′-CTACTCAGGAGAGCGTTCACCG-3′) (50); and 23S rRNA (5′-CGTCCTTCATCGCCTCTGACT-3′). The oligonucleotide sequence for the primer extension reaction on the nirB mRNA is 5′-AGCGATGCGCGGTTCTTCAC-3′. The primers used to generate a PCR sequencing template for nirB are 5′-TCAGCCGTCACCGTCAGCAT-3′ and 5′-CGCGTTCGCCGACCAGAAC-3′.
Figure 4.

Northern analysis of specific transcripts. Northern blots were conducted as described in ‘Materials and Methods’ section. For each blot with the exception of dnaK, the relative quantity values of total signal from the bands shown (RQ) represent an average of at least two independent experiments. The RQ values have been normalized based on an rRNA loading control appropriate to the gel used (each nylon membrane was stripped of the original oligonucleotide probe used as described in ‘Materials and Methods’ section, then hybridized with oligonucleotides specific for either 23S rRNA in the case of dnaK and ompF, and 5S rRNA for remaining blots). Standard deviations (±) are shown below the RQ values where appropriate. All northern blots shown are representative of multiple independent replicates which have been performed (data not shown). Ribonucleotide size estimates are indicated with arrows for each band shown, with the exception of ryhB for which the annotated length is shown.
RESULTS
Analysis of the E. coli transcriptome in the absence of either RNase E or RNase III
The construction of a tiling microarray at 20 nt resolution provided an opportunity to determine the impact of both RNase E and RNase III on every transcript generated from the entire genome. Total steady state RNA from MG1693 (wild type), SK3564 (rneΔ1018/rng-219) and SK4455 (rnc-14::ΔTn10) was directly labeled with either Cy3 or Cy5 fluorescent dyes and hybridized to custom DNA tiling microarrays (see ‘Materials and Methods’ section). Duplicate arrays were performed with biological replicates and dye swaps to minimize experimental artifacts. Normalized log2 binding ratios were mapped to the genome and visualized using the Integrated Genome Browser (IGB) (51), while peaks associated with significant log2 ratios were identified using the PeakFinder program (44).
Initial validation of microarray data
For the initial validation of the microarray data, we examined the steady-state levels of the rne and rng mRNAs isolated from SK3564 (rneΔ1018/rng-219). As expected, there was both a significant decrease in the rne mRNA along with a concomitant increase in the rng transcript (data not shown). As a second internal control, we examined the rpsO pnp operon that encodes ribosomal protein S15 and polynucleotide phosphorylase, respectively. It has previously been shown that the pnp transcript is significantly stabilized in the absence of RNase III (52–54), while the rpsO mRNA is very dependent on RNase E for its degradation (13,17,22,25). As shown in Figure 1, there was increased abundance for the rpsO mRNA in SK3564 but no significant change in SK4455. In contrast, in the absence of RNase III the pnp transcript showed a significant increase in abundance (Figure 1). Finally, we observed that changes in the steady state levels of six additional mRNAs (rpsT, cspE, htpG, glpQ, raiA and trxA) were in excellent agreement with previously published results obtained from direct measurements of their half-lives in the RNase E mutants (Table 1).
Figure 1.

Microarray data for the rpsO pnp operon. Changes in the steady-state levels of the rpsO and pnp mRNAs in RNase E and RNase III deletion mutants. The image presented was obtained from a screen shot of the Integrated Genome Browser program (51). Labels for the data displayed are located on the far left of the image and identify all features within the same horizontal plane. Gene names appear above or below the horizontal bar indicating their location on the genome relative to actual nucleotide coordinates, which are displayed at the center of the graph. Objects above the genome coordinate line are on the forward strand, while objects below the coordinate line are on the reverse strand. Black arrows indicate the direction of transcription. CDSs are colored blue, while red indicates a sRNA. The actual array data are displayed as a series of vertical lines representing the log2 ratio of fluorescence between the mutant and wild-type strains along a horizontal that intersects the label on the far left designating the mutant strains and strand (+ or −). The horizontal line in the array data is equal to a log2 ratio of 0, with vertical lines going above or below the baseline, representing changes in the log2 ratio of greater or less than 0 for each probe. Vertical lines above the baseline indicate higher RNA abundance in the mutant versus wild type, while line extending below the baseline indicate lower RNA abundance in the mutant versus wild type. Maximum and minimum peak heights displayed are equivalent to log2 ratios of ±3.
Table 1.
Comparison of array data with published experiments that employed various RNase E mutants
| Gene/operon | Published observation in absence of RNase E | References | Observed change in steady-state RNA levels on array |
|---|---|---|---|
| rpsT | Increased half-life | (22,58) | Increased abundance of transcript |
| rpsO | Increased half-life | (13,22) | Increased abundance of transcript |
| cspE | Increased half-life | (58) | Increased abundance of transcript |
| htpG | Increased half-life | (58) | Increased abundance of transcript |
| glpQ | Increased half-life | (58) | Increased abundance of transcript |
| raiA | No change in half-life | (22) | No change in transcript abundance |
| trxA | No change in half-life | (22) | No change in transcript abundance |
| csrC | Increased half-life | (84) | Increased abundance of transcript |
| rrl rrs | Increased stability of spacer region between 23S and 5S sequences | (22) | Increased abundance of spacer region between mature 23S and 5S rRNA sequences |
| pheU | Increased abundance of full-length tRNA precursor | (3) | Increased abundance of terminator region |
| secG leuU | Increased half-life of secG mRNA and stabilization of leuU terminator region | (5) | Increased abundance of secG mRNA and leuU terminator region |
| glyW cysT leuZ | Increased abundance of polycistronic transcript | (3,19) | Increased abundance of spacer regions |
| tyrT tyrV tpr | Increased abundance of polycistronic transcript | (3,19) | Increased abundance of spacer regions |
| metT leuW glnU glnW metU glnV glnX | Increased abundance of polycistronic transcript | (5) | Increased abundance of spacer regions |
| argX hisR leuT proM | Increased abundance of polycistronic transcript | (3,19) | Increased abundance of spacer regions |
| valV valW | No change in abundance of polycistronic transcript | (4) | No change in abundance of polycistronic transcript |
| leuQ leuP leuV | No change in abundance of polycistronic transcript | (4) | No change in abundance of polycistronic transcript |
| metV metW metZ | No change in abundance of polycistronic transcript | (4) | No change in abundance of polycistronic transcript |
In the case of mRNAs, published data relates to observed changes in individual half-lives. For tRNA transcripts, the published data relate to whether inactivation of RNase E led to changes in the levels of the full-length mono- or polycistronic transcripts.
It is important to note that changes in probe signal intensity between mutant and wild type strains on a tiling microarray reflect a change in steady-state RNA levels and do not necessarily indicate alterations in RNA stability, since changes in transcription could also result in abundance differences. However, if changes in abundance are observed in externally or internally transcribed spacer regions flanking the mature sequences in non-coding RNAs (such as tRNAs, rRNAs and sRNAs), while the abundance of the mature sequences is unchanged, then transcription likely has not been affected and the resulting increases or decreases in RNA levels arise from either stabilization or destabilization of that region of an RNA transcript.
For example, for all seven rRNA operons in E. coli, which are known to undergo rapid initial maturation by RNase III to separate the 16S and 23S rRNAs from a larger 30S precursor (55,56), in the absence of RNase III we saw significant increases in the spacer regions of the rRNA operons but not in the abundance of the mature 16S and 23S species (Figure 2, data not shown). Interestingly, small increases in the spacer regions were also observed in the absence of RNase E (Figure 2, data not shown), which may indicate a slight stabilization of the 30S rRNA precursor. Similar increases in the abundance of either intergenic regions, upstream or downstream sequences were also detected for a variety of tRNA transcripts (pheU, glyW cysT leuZ, argX hisR leuT proM) (data not shown) that have been shown to be dependent on RNase E for their initial processing (3,19).
Figure 2.

Microarray data for the rrnE ribosomal RNA operon. Data are presented as described in Figure 1. Green bars represent rRNA genes, while orange bars indicate tRNA genes.
Detailed analysis of specific transcripts
Although the preliminary analysis of the array data showed the expected changes in the steady state levels for some specific mRNAs, we chose a number of additional transcripts to examine in more detail in order to better correlate the changes observed on the arrays with the actual alterations in steady state RNA levels. For example, the cspE mRNA, which encodes a transcription anti-terminator and RNA stability regulatory protein, has previously been shown to be more stable in strains carrying a temperature-sensitive allele of RNase E (57–59). Interestingly, our array data showed increased abundance of the cspE mRNA in both SK3564 (4.0-fold) and SK4455 (2.2-fold) versus the wild type control (Figure 3). When northern analysis was used to verify the abundance of the cspE mRNA in wild type, SK3564 and SK4455 strains, there were 3.4-fold and 1.3-fold increases in the full-length cspE mRNA in the SK3564 and SK4455, respectively (Figure 4; Table 2).
Figure 3.

Microarray data for cspE and adjacent genes. Data are presented as described in Figure 1.
Table 2.
Comparison of transcript abundance between wild-type and mutant strains using array and northern data
| Gene | Mutant/wild type | Fold change from array | Fold change from northern analysis |
|---|---|---|---|
| cspE | ΔRNase E | 4.0 | 3.4 |
| ΔRNase III | 2.2 | 1.3 | |
| dnaK | ΔRNase E | 2.0 | 2.3 |
| ΔRNase III | 2.0 | 1.3 | |
| ompF | ΔRNase E | −1.6 | −1.3 |
| ΔRNase III | 2.0 | 2.7 | |
| ryhB | ΔRNase E | 7.6 | 20.4 |
| ΔRNase III | 5.4 | 3.7 | |
| ssrS | ΔRNase E | ND | −1.6 |
| ΔRNase III | ND | 1.1 |
Positive values indicate higher abundance in the mutant, while negative values indicate higher abundance in wild type. Fold changes from the array data were calculated by averaging the ratios of every oligonucleotide probe across the genomic feature (see ‘Materials and Methods’ section). Fold change numbers from the northern analyses were calculated as described in the ‘Materials and Methods’ section. ND, not determined due to the nature of the abundance change of ssrS (see Supplementary Figure S6).
The dnaK mRNA (60), which encodes heat shock protein 70, appeared to be significantly more abundant in both mutants compared with the wild type control, while the downstream dnaJ transcript was more abundant in only the RNase III strain (Supplementary Figure S1). The dnaK mRNA is transcribed from three separate promoters to produce a dnaK dnaJ dicistronic transcript, which also includes the putative tpke11 sRNA between the two coding regions (61–63). Northern analysis showed that dnaK mRNA was 2.3-fold more abundant in SK3564 and 1.3-fold more abundant in SK4455 than in the wild type control (Figure 4; Table 2). The primary band detected on the northern blot corresponded to a size of 2.1-kb, which contained only the coding region of dnaK with additional nucleotides at both the 5′- and 3′-ends. Additional mRNAs that were tested included yncL (Supplementary Figure S2) and yncE (Supplementary Figure S3). In both cases, the northern data confirmed the changes observed on the microarrays (data not shown).
The ompF mRNA, which encodes the outer membrane porin F protein, has a single known promoter and is thought to be produced as a monocistronic transcript (64,65). The array data showed that in SK3564 the mRNA was actually less abundant with a fold change of −1.6 (the negative inverse of the linear ratio between mutant and wild type of 0.63. See ‘Materials and Methods’ section) as compared with the wild type control (Figure 5). In contrast, in the RNase III mutant, the ompF mRNA was 2.0-fold more abundant compared with the wild type control (Figure 5). Northern analysis of ompF confirmed the array data, showing that there was 2.7-fold more ompF in SK4455 and 1.3-fold less in SK3564 than in wild type control (Figure 4; Table 2). Although the decrease in the ompF mRNA was initially surprising, it has been shown that the micF sRNA is a negative regulator of ompF stability and is itself regulated by RNase E (66,67). In fact, there was a significant increase (4.8-fold) in the amount of the micF sRNA in the absence of RNase E (Figure 6).
Figure 5.

Microarray data for ompF and adjacent genes. Data are presented as described in Figure 1.
Figure 6.

Microarray data for the micF sRNA. Data are presented as described in Figure 1.
The sRNA rhyB has been shown to be more abundant in both RNase E and RNase III mutant backgrounds (10). These results were confirmed by our array data and subsequent northern blotting (Supplementary Figure S4; Figure 4; Table 2). Also, 6S RNA, a sRNA involved in the regulation of stationary phase transcription, showed an increase in abundance at its 5′-end in SK3564 in both the array data and northern analysis as compared with very little change in the SK4455 (Supplementary Figure S5; Figure 4; Table 2), which was in agreement with previous 6S RNA biogenesis studies (18).
Genome-wide impact of RNase E and RNase III deletions
In order to determine the transcriptome-wide impact of eliminating either RNase E or RNase III, we established a threshold of significance based on our previous comparisons between the array data and published observations, in which the ratio of signal intensity between mutant and wild type of at least two contiguous oligonucleotide probes (with respect to the genome coordinates, not physical location) had to change by at least ±1.5-fold (linear ratio comparing mutant versus wild type, not a log2 ratio) in order for a difference between the mutant and wild type strain to be considered significant. These criteria for significance, in the vast majority of cases, excluded both experimental noise (due to background non-specific hybridization on the microarray slides) and transcripts that were known to be unaffected in these mutants, from being included in the list of significant changes. We also ensured that our selection thresholds remained suitably sensitive to reliably include transcripts that have been shown to change in abundance in these mutants, even if the abundance changes were relatively small.
Using the PeakFinder program (44) with the thresholds described above, we generated lists of genomic coordinates that had significantly different abundances of RNA when comparing our mutants with a wild type control. The list generated using data from SK3564 included 1520 CDSs (35%) that contained at least one positive peak, indicating higher RNA abundance in the RNase E deletion strain compared with the wild type control (Figure 7; Supplementary Table S1). Furthermore, 35 ncRNAs (Supplementary Table S1) also showed increased in abundance in the RNase E deletion strain. In contrast, 1096 CDSs (25%) contained at least one negative peak, along with 12 ncRNAs (Supplementary Table S1). Also of interest was the observation that in many messages, including the clpX, lon and ppiD mRNAs, there were differential changes from the 5′- to 3′-end of each transcript (Supplementary Figure S6; data not shown).
Figure 7.

Characterization of transcriptome changes in the absence of RNase E and RNase III. The data within the graph is derived from the number of specific genomic features in each strain that contain a region of at least 100 nt changed by at least 1.5-fold, versus the total number of that gene feature annotated in the genome (see ‘Materials and Methods’ section). Dark grey bars that extend above the 0% X-axis indicate the number of a specific type of genomic feature which contain an area of increased abundance, versus the number of total features of that type. Light grey bars that extend below the 0% X-axis indicate the number of a specific type of genomic feature that contain an area of decreased abundance, versus the number of total features of that type. The list of genomic features affected in these strains can be found in Supplementary Table S1. The two annotated tmRNA features are not included in this figure.
Using the same criteria with data from SK4455, 391 CDSs (9%) containing at least one peak of higher abundance and 120 CDS (3%) containing at least one peak of lower abundance were detected (Figure 7). Furthermore, 11 ncRNAs were more abundant in SK4455 than in the wild type control, with 4 additional ncRNAs, showing decreased abundance.
While the overall number of RNA abundance changes between the two ribonuclease mutants was very different, there were a significant number of genomic features, which were shared between both the SK3564 and SK4455 lists of affected genes (Supplementary Table S1). The number of shared RNA abundance changes within total genomic features included: 10% of the CDSs and 21% of annotated ncRNAs.
To obtain gene expression array profiling information about the overall abundance of a particular genomic feature, the RNA abundance ratios corresponding to each genomic feature that contained a significant change (as determined by our criteria described above) were averaged (see ‘Material and Methods’ section) (Supplementary Table S1). We observed that many genomic features only had a higher or lower RNA abundance in the mutant versus wild type in a small region of the feature, which may in fact still affect the functionality of the RNA species. Accordingly, in order to permit a more accurate numerical view of the changes in RNA abundance with these mutants, we also included in Supplementary Table S1 a greatest fold change value, which was simply the greatest change in average binding ratio within the feature as detected and calculated by PeakFinder (44). The calculation of both the average change and greatest change (along with the location of the greatest change) allows perspective on which genomic features were affected uniformly (where the difference between the average and greatest values is minimal) or non-uniformly (where the difference between the average and greatest values is appreciable) and provides information about which area(s) of a transcript were most highly affected. Additionally, some genomic features contained both a peak of higher RNA abundance and a peak of lower abundance and are so noted in Supplementary Table S1.
RNase E and RNase III do not significantly affect polycistronic mRNAs
A distinguishing feature of prokaryotic organisms is the presence of significant numbers of polycistronic transcripts. It is widely assumed that larger polycistronic transcripts are rapidly processed into smaller species by endonucleolytic cleavages. Accordingly, it was of considerable interest to determine if the absence of either RNase E or RNase III led to differential effects on individual CDSs within polycistronic transcripts. For example, as shown in Figure 1 for the dicistronic rpsO pnp transcript, loss of RNase E activity led to the stabilization of the complete rpsO mRNA, while inactivation of RNase III increased the steady state level of every oligonucleotide probe specific for pnp but not rpsO. In fact, for a number of short polycistronic operons (2–3 CDSs), we observed situations where inactivation of RNase E and/or RNase III each led to increased steady state levels of both CDSs (groS groL; Table 3), decreased steady-state levels (cysD cysN cysC; Table 3) or as in the case of the lac operon, increased steady-state levels in the absence of RNase E but no change in the absence of RNase III (Table 3). However, for the vast majority of larger polycistronic transcripts examined (4–12 CDSs), there was little or no change in the steady-state levels of any of the CDSs within the larger transcripts or increased steady-state levels of the full-length transcripts (Table 3).
Table 3.
Changes in steady-state levels of individual ORFs within representative polycistronic transcripts in the absence of either RNase Ea or RNase IIIb
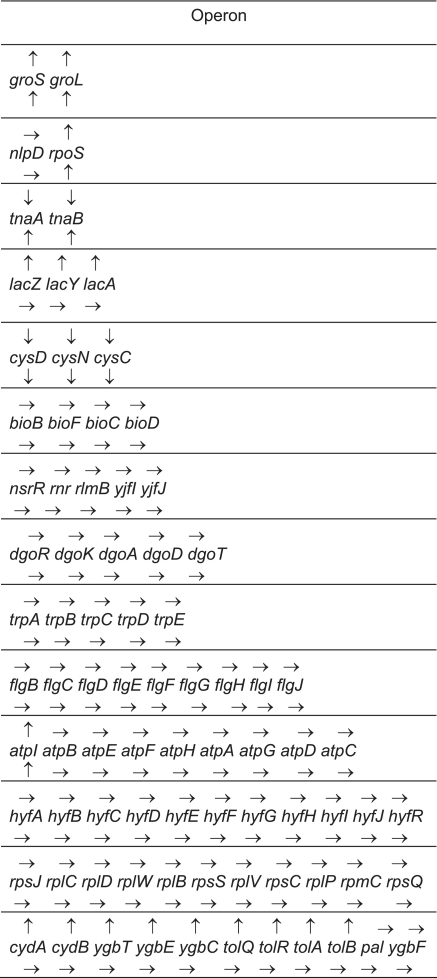 |
Arrows over the gene name indicate the steady-state abundance of the mRNA in the absence of RNase E versus wild type, while arrows under the gene name indicate the abundance changes in the absence of RNase III versus wild type. Upward arrows indicated increased steady-state levels in the mutant compared with the wild type control. Downward arrows indicated decreased steady-state levels in the mutant versus the wild type control. Horizontal arrows mean no significant change in the steady-state RNA levels between the mutant and wild type control. In order to be included in this table, every oligonucleotide probe for a particular ORF had to be significantly increased, decreased or unchanged.
arneΔ1018/rng-219.
brnc-14::ΔTn10.
Identification of novel RNA transcripts
Microarrays have been used to identify novel sRNAs in microbial genomes by comparing total RNA harvested from wild type cells grown at various stages of their growth cycle, grown under different stress or nutrient-starvation conditions or RNA coimmunoprecipitated with antibodies raised against the RNA binding protein Hfq (68–71). Since it has been shown that RNase E is involved in the degradation of some microbial sRNAs (10), we hypothesized that the array using RNA from the RNase E deletion mutant could permit the detection of both annotated and potential new sRNAs as well as other novel transcripts. Consistent with this hypothesis was the fact that 47/63 (75%) of the annotated ncRNAs in the NCBI database (see ‘Materials and Methods’ section) showed at least a ±1.5-fold change in the RNase E deletion mutant relative to the wild type control (Supplementary Table S1). This observation highlighted the potential utility of tiling array analysis of ribonuclease mutant strains in screening for novel transcripts.
The array data also facilitated the detection of RNase E-dependent sRNAs under different cell growth conditions. For example, based on published data using wild-type cells, ryhB is only detected in either stationary phase or cells that have been grown in minimal medium (72). Yet in exponentially growing cells, the deletion of RNase E resulted in a >20-fold increase in the ryhB sRNA compared with the wild-type control (Figure 4; Supplementary Figure S2; Table 2).
To extend this analysis, we manually mined the entire genome for unannotated regions that contained at least two adjacent oligonucleotide probes whose abundance was increased by ≥1.5-fold in the RNase E deletion strain compared with the wild type control, without regard to the location of currently annotated ncRNAs. In this manner, we identified a total of 328 loci (Supplementary Table S2). Within this list were 37 annotated sRNAs, none of which were placed on the list intentionally as the data were compiled without regard to the location of annotated sRNAs. Of the remaining newly identified loci, 113 were located in intergenic regions, 156 were antisense to an annotated gene, 11 overlapped with the putative 5′-end of a gene and 14 overlapped with the putative 3′-end of a gene. Additionally, we detected 74 loci whose abundance decreased in the absence of RNase E by ≤0.67 (the negative inverse of which gives a -1.5-fold change in SK3564 versus wild type). Of these loci, 11 were located within intergenic regions, 62 were antisense to an annotated gene and none overlapped with either the putative 5′- or 3′-end of a gene. Furthermore, two annotated sRNAs, isrA and fnrS, fell into this category (Supplementary Table S2).
Also of interest is candidate 62 in Supplementary Table S2, which is a putative small open reading frame (ORF) located on the opposite strand between genes ompF and asnS (Figure 5) named C0240 by Tjaden et al. (69,73,74). Based on the identification of C0240 in our list of potentially novel transcripts, it is possible that other loci within Supplementary Table S2 are also small ORFs, which remain to be validated. It should also be noted that the array data from the RNase E deletion mutant were utilized to assist in the development of a computational algorithm for de novo identification of non-coding RNAs in prokaryotes (75).
Identification of RNase III cleavage sites based on array data
RNase III cleavage sites, which are associated with certain stem-loop structures, are better characterized than those associated with many other ribonucleases (76). We hypothesized that based on the resolution of the tiling array data we might be able to predict the location of RNase III cleavage sites. For example, there is a sharp change in the RNA abundance ratios between the rpsO and pnp transcripts (Figure 1), which directly corresponds to the known RNase III cleavage site in the intercistronic region between these two genes (77). Accordingly, we scanned the array data derived from SK4455 for CDSs which showed either uniform increased or decreased abundance in the absence of RNase III. The 5′-UTRs for a number of selected CDSs were then folded using RNAstar (78) to determine if a hairpin could form that might be a suitable recognition sequence for RNase III. Subsequently, primer extension experiments were carried out to map potential RNase III cleavages. In the case of nirB, the gene encoding the large subunit of nitrite reductase, the steady-state level of the entire CDS was significantly lower in the absence of RNase III and it appeared that the decrease in abundance mapped closely to the start of translation (Figure 8A). Surprisingly, primer extension analysis showed an RNase III-dependent cleavage 61 nt downstream of the AUG translation start codon (Figure 8B). The intensity of the extension product corresponding to the putative RNase III cleavage site was shown to be dependent on the amount of RNA loaded in each lane in both the wild type and RNase E deletion strains (data not shown), indicating that this extension product was not an experimental artifact. The location of this RNase III cleavage site is unusual in that it is the first report of RNase III cutting within a coding sequence in E. coli.
Figure 8.

Transcriptional analysis of the nirB mRNA. (A) Microarray data for nirB and adjacent genes. Data are presented as described in Figure 1. (B) Primer extension analysis of the nirB transcript. The primer extension was performed as described in the ‘Materials and Methods’ section. The transcription start site of the nirB operon transcript is indicated by an open circle. The diamond denotes a cleavage site found in all three strains located 2 nt upstream of the AUG translation start site, while the open triangle marks the start of the translation start codon in the DNA sequencing ladder. The asterisk (bottom right) indicates a major 5′ terminus, located within the nirB coding sequence that is not seen in the absence of RNase III. The putative RNase III cleavage site (in bold/underlined) is shown in the expanded sequence on the bottom left of the figure.
RNase III affects the regulation of cysteine biosynthesis
As noted above, in both the RNase E and RNase III arrays there were a significant number of CDSs in which steady-state RNA levels were reduced in the absence of either ribonuclease. In the case of RNase III, a systematic analysis of the CDSs identified 12 genes involved in cysteine metabolism, 4 associated with sulfate uptake (cysP cysU cysW cysA) and 8 involved in de novo cysteine biosynthesis (cysD cysN cysC cysJ cysI cysH cysK cysM) (data not shown). The 12 genes are organized in 3 polycistronic and 1 monocistronic operons (cysD cysN cysC, cysJ cysI cysH, cysP cysU cysW cysA cysM and cysK). However, bioinformatic analysis of the leader regions and the first CDS of each transcript did not reveal any significant regulatory sequence motif.
Gene pathway analysis
To better understand how inactivation of RNase E and RNase III impacted gene networks and pathways within the cell, we obtained the KEGG pathway annotations (79) for each genomic feature affected in both SK3564 and SK4455. Unfortunately, due to the relatively large number of genes that were affected in the strains, in addition to the large number of genes that remain unannotated in the KEGG pathway database, we were unable to draw any meaningful conclusions about changes in the metabolic and non-metabolic pathways in the cell (data not shown). However, gene ontology (GO) analysis (46) of the data derived from SK4455 revealed that seven GO terms were significantly affected (P ≤ 0.1) in the absence of RNase III versus the wild type control (Table 4). Among the seven GO terms that were changed was the assimilation of sulfate, which was also indicated by the changes in RNA abundance of 12 cys genes noted above. The pathway changes outlined in Table 4 also showed that in the absence of RNase III there were significant changes in the heat shock pathway, iron transport, enterobactin production, the membrane fraction, cytosol and unfolded protein binding activity. Changes in the membrane components may help to explain a rather odd but reproducible observation that cells deficient in RNase III tend to be more difficult to lyse than wild type cells, or for that matter most other ribonuclease mutant strains (data not shown).
Table 4.
Gene ontology pathways affected in the absence of RNase III
| Ontology type | GO term affected | Definition of GO term | P-value | GO ID |
|---|---|---|---|---|
| Biological process | Response to heat | A change in state or activity of a cell or an organism (in terms of movement, secretion, enzyme production, gene expression, etc.) | 0.0007 | GO:0009408 |
| Biological process | Iron ion transport | The directed movement of iron (Fe) ions into, out of, within or between cells by means of some external agent such as a transporter. | 0.007 | GO:0006826 |
| Biological process | Enterobactin biosynthetic process | The chemical reactions and pathways resulting in the formation of enterobactin, a catechol-derived siderochrome of enterobacteria; 2,3-dihydroxy-N-benzoyl-l-serine and a product of the shikimate pathway. | 0.03 | GO:0009239 |
| Cellular component | Membrane fraction | That fraction of cells, prepared by disruptive biochemical methods, that includes the plasma and other membranes. | 0.03 | GO:0005624 |
| Cellular component | Cytosol | The part of the cytoplasm that does not contain organelles but which does contain other particulate matter, such as protein. | 0.03 | GO:0005829 |
| Molecular function | Unfolded protein binding | Interacting selectively and non-covalently with an unfolded protein. | 0.07 | GO:0051082 |
| Molecular function | Sulfate assimilation | The pathways by which inorganic sulfate is processed and incorporated into sulfated compounds. | 0.08 | GO:0000103 |
The following pathways are enriched for changes in SK4455 (rnc-14) versus wild type at P ≤ 0.1 (see ‘Materials and Methods’ section).
DISCUSSION
Even though microarrays have been previously used to examine changes in steady-state levels of E. coli ORFs (23,80), the data presented here demonstrate that tiling microarrays, based on their 20 nt resolution, provide a much more detailed perspective on the in vivo roles of ribonucleases on E. coli RNA metabolism. The array data were in excellent agreement with previously published results regarding changes in the steady-state levels of specific mRNAs in either RNase E or RNase III mutants (Figures 1, 3 and 6; Supplementary Figure S1; Table 1). Furthermore, northern analysis of additional CDSs and sRNAs demonstrated that changes in steady-state RNA levels, either up or down, relative to a wild type control were accurately reflected by the array data (Figures 3–5; Supplementary S1, S4 and S5; Table 1).
The RNA abundance changes in the CDSs of SK3564 were largely in agreement with previous CDS expression array studies using a different RNase E mutant (23). However, the mutant used in this study, along with the resolution of the arrays, facilitated a much more comprehensive view of the transcriptome changes in the absence of RNase E. This is particularly important as the transcriptome of RNase III-deficient mutants in E. coli had not been previously examined. Additionally, our data suggest that RNase III has a much greater role in the regulation of the transcriptome than previously thought. Furthermore, there also seems to be a considerable level of redundancy, and/or cooperation between RNase E and RNase III activity on a significant number of transcripts, as demonstrated by the 51 CDSs and 9 ncRNAs which were affected by at least ±1.5-fold in both mutants versus the wild type control (Supplementary Table S3). Interestingly, for 11 transcripts, their abundance was higher than wild type in one mutant, but lower than wild type in the other. However, additional experiments are needed to verify whether the endoribonucleases are involved in the same pathway.
It is important to note that the changes in abundance of many annotated ncRNAs (Supplementary Tables S1 and S2) highlight the complexity associated with interpreting some of the data derived from ribonuclease mutants. Although increases in RNA steady-state levels in the absence of a ribonuclease are easily explained by the loss of degradative capacity, we observed that many CDSs actually showed significantly reduced abundance in the RNase E and RNase III mutants compared with wild type control. It is likely that some of these decreases in mRNA abundance were due to changes in the amount of regulatory RNAs in the mutant strains, as demonstrated by the decrease in abundance of ompF mRNA in SK3564 (Figure 5). In this case, an explanation for the decrease in abundance in SK3564 versus wild type may be related to the increase in the steady-state level of the micF sRNA (Figure 6), which is known to regulate the stability of the ompF mRNA. Similarly, we hypothesize that for many other mRNAs changes in sRNA abundance related to inactivation of either RNase E or RNase III indirectly leads to reduced steady-state levels of specific target mRNAs.
Thus, it seems likely that many of the changes observed, with both the RNase E and RNase III arrays, are secondary effects that can be associated with changes in the steady-state levels of various sRNAs or other unidentified regulatory factors. Since some of the annotated sRNAs have already been shown to have multiple targets (49), when coupled with the likelihood of additional sRNAs (Supplementary Table S2), it seems quite probable that a significant portion of the E. coli transcriptome is affected by the abundance of sRNAs.
It is also important to note that there were some cases where increased abundance of a specific sRNA in either the RNase E or RNase III mutant was not accompanied by a decrease in the level of its known mRNA target. There could be a number of possible reasons for this type of result. The most likely explanation for this type of observation is that the abundance of the target mRNA is relatively low such that a significant increase in sRNA level would have little effect on its abundance. It is also possible that the sRNAs affected in either mutant are dependent on the missing ribonuclease for their processing. In the absence of processing, the full-length sRNA may not be functionally active. Additionally, it is possible that under conditions where there is a significant increase in the levels of a large number of sRNAs compared with wild-type cells, that the amount of Hfq [an RNA binding protein required for the function of many sRNAs (70)] may be rate limiting. If this were the case, changes in sRNA levels would not be expected to efficiently affect mRNA target levels.
Overall, our observations provide further support for the notion that the control of RNA stability is in fact a very important aspect of gene regulation in E. coli. Our results also re-emphasize that the changes in steady-state levels of a particular RNA species in the absence of a particular ribonuclease are not adequate proof that the enzyme is directly involved in its processing, maturation or decay. Although our data show that the analysis of ribonuclease mutants can be challenging, they also highlight the sophistication and complexity of post-transcriptional regulation with regard to gene expression in E. coli and most likely in other prokaryotes.
Another important finding regarding sRNAs in E. coli relates to the fact that even though the RNA used in this study was isolated from exponentially growing cells, we were able to detect changes in the steady-state levels of 75% of the annotated ncRNAs in SK3564 and 22% in SK4455 (Supplementary Table S1). This fact is of considerable interest because many of these sRNA species have been shown to be present primarily in either early or late stationary phase cultures. Thus, the arrays will be useful for examining the effects of ribonuclease mutants on the transcriptome during all phases of growth. For example, manual analysis of the data indicated the possibility of 363 additional novel transcripts (excluding annotated sRNAs and a putative ORF, Supplementary Table S2).
Another somewhat surprising observation was the failure to observe significant changes in the steady-state levels of large polycistronic mRNAs (Table 3). It has been assumed that RNase E is responsible for separating polycistronic operons into smaller but functional mRNAs that have differential half-lives. While this notion may be true for a small fraction of polycistronic operons, the more likely scenario is that RNase E is not involved in establishing differential intra-operon half-lives. In fact, mRNAs within large operons may be grouped in such a way to guarantee equal mRNA copy numbers. Such a hypothesis is already widely accepted when dealing with mRNAs encoding ribosomal proteins, and may apply to other classes of mRNAs as well. This could be especially true for mRNAs which together encode multi-subunit enzymes that require the production of each subunit in stoichiometric amounts.
The analysis of the array data also produced some rather unexpected observations. For example, the array showed that the nirB mRNA was less abundant in SK4455 than the wild type control (Figure 8A). However, when we tested the transcript for an RNase III cleavage site because of a predicted hairpin within the 5′-UTR, we detected a strong RNase III cleavage site (Figure 8B), but to our surprise the cleavage occurred at a site well within the coding sequence that would functionally inactivate the mRNA. Since functionally inactive mRNAs tend to have shorter half-lives because they are no longer being translated, it is not clear at this time why inactivating RNase III actually led to reduced abundance of the nirB message. It is possible that the nirB observations are another case of indirect effects, since there was also reduced abundance of the transcript in the absence of RNase E (Figure 8A).
Another example of potential indirect effects associated with removing an important ribonuclease involved the decreased steady-state levels of the 12 genes associated with cysteine metabolism in the absence of RNase III. These 12 cysteine-related genes are found in four different operons (three are polycistronic), and none of them seem to have an RNase III cleavage site within their 5′- or 3′-UTRs. It is thus likely in this case that the inactivation of RNase III leads to changes of a common factor (either a protein or perhaps a sRNA) that coordinately regulates the expression of these genes, rather than direct RNase III cleavages of the mRNAs themselves.
Additionally, while the mapping of promoter initiation sites in E. coli has been performed using a number of highly effective techniques (81,82), because of the plethora of transcriptome changes in the ribonuclease mutants, we suspect that tiling microarray data could allow for the detection of approximate locations of alternative transcription start sites within the transcriptome. By examining the array data near the 5′-ends of a few messages with experimentally verified transcription start sites (data not shown), we found that we could accurately predict the known start sites within a 60-nt window. We believe that our data may prove valuable as a preliminary screen for groups interested in genome-wide promoter usage determination.
It is already clear from the data presented here that the post-transcriptional regulation of gene expression in E. coli is far more complex than previously envisioned. As the transcriptomes of more ribonuclease mutants are analyzed, a more detailed picture of the complexity within transcriptome regulatory networks will begin to emerge and may begin to answer some of the questions which have plagued the prokaryotic RNA field since its inception. It should also be noted that in the time since our tiling microarray data sets were generated, the RNAseq method of transcriptome analysis has been published and widely adopted. While RNAseq is a powerful method for transcriptome analysis (83), the associated costs are prohibitive for many researchers and therefore tiling microarray technology will continue to have a place in the analysis of many transcriptomes.
Finally, it is important to note that when we employed Peakfinder (40) to detect areas of significant changes, we used thresholds, which in the vast majority of cases, excluded both experimental noise (due to background non-specific hybridization on the microarray slides) and transcripts that were known to be unaffected in these mutants, from being included in the list of significant changes. We also ensured that our selection thresholds remained suitably sensitive to reliably include transcripts that have been shown to change in abundance in these mutants, even if the abundance changes were relatively small. Although it is possible that some of the 1.5-fold changes may not be biologically relevant, several such changes were validated using northern blot analysis (Table 2; data not shown). In fact, any change observed on a tiling array needs to be verified by an independent method of analysis. Thus, based on the nature of tiling array technology, no set of criteria will be perfect and the data reported here probably contain a very limited number of both false-positive and false-negative results. For example, regions of the genome that are repetitive such as related tRNA operons may have some level of cross-hybridization. Therefore, the use of tiling array data for these types of genomic features has the potential to be misleading and has not been a focus of this study. In spite of these caveats, the data from this study have allowed for a remarkably detailed overview of the transcriptome-wide impacts caused by the absence of either RNase E or RNase III, and will prove to be of value to anyone interested in these enzymes or the effects they have on specific transcripts.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) (GM57220 and GM81554) to S.R.K in part. NIH predoctoral traineeship from National Institute of General Medical Sciences (NIGMS) (GM07106) to M.B.S in part. Funding for open access charge: NIH grant GM81554.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank D. Court for his gift of strains and antibodies.
REFERENCES
- 1.Srivastava AK, Schlessinger D. Mechanisms and regulation of bacterial ribosomal RNA processing. Ann. Rev. Microbiol. 1990;44:105–129. doi: 10.1146/annurev.mi.44.100190.000541. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Deutscher MP. Maturation pathways for E.coli tRNA precursors: a random multienzyme process in vivo. Cell. 1996;86:503–512. doi: 10.1016/s0092-8674(00)80123-3. [DOI] [PubMed] [Google Scholar]
- 3.Ow MC, Kushner SR. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 2002;16:1102–1115. doi: 10.1101/gad.983502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohanty BK, Kushner SR. Ribonuclease P processes polycistronic tRNA transcripts in Escherichia coli independent of ribonuclease E. Nucleic Acids Res. 2007;35:7614–7625. doi: 10.1093/nar/gkm917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohanty BK, Kushner SR. Rho-independent transcription terminators inhibit RNase P processing of the secG leuU and metT tRNA polycistronic transcripts in Escherichia coli. Nucleic Acids Res. 2008;36:364–375. doi: 10.1093/nar/gkm991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohanty BK, Kushner SR. Processing of the Escherichia coli leuX tRNA transcript, encoding tRNAleu5, requires either the 3′-5′ exoribonuclease polynucleotide phosphorylase or RNase P to remove the Rho-independent transcription terminator. Nucleic Acids Res. 2010;38:5306–5318. doi: 10.1093/nar/gkp997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutscher MP. Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res. 2006;34:659–666. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kushner SR. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Böck A, Curtis R III, Gross CA, Kaper JB, Neidhardt FC, Nyström T, Rudd KE, editors. Washington, DC: American Society for Microbiology Press; 2007. L., S. C. [Google Scholar]
- 9.Lundberg U, Altman S. Processing of the precursor to the catalytic RNA subunit of RNase P from Escherichia coli. RNA. 1995;1:327–334. [PMC free article] [PubMed] [Google Scholar]
- 10.Masse R, Escorcia FE, Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003;17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Pandit S, Deutscher MP. 3′ Exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc. Natl Acad. Sci. USA. 1998;95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpousis AJ. The RNA degradosome of Escherichia coli: An mRNA-dgrading machine assembled on RNase E. Ann. Rev. Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 13.Ow MC, Liu Q, Kushner SR. Analysis of mRNA decay and rRNA processing in Escherichia coli in the absence of RNase E-based degradosome assembly. Mol. Microbiol. 2000;38:854–866. doi: 10.1046/j.1365-2958.2000.02186.x. [DOI] [PubMed] [Google Scholar]
- 14.Ono M, Kuwano M. A conditional lethal mutation in an Escherichia coli strain with a longer chemical lifetime of mRNA. J. Mol. Biol. 1979;129:343–357. doi: 10.1016/0022-2836(79)90500-x. [DOI] [PubMed] [Google Scholar]
- 15.Arraiano CM, Yancey SD, Kushner SR. Stabilization of discrete mRNA breakdown products in ams pnp rnb multiple mutants of Escherichia coli K-12. J. Bacteriol. 1988;170:4625–4633. doi: 10.1128/jb.170.10.4625-4633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackie GA. Specific endonucleolytic cleavage of the mRNA for ribosomal protein S20 of Escherichia coli requires the products of the ams gene in vivo and in vitro. J. Bacteriol. 1991;173:2488–2497. doi: 10.1128/jb.173.8.2488-2497.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regnier P, Hajnsdorf E. Decay of mRNA encoding ribosomal protein S15 of Escherichia coli is initiated by an RNase E-dependent endonucleolytic cleavage that removes the 3′ stabilizing stem and loop structure. J. Mol. Biol. 1991;187:23–32. doi: 10.1016/0022-2836(91)90542-e. [DOI] [PubMed] [Google Scholar]
- 18.Kim KS, Lee Y. Regulation of 6S RNA biogenesis by switching utilization of both sigma factors and endoribonucleases. Nucleic Acids Res. 2004;32:6057–6068. doi: 10.1093/nar/gkh939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Deutscher MP. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA. 2002;8:97–109. doi: 10.1017/s1355838202014929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghora BK, Apirion D. Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell. 1978;15:1055–1066. doi: 10.1016/0092-8674(78)90289-1. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Pandit S, Deutscher MP. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 1999;18:2878–2885. doi: 10.1093/emboj/18.10.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ow MC, Perwez T, Kushner SR. RNase G of Escherichia coli exhibits only limited functional overlap with its essential homologue, RNase E. Mol. Microbiol. 2003;49:607–622. doi: 10.1046/j.1365-2958.2003.03587.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee K, Bernstein JA, Cohen SN. RNase G complementation of rne null mutation identified functional interrelationships with RNase E in Escherichia coli. Mol. Microbiol. 2002;43:1445–1456. doi: 10.1046/j.1365-2958.2002.02848.x. [DOI] [PubMed] [Google Scholar]
- 24.Wachi M, Umitsuki G, Shimizu M, Takada A, Nagai K. Escherichia coli cafA gene encodes a novel RNase, designated as RNase G, involved in processing of the 5′ end of 16S rRNA. Biochem. Biophys. Res. Commun. 1999;259:483–488. doi: 10.1006/bbrc.1999.0806. [DOI] [PubMed] [Google Scholar]
- 25.Chung D-H, Min Z, Wang B-C, Kushner SR. Single amino acid changes in the predicted RNase H domain of E. coli RNase G lead to the complementation of RNase E mutants. RNA. 2010;16:1371–1385. doi: 10.1261/rna.2104810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunn JJ, Studier FW. T7 early RNAs and Escherichia coli ribosomal RNAs are cut from large precursor RNAs in vivo by ribonuclease III. Proc. Natl Acad. Sci. USA. 1973;70:3296–3300. doi: 10.1073/pnas.70.12.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsunaga J, Simons EL, Simons RW. RNase III autoregulation: Structure and function of rncO, the posttranscriptional "operator". RNA. 1996;2:1228–1240. [PMC free article] [PubMed] [Google Scholar]
- 28.Drider D, Condon C. The continuing story of endoribonuclease III. J Mol. Microbiol. Biotechnol. 2004;8:195–200. doi: 10.1159/000086700. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson A. Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev. 1999;23:371–390. doi: 10.1111/j.1574-6976.1999.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 30.Babitzke P, Granger L, Olszewski J, Kushner SR. Analysis of mRNA decay and rRNA processing in Escherichia coli multiple mutants carrying a deletion in RNase III. J. Bacteriol. 1993;175:229–239. doi: 10.1128/jb.175.1.229-239.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bardwell JCA, Regnier P, Chen S-M, Nakamura Y, Grunberg-Manago M, Court DL. Autoregulation of RNase III operon by mRNA processing. EMBO J. 1989;8:3401–3407. doi: 10.1002/j.1460-2075.1989.tb08504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talkad V, Achord D, Kennell D. Altered mRNA metabolism in ribonuclease III-deficient strains of Escherichia coli. J. Bacteriol. 1978;135:528–541. doi: 10.1128/jb.135.2.528-541.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afonyushkin T, Vecerek B, Moll I, Blasi U, Kaberdin VR. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res. 2005;33:1678–1689. doi: 10.1093/nar/gki313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Resch A, Afonyushkin T, Lombo TB, McDowall KJ, Blasi U, Kaberdin VR. Translational activation by the noncoding RNA DsrA involves alternative RNase III processing in the rpoS 5′-leader. RNA. 2008;14:454–459. doi: 10.1261/rna.603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khodursky AB, Peter BJ, Cozzarelli NR, Botstein D, Brown PO, Yanofsky C. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. Proc. Natl Acad. Sci. USA. 2000;97:12170–12175. doi: 10.1073/pnas.220414297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 2001;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohanty BK, Kushner SR. Genomic analysis in Escherichia coli demonstrates differential roles for polynucleotide phosphorylase and RNase II in mRNA abundance and decay. Mol. Microbiol. 2003;50:645–658. doi: 10.1046/j.1365-2958.2003.03724.x. [DOI] [PubMed] [Google Scholar]
- 38.Takiff HE, Chen S, Court DL. Genetic analysis of the rnc operon of Escherichia coli. J. Bacteriol. 1989;171:2581–2590. doi: 10.1128/jb.171.5.2581-2590.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Seed B. Selection of oligonucleotide probes for protein coding sequences. Bioinformatics. 2003;19:796–802. doi: 10.1093/bioinformatics/btg086. [DOI] [PubMed] [Google Scholar]
- 40.O'Hara EB, Chekanova JA, Ingle CA, Kushner ZR, Peters E, Kushner SR. Polyadenylylation helps regulate mRNA decay in Escherichia coli. Proc. Natl Acad. Sci. USA. 1995;92:1807–1811. doi: 10.1073/pnas.92.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohanty BK, Giladi H, Maples VF, Kushner SR. Analysis of RNA decay, processing, and polyadenylation in Escherichia coli and other prokaryotes. Methods Enzymol. 2008;447:3–29. doi: 10.1016/S0076-6879(08)02201-5. [DOI] [PubMed] [Google Scholar]
- 42.Hiley SL, Babak T, Hughes TR. Global analysis of yeast RNA processing indentifies new targets of RNase III and uncovers a link between tRNA 5′ end processing and tRNA splicing. Nucleic Acids Res. 2005;33:3048–3056. doi: 10.1093/nar/gki608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, et al. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 2009;37:D464–D470. doi: 10.1093/nar/gkn751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2004;2:E259. doi: 10.1371/journal.pbio.0020259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Royal Stat. Soc. Ser. B Method. 1995;57:289–300. [Google Scholar]
- 48.Vincze E, Bowra S. Northerns revisited: a protocol that eliminates formaldehyde from the gel while enhancing resolution and sensitivity. Anal. Biochem. 2005;342:356–357. doi: 10.1016/j.ab.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 49.Masse R, Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl Acad. Sci. USA. 2002;99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babitzke P, Kushner SR. The Ams (altered mRNA stability) protein and ribonuclease E are encoded by the same structural gene of Escherichia coli. Proc. Natl Acad. Sci. USA. 1991;88:1–5. doi: 10.1073/pnas.88.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beran RK, Simons RW. Cold-temperature induction of Escherichia coli polynucleotide phosphorylase by reversal of its autoregulation. Mol. Microbiol. 2001;39:112–125. doi: 10.1046/j.1365-2958.2001.02216.x. [DOI] [PubMed] [Google Scholar]
- 53.Robert-Le Meur M, Portier C. Polynucleotide phosphorylase of Escherichia coli induces the degradation of its RNase III processed messenger by preventing its translation. Nucleic Acids Res. 1994;22:397–403. doi: 10.1093/nar/22.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robert-Le Meur M, Portier C. Escherichia coli polynucleotide phosphorylase expression is autoregulated through an RNase III-dependent mechanism. EMBO J. 1992;11:2633–2641. doi: 10.1002/j.1460-2075.1992.tb05329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King TC, Sirdeshmukh R, Schlessinger D. RNase III cleavage is obligate for maturation but not for function of Escherichia coli pre-23S rRNA. Proc. Natl Acad. Sci. USA. 1984;81:185–188. doi: 10.1073/pnas.81.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srivastava AK, Schlessinger D. Processing pathway of Escherichia coli 16S precursor rRNA. Nucleic Acids Res. 1989;17:1649–1663. doi: 10.1093/nar/17.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uppal S, Akkipeddi VS, Jawali N. Posttranscriptional regulation of cspE in Escherichia coli: involvement of the short 5′-untranslated region. FEMS Microbiol. Lett. 2008;279:83–91. doi: 10.1111/j.1574-6968.2007.01009.x. [DOI] [PubMed] [Google Scholar]
- 58.Perwez T, Kushner SR. RNase Z in Escherichia coli plays a significant role in mRNA decay. Mol. Microbiol. 2006;60:723–737. doi: 10.1111/j.1365-2958.2006.05124.x. [DOI] [PubMed] [Google Scholar]
- 59.Bae WH, Xia B, Inouye M, Severinov K. Escherichia coli CspA-family RNA chaperones are transcription antiterminators. Proc. Natl Acad. Sci. USA. 2000;97:7784–7789. doi: 10.1073/pnas.97.14.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mogk A, Deuerling E, Vorderwulbecke S, Vierling E, Bukau B. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol. Microbiol. 2003;50:585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- 61.Wade JT, Roa DC, Grainger DC, Hurd D, Busby SJ, Struhl K, Nudler E. Extensive functional overlap between sigma factors in Escherichia coli. Nat. Struct. Mol. Biol. 2006;13:806–814. doi: 10.1038/nsmb1130. [DOI] [PubMed] [Google Scholar]
- 62.Zhou YN, Kusukawa N, Erickson JW, Gross CA, Yura T. Isolation and characterization of Escherichia coli mutants that lack the heat shock sigma factor sigma 32. J. Bacteriol. 1988;170:3640–3649. doi: 10.1128/jb.170.8.3640-3649.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cowing DW, Bardwell JC, Craig EA, Woolford C, Hendrix RW, Gross CA. Consensus sequence for Escherichia coli heat shock gene promoters. Proc. Natl Acad. Sci. USA. 1985;82:2679–2683. doi: 10.1073/pnas.82.9.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP. Crystal structures explain functional properties of two E. coli porins. Nature. 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 65.Mitchell JE, Zheng D, Busby SJ, Minchin SD. Identification and analysis of ‘extended -10' promoters in Escherichia coli. Nucleic Acids Res. 2003;31:4689–4695. doi: 10.1093/nar/gkg694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andersen J, Forst SA, Zhao K, Inouye M, Delihas N. The function of micF RNA. micF. RNA is a major factor in the thermal regulation of OmpF protein in Escherichia coli. J. Biol. Chem. 1989;264:17961–17970. [PubMed] [Google Scholar]
- 67.Schmidt M, Delihas N. micF RNA is a substrate for RNase E. FEMS Microbiol. Lett. 1995;133:209–213. doi: 10.1111/j.1574-6968.1995.tb07886.x. [DOI] [PubMed] [Google Scholar]
- 68.Landt SG, Abeliuk E, McGrath PT, Lesley JA, McAdams HH, Shapiro L. Small non-coding RNAs in Caulobacter crescentus. Mol. Microbiol. 2008;68:600–614. doi: 10.1111/j.1365-2958.2008.06172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C. Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res. 2002;30:3732–3738. doi: 10.1093/nar/gkf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 71.Hu Z, Zhang A, Storz G, Gottesman S, Leppla SH. An antibody-based microarray assay for small RNA detection. Nucleic Acids Res. 2006;34:e52. doi: 10.1093/nar/gkl142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 73.Rivas E, Klein RJ, Jones TA, Eddy SR. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001;11:1369–1373. doi: 10.1016/s0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- 74.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tran TT, Zhou F, Marshburn S, Stead M, Kushner SR, Xu Y. De novo Computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics. 2009;25:2897–2905. doi: 10.1093/bioinformatics/btp537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pertzev AV, Nicholson AW. Characteization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Res. 2006;34:3708–3721. doi: 10.1093/nar/gkl459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Regnier P, Portier C. Initiation attenuation and RNase III processing of transcripts from the Escherichia coli operon encoding ribosomal protein S15 and polynucleotide phosphorylase. J. Mol. Biol. 1986;187:23–32. doi: 10.1016/0022-2836(86)90403-1. [DOI] [PubMed] [Google Scholar]
- 78.Gultyaev AP, van Batenburg FHD, Pleij CWA. The computer simulation of RNA folding pathways using a genetic algorithm. J. Mol. Biol. 1995;250:37–51. doi: 10.1006/jmbi.1995.0356. [DOI] [PubMed] [Google Scholar]
- 79.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bernstein JA, Lin P-H, Cohen SN, Lin-Chao S. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl Acad. Sci. USA. 2004;101:2748–2763. doi: 10.1073/pnas.0308747101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon JJ, Towsey MW, Hogan JM, Mathews SA, Timms P. Improved prediction of bacterial transcription start sites. Bioinformatics. 2006;22:142–148. doi: 10.1093/bioinformatics/bti771. [DOI] [PubMed] [Google Scholar]
- 82.Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, Taboada B, Jimenez-Jacinto V, Salgado H, Juarez K, Contreras-Moreira B, et al. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS ONE. 2009;4:e7526. doi: 10.1371/journal.pone.0007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Bakel H, Nislow C, Blencowe BJ, Hughes TR. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010;8:e1000371. doi: 10.1371/journal.pbio.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suzuki K, Babitzke P, Kushner SR, Romeo T. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 2006;20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.