

Abstract
The identification of genes linked to neurodegenerative diseases such as Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and Parkinson's disease (PD) has led to the development of animal models for studying mechanism and evaluating potential therapies. None of the transgenic models developed based on disease-associated genes have been able to fully recapitulate the behavioral and pathological features of the corresponding disease. However, there has been enormous progress made in identifying potential therapeutic targets and understanding some of the common mechanisms of neurodegeneration. In this review, we will discuss transgenic animal models for AD, ALS, HD and PD that are based on human genetic studies. All of the diseases discussed have active or complete clinical trials for experimental treatments that benefited from transgenic models of the disease.
Keywords: ALS, PD, AD, HD, Huntington's disease, Parkinson's disease, Amyotrophic lateral sclerosis, Alzheimer's disease, Transgenic, Animal model, Neurodegeneration, Genetic linkage
Introduction
Genes have been identified as risk factors for the cause of neurodegenerative diseases through studies of families and populations with patterns of inherited neurodegenerative diseases. Once a pedigree analysis has identified a pattern of inheritance, linkage studies can identify chromosomal loci where putative disease genes may be located. Sequencing of putative genes from members of an established disease pedigree can potentially identify a specific mutation that follows the disease inheritance pattern. Once a gene and mutation have been identified, transgenic animals can be generated to model the human disease or provide insight into the gene's function (see Fig. 1 for overview). In this review, we discuss transgenic animal models of several prominent neurodegenerative diseases that are based on genes identified through human gene linkage studies. The common symptoms and pathological changes of Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and Parkinson's disease (PD) and their associated loci and genes are summarized in Table 1.
Fig. 1.
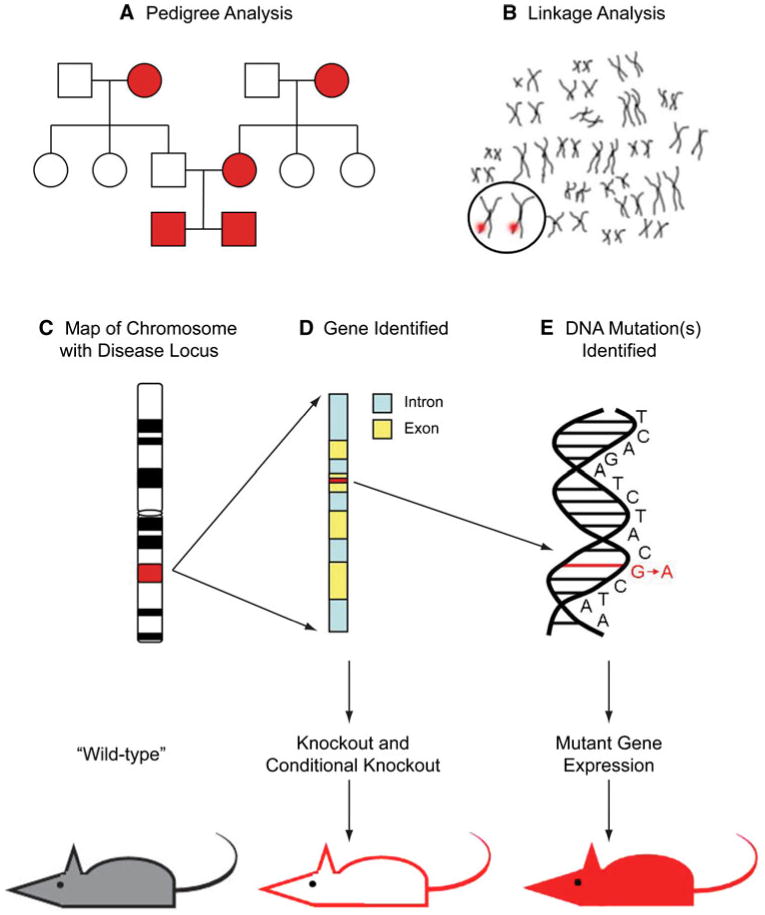
Generation of transgenic mouse models based on inheritance patterns of human disease. Families or populations with traceable lineages of a disease are subjected to pedigree analysis (a) and individuals are subjected to gene linkage studies to identify associated chromosomes (b). Further molecular analyses link genetic markers to specific region of chromosome, or locus (c) and potentially identify a disease-related gene (d). Sequence analysis of putative gene(s) from diseased and non-diseased members of the pedigree can reveal inherited mutation(s) in the gene (e). Once the gene and mutation have been identified in affected pedigree members, the mouse ortholog of the human gene can be knocked out completely or conditionally (d arrows). Also, the mutated human gene can be introduced into the mouse as a replacement for, or in addition to the mouse ortholog (e arrows). Transgenic mice generated based on the identified gene and its mutants are an important tool when studying behavioral and pathological phenocopy of human disease and when evaluating potential therapeutics for disease
Table 1. Neurodegenerative diseases: symptoms, pathology and associated genetic linkage.
| Disease | Symptoms | Pathology | Gene map locus | Locus name | Associated gene |
|---|---|---|---|---|---|
| Alzheimer's disease (AD) | Dementia | Amyloid-containing plaques in cortex | 21q21 | AD1 | Amyloid precursor protein |
| Apraxia | Neurofibrillary tangles | 19q13.2 | AD2 | Apolipoprotein E (APOE) | |
| Aphasia | 14q24.3 | AD3 | Presenilin-1 (PSEN1) | ||
| Depression, anxiety and delusions | 1q31–q42 | AD4 | Presenilin-2 (PSEN2) | ||
| Short attention span | 12p11.23– q13.12 | AD5 | Not yet determined | ||
| Visuospatial navigation deficits | 10q24 | AD6 | Not yet determined | ||
| 10p13 | AD7 | Not yet determined | |||
| 20p | AD8 | Not yet determined | |||
| 19p13.2 | AD9 | Not yet determined | |||
| 7q36 | AD10 | Not yet determined | |||
| 9q22.1 | AD11 | Not yet determined | |||
| 8p12–q22 | AD12 | Not yet determined | |||
| 1q21 | AD13 | Not yet determined | |||
| 1q25 | AD14 | Not yet determined | |||
| 3q22–q24 | AD15 | Not yet determined | |||
| Xq21.3 | AD16 | Not yet determined | |||
| Amyotrophic lateral sclerosis (ALS) | Muscle weakness and wasting | Loss of lower motor neurons in ventral horn of spinal cord | 21q22.1 | ALS1 | Superoxide dismutase-1 (SOD1) |
| Muscle fasciculations and cramping | Degeneration of corticospinal tracts | 2q33 | ALS2 | Alsin | |
| Impaired limb dexterity | Neurodegeneration in primary motor cortex | 18q21 | ALS3 | Not yet determined | |
| Respiratory failure | Reactive astrocytes in motor cortex and spinal cord | 9q34 | ALS4 | Senataxin (SETX) | |
| Dysphagia and aspiration | Cytoplasmic inclusions in degenerating neurons | 15q15.1–q21.1 | ALS5 | Not yet determined | |
| Hypophonic speech | 16q12 | ALS6 | Fused in sarcoma (FUS) | ||
| 20p13 | ALS7 | Not yet determined | |||
| 20q13.3 | ALS8 | Vesicle-associated membrane protein-associated protein B (VAPB) | |||
| 14q11.2 | ALS9 | Angiogenin (ANG) | |||
| 1p36.2 | ALS10 | TAR DNA Binding protein (TARDBP) | |||
| 6q21 | ALS11 | Homolog of yeast FIG 4 | |||
| 10p15–p14 | ALS12 | Optineurin | |||
| Huntington's disease (HD) | Choreiform movements | Loss of neurons in caudate and putamen | 4pl6.3 | ITI5 | Huntingtin |
| Depression | Gliosis in striatum | ||||
| Dementia | |||||
| Emotional incontinence | |||||
| Parkinson's disease (PD) | Resting tremor | Loss of neurons in substantia nigra | 4q21 | PARK1 | Synuclein (SNCA) |
| Muscle rigidity | Cytoplasmic inclusions in degeneration neurons (Lewy bodies) | 6q25.2–q27 | PARK2 | Parkin | |
| Bradykinesia | 2p13 | PARK3 | Not yet determined | ||
| Postural instability | 4q21 | PARK4 | Synuclein (SNCA) | ||
| Cognitive decline | 4p14 | PARK5 | Ubiquitin C-ternubak esterase L1 (UCHL1) | ||
| Depression | 1p36 | PARK6 | PTEN-induced putative kinase1 (PINK1) | ||
| Physical discomfort | 1p36 | PARK7 | DJ1 | ||
| 12q12 | PARK8 | Leucine-rich repeat 2 (LRRK2) | |||
| 1p36 | PARK9 | ATP13A2 | |||
| 1p32 | PARK10 | Not yet determined | |||
| 2q37.1 | PARK11 | GRB10-interacting GYF protein (GIGYF2) | |||
| Xq21–q25 | PARK12 | Not yet determined | |||
| 2p12 | PARK13 | HTRA serine peptidase 2 (HTRA2) | |||
| 18q11 | PARK14 | Not yet determined | |||
| 22q12–q13 | PARK15 | FBX07 | |||
| 1q32 | PARK16 | Not yet determined |
Gene map locus, locus name and associated gene information based on Online Mendelian Inheritance in Man (OMIM) database at http://www.ncbi.nlm.nih.gov/omim/
Alzheimer's disease
Alzheimer's disease is the most prevalent of neurodegenerative diseases that causes progressive memory loss and dementia in affected patients. Diagnosis of AD occurs post-mortem by confirming the presence of neurofibrillary tangles (NFT) and amyloid plaques which are found in the several brain regions including the subiculum and entorhinal cortex. The NFT are intraneuronal microtubule bundles comprised hyperphosphorylated forms of microtubule associated protein tau (MAPT). The amyloid plaques are extracellular deposits primarily consisting of the amyloid β peptide. To date, 16 genes or loci have been identified for AD (OMIM 104300). In this review, we will briefly describe several genes that have been used to generate transgenic models of AD.
Amyloid beta A4 precursor protein (APP; AD1)
Pedigree analysis of a family with early onset autosomal dominant AD identified a mutation (V717F) in the APP gene (Murrell et al. 1991). Using the APP V717F mutation, the first transgenic mouse model of AD (PDAPP) was developed (Games et al. 1995). The PDAPP mouse exhibited plaque pathology by 6–9 months of age but did not show NFT pathology or substantial cell loss. Analysis of Swedish families with early onset AD identified a double mutation (K670N and M671L) in the APP gene (Mullan et al. 1992). A transgenic mouse (Tg2576) was generated based on the double mutation in APP and these mice had plaque pathology at 9 months of age and cognitive deficits. No NFT pathology or cell loss was observed (Hsiao et al. 1996). A second laboratory generated a transgenic mouse (APP23) based on the Swedish double-mutant but used a different promoter to express the transgene (Sturchler-Pierrat et al. 1997). Similar to Tg2576, the APP23 developed plaques, around 6 months of age, and had cerebrovascular amyloid and some neuronal loss (Sturchler-Pierrat et al. 1997; Calhoun et al. 1998, 1999). A mutation in APP (E693Q) was identified in patients with hereditary cerebral hemorrhage with amyloidosis of Dutch type (HCHWA-D), an autosomal dominant form of amyloidosis (Levy et al. 1990). The E693Q mutant form of APP was used to generate the APP-Dutch transgenic mouse which developed congophilic amyloid angiopathy, but did not show other phenotypes consistent with AD (Herzig et al. 2004).
Additional APP mutant-based transgenic mice and genetic crosses with other AD gene-based mutants (some described below) have been studied and these models have allowed hundreds of studies evaluating therapeutics and investigating the mechanisms of Alzheimer's disease (Dodart and May 2005; McGowan et al. 2006; Duyckaerts et al. 2008; Woodruff-Pak 2008; Howlett and Richardson 2009). In addition to mouse models based on mutations found in human genes, there are non-transgenic models of AD in the rat, rabbit, dog and primate that offer the ability to conduct complementary studies for the evaluation of therapeutics and the understanding of disease mechanisms (Woodruff-Pak 2008). However, mouse models of AD based on human mutations in APP and other AD genes remain the most widely used for evaluating potential therapeutics.
Apoliprotein E (APOE; AD2)
The epsilon 4 allele of apolipoprotein E (APOE-E4) gene was detected at a high frequency in a population of patients with late-onset familial AD compared to spousal controls (Saunders et al. 1993). Another study identified APOE-E4 as a highly significant risk factor for AD (Corder et al. 1993). Although APOE-E4 remains the highest risk factor identified for sporadic AD (Bertram and Tanzi 2005), no transgenic animals based on APOE-E4 allele have produced an AD phenotype. However, replacement of the mouse APOE gene with human APOE4 allele causes cognitive impairments (Raber et al. 1998), decreased excitatory synaptic transmission and reduction in dendritic density and complexity (Wang et al. 2005a; Dumanis et al. 2009), There is also strong evidence that APOE-E4 increases deposition of Aβ peptide in amyloid plaques and evidence that APOE can influence tau phosphorylation (Small and Duff 2008). Mice expressing human APOE4 allele in a mouse APOE4 null background exhibited increased insoluble APOE protein in parallel with an increased Aβ burden as they aged (Bales et al. 2009). Overall, the identification of APOE as a risk factor for AD may be most useful for diagnostic purposes to identify patients who may benefit from a therapy aimed at reducing Aβ (Lambert and Amouyel 2007; Bird 2008). As more studies unravel the mechanistic actions of APOE in AD and other diseases, better therapeutic strategies may be developed.
Presenilin 1 (PSEN1; AD3)
Mutations in the presenilin 1 gene on chromosome 14 (Sherrington et al. 1995) is the most common cause of autosomal dominant forms of familial AD. To date, 175 mutations in PSEN1 have been identified (AD and FTD Mutation Database; http://www.molgen.ua.ac.be/ADMutations). The PSEN1 gene encodes a protein component of the proteolytic gamma-secretase complex whose activity in combination with beta-secretase generates Aβ from APP. Using two mutations, M146L (Sherrington et al. 1995) and M146V (Haltia et al. 1994), identified in PSEN1, two transgenic mouse strains were generated (Duff et al. 1996). Both strains exhibited increased Aβ peptide in the brain but no obvious plaque pathology was reported. Additional mutations in PSEN1 can influence Aβ processing (Brouwers et al. 2008) but, to date, no mutant of PSEN1 alone provides a suitable phenocopy of AD.
Presenilin 2 (PSEN2; AD4)
Through linkage studies of autosomal dominant forms of early-onset AD, the gene encoding presenilin 2 on chromosome 1 was identified (Levy-Lahad et al. 1995a, b). To date, 14 mutations in PSEN2 have been identified (AD and FTD Mutation Database; http://www.molgen.ua.ac.be/ADMutations). Similar to PSEN1, PSEN2 is a protein component of the proteolytic gamma secretase complex important for processing APP (Brouwers et al. 2008). Unlike mice lacking PSEN1, PSEN2 null mice are viable with no obvious defects and no significant differences in processing APP (Donoviel et al. 1999; Herreman et al. 1999). Using the N141I mutation in PSEN2, transgenic mice were generated and found to have higher levels of insoluble Aβ peptide compared to control mice (Sawamura et al. 2000). Although PSEN2 can influence Aβ peptide processing, no transgenic animals based on PSEN2 alone recapitulate AD phenotype. The relevance of presenilin-based transgenics is discussed in a recent review (Elder et al. 2010).
Microtubule-associated protein tau (MAPT)
The presence of NFTs in post-mortem brain is one of the defining pathologies of AD. A major component of the NFTs is MAPT and changes in its phosphorylation, cellular distribution, and structure can lead to tau pathologies or “tauopathies” such as that seen in AD (Brunden et al. 2008; Garcia-Sierra et al. 2008; Honson and Kuret 2008; Takashima 2008). Linkage studies identified mutations in human chromosome 17q21.1 that are correlated to frontotemporal dementia. To date, 44 mutations in MAPT have been identified (AD and FTD Mutation Database; http://www.molgen.ua.ac.be/ADMutations). There have been several transgenic mice generated based on mutations in the human MAPT gene that have provided clear evidence for mutant tau in NFT pathology and dementia (McGowan et al. 2006). Although the mechanism by which mutations in MAPT lead to neurodegeneration remains unknown, therapeutic strategies targeting the phosphorylation status of tau are being developed (Gong and Iqbal 2008).
Combined transgenic models
Although none of the transgenic rodent models based on single gene mutations have been able to fully recapitulate the features of AD, combinations of transgenes have provided novel transgenic models that have a progressive pathology with behavioral deficits. For example, mice containing the P301L mutation in MAPT were crossed with the amyloid precursor protein mutant Tg2576 to generate “TAPP” mice. The TAPP mice exhibited increased NFTs in the limbic system and olfactory cortex (Lewis et al. 2001). Using an alternative approach, Oddo et al. (2003a, b) microinjected the transgenes for APP (Swe) and Tau (P301L) into single-cell embryos from homozygous PS1 (M146V) mice to create a triple transgenic mouse (3xTg-AD). The 3xTg-AD mice progressively develop synaptic dysfunction, APP-containing plaques and NFTs (Oddo et al. 2003a, b). The 3xTg-AD mouse has thus been the most widely used model of AD for evaluating potential therapies, examining environmental vulnerabilities and studying disease mechanism (Gimenez-Llort et al. 2007; Foy et al. 2008). Overall, there has been significant progress in understanding disease mechanisms for AD using transgenic rodent models that are based on mutated human genes in AD patients.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis is a neurodegenerative disease that results from the progressive loss of motor neurons in brain and spinal cord. Onset of disease typically occurs in middle adulthood but forms with juvenile onset also occur. Symptoms include asymmetrical muscle weakness and muscle fasciculations. The disease progresses rapidly after onset leading to paralysis and eventually death within 5 years. Most cases of ALS are sporadic with an estimated 10% of cases exhibiting a pattern of inheritance. To date, molecular genetic studies have identified the corresponding genes for nine of the twelve ALS loci defined by linkage studies (Table 1).
Superoxide dismutase (SOD1; ALS1)
The first gene associated with ALS was the superoxide dismutase-1 (SOD1) gene encoding an enzyme capable of inactivating superoxide radicals (Rosen et al. 1993). Approximately 20% of the familial forms of ALS can be attributed to mutations in the SOD1 gene, many of which follow an autosomal dominant inheritance pattern. The identification of SOD1 as a genetic factor for ALS led to the development of SOD1 transgenic animals. Gurney et al. (1994) reported that mice over-expressing a human SOD1 allele containing a G93A substitution developed spinal cord motor neuron loss and related paralysis. Following that initial study with the G93A variant, 13 additional transgenic mice have been made that produced a broad range of outcomes but all exhibit some characteristics of the disease (Ripps et al. 1995; Wong et al. 1995; Bruijn et al. 1997; Wang et al. 2002, 2003, 2005b; Tobisawa et al. 2003; Jonsson et al. 2004, 2006; Chang-Hong et al. 2005; Watanabe et al. 2005; Deng et al. 2006). A recent publication has extensively reviewed and summarized the numerous studies related to SOD1 transgenic mice including therapeutic testing with these models (Turner and Talbot 2008). For the current review, we will focus on the remaining genes that have been implicated in ALS.
Alsin (ALS2)
Two deletion mutations in the ALS2 gene were identified from pedigree analysis of an autosomal recessive inherited form of juvenile onset ALS (Yang et al. 2001). Alsin, the protein encoded by the ALS2 gene, has been shown to be important for the survival and growth of neurons, as well as neurite outgrowth (Tudor et al. 2005; Jacquier et al. 2006). Although the primary sequence contains several functional domains implicating a role in guanine nucleotide exchange, the precise structural or enzymatic function of this protein remains unknown. To date, at least five independently generated ALS2 knockout mice have been characterized (Cai et al. 2005; Devon et al. 2006; Hadano et al. 2006; Yamanaka et al. 2006; Deng et al. 2007) Although each of these knockout alleles produced viable homozygous mice, the resulting animals failed to show any neuropathological features consistent with motor neuron degeneration. However, subsequent studies with these variants did provide several insights into the function of alsin. These insights will be discussed here only briefly, since a recent review by Cai et al. (2008) provides an in-depth comparison of their construction and phenotypic characterization. The first report demonstrated that ALS2 null mice or neurons derived from ALS2 null mice were more vulnerable to oxidative stress. Interestingly, this allele did not enhance the spinal cord degeneration or paralysis phenotype of SOD1 (G93A) mice (Lin et al. 2007). Age-related loss of Purkinje cells and disturbance of spinal motor neurons was detected by electrophysiological and immunohistochemical analyses in a second variant (Hadano et al. 2006). Devon et al. (2006) showed that their ALS2 null mice were hypoactive, had smaller than normal cortical motor neurons and disrupted endosomal fusion activity. Lai et al. (2006) discovered that AMPA receptor trafficking was disrupted in ALS2 null mice and this rendered neurons more vulnerable to glutamate-mediated excitotoxicity. Finally, several groups have shown that ALS2 is required for proper axonal development and function (Yamanaka et al. 2006; Deng et al. 2007; Otomo et al. 2008).
Although each variant has provided some insight into the functions of ALS2 in motor neurons, no single ALS2 null allele has completely reproduced the behavioral and pathological features of ALS. While it is difficult to speculate on the precise reason for this shortfall, it is possibly due to nuances in the ALS2 genomic sequence (i.e. alternate splicing) as it spans eighty kilobases, but ultimately produces a three kilobase transcript. Recreating such structural complexity is difficult with current techniques. Nonetheless, future studies may benefit from the development of transgenic mice harboring human mutant forms of ALS2 in an ALS2 null background either alone or in conjunction with other predisposing genes.
Senataxin (SETX; ALS4)
Senataxin is the affected gene that causes the autosomal dominant familial form of juvenile ALS associated with the ALS4 locus (Chance et al. 1998; Chen et al. 2004). Mutations in SETX have been previously linked to the autosomal recessive disease ataxia with oculomotor apraxia type 2 (AOA2). SETX contains a conserved superfamily I DNA/RNA helicase domain that suggests this protein has a nuclear function related to nucleic acid processing (Chen et al. 2004). Although in vitro studies have demonstrated that cells carrying the AOA2 type mutations have increased sensitivity to oxidative DNA damage, similar studies with ALS4-related alleles have not been reported (Suraweera et al. 2007). The development of ALS4/SETX transgenic animals is needed to understand the role of ALS4 mutants in motor neuron disease.
Vesicle-associated membrane protein-associated protein B (VAPB; ALS8)
The gene encoding vesicle-associated membrane protein-associated protein B (VAPB), corresponds to ALS8, an autosomal dominant adult-onset form of ALS. The VAPB gene was identified as a putative ALS gene from a pedigree analysis of a large Brazilian family (Nishimura et al. 2004) and a specific mutation, P56S, was linked to 200 diseased individuals out of 1500 in the 8 pedigrees studied (Nishimura et al. 2005). A study of post-mortem spinal cord tissue from ALS patients determined that VAPB mRNA expression was decreased compared to control tissue (Anagnostou et al. 2008). Transgenic flies expressing the corresponding mutation in Drosophila VAPB produced hallmarks of human ALS including locomotion defects, neuronal death and aggregate formation (Chai et al. 2008). In yeast and neuronal cells, expression of the P56S mutant increased ER stress and vulnerability to ER stress, respectively (Suzuki et al. 2009). Although the development of a transgenic rodent model has yet to be reported, the ability to study the ALS8 phenotypes in flies and yeast should prove to be powerful tools when screening for genetic modifiers of disease or therapeutic drugs.
Angiogenin (ANG; ALS9)
Based on an association between the ANG gene and a frequently occurring single nucleotide polymorphism in a population of ALS patients (Greenway et al. 2004), a more comprehensive study was conducted which identified seven missense mutations in patients with both sporadic ALS and familial autosomal dominant ALS9 (Greenway et al. 2006). Additional mutations in ANG have been identified in various populations with ALS (Wu et al. 2007; Conforti et al. 2008; Gellera et al. 2008; Paubel et al. 2008). Similar to activities described above for ALS2/alsin, cell culture studies have shown that mutant forms of human ANG impeded neurite extension and promoted degeneration (Subramanian et al. 2008). ANG promotes survival of motor neurons in vitro and in vivo and angiogenin delivery to SOD1 (G93A) mice increased lifespan and motor neuron survival (Kieran et al. 2008). Collectively, angiogenin affects motor neuron survival and neuritic growth, and the presence of angiogenin in serum may be a useful biomarker for ALS. The development of animal models based on human mutant forms of ANG may provide a means to evaluate therapeutic and diagnostic approaches to ALS.
TAR DNA binding protein (TARDBP; ALS10)
Several studies have confirmed that mutations in the TARDBP gene are associated with familial ALS10-type forms of ALS (Kabashi et al. 2008; Kuhnlein et al. 2008; Rutherford et al. 2008; Sreedharan et al. 2008; Van Deerlin et al. 2008; Corrado et al. 2009) and sporadic ALS (Daoud et al. 2009). The TARDBP protein contains two RNA-binding motifs which are involved in alternative mRNA splicing (Bose et al. 2008). A recent publication by Kraemer et al. reported that while homozygous TARDBP-null mice (−/−) die as embryos around day 7.5, heterozygous (+/−) mice exhibit signs of motor defects and muscle weakness (Kraemer et al. 2010). Another report by Kabashi et al. (2010) employs a zebrafish model to express the mutant alleles associated with the human disease state. These transgenic fish displayed swimming deficits and defects in axonal elongation and branching. The ability of the zebrafish model to phenocopy the human disease state and its feasibility as a high throughput system makes it an ideal platform for experimental therapeutics.
In conclusion, the available repertoire of transgenic animals modeling non-SOD1-based ALS has only begun to develop, and new genes and loci are still being added. While this manuscript was in preparation, the causative gene was identified for ALS6 and two new loci, ALS11 and ALS12, have been identified (Chow et al. 2009; Vance et al. 2009; Maruyama et al. 2010). With continued growth and characterization of the ALS loci and associated genes, these tools may prove to be as powerful as the SOD1 animal collection, which has produced several therapeutic strategies (e.g. arimoclomal, ceftriaxone, IGF-1, HDAC inhibitors) that are now in clinical trials (http://www.clinicaltrials.gov). Future studies will no doubt lead to the identification of more ALS loci and give rise to even more transgenic animal models to be used in our effort to understand this complicated degenerative disease.
Huntington's disease
Huntington's disease is an autosomal dominant progressive neurodegenerative disorder. HD was found to be an inherited disease already in nineteenth century and it was named after George Huntington who characterized the disease (Huntington 1872). Distinctive symptoms for HD are chorea, dystonia, incoordination, weight loss, cognitive difficulties and psychiatric problems. The onset of symptoms is usually midlife, but it can vary from early childhood to old age. HD is fatal usually within 15–20 years after the onset. Pathologically, HD is characterized by neuronal cell loss and atrophy most prominently in caudate and putamen (Vonsattel et al. 1985). Within the striatum neurodegeneration occurs in medium spiny neurons. Enkephalin containing striatal medium spiny neurons projecting to the external globus pallidus are more vulnerable than those containing substance P projecting to the internal globus pallidus (Reiner et al. 1988).
Huntingtin (IT15)
Unlike the other neurodegenerative disorders described in this review, HD is linked to a specific genetic deficit found on chromosome 4 (Gusella et al. 1983) and the gene, IT15, was found to have a CAG repeat longer than normal (HDCRG 1993). The expansion of the CAG/polyglutamine repeat is at the N-terminus of huntingtin protein; the normal allele sizes are 6–37 CAG repeats and expanded HD allele sizes are 35–121 repeats. The length of the repeat correlates with age of onset and the longest repeats are detected in juvenile cases (HDCRG 1993). Patients having CAG repeats 30–40 are rare, and individuals without symptoms have been found to have 36, 37, 38 and 39 repeats (Rubinsztein et al. 1996).
Huntingtin is a 350 kDa protein and is expressed at high levels in neurons of the brain and in testis of adults (Bennett et al. 2009). Huntingtin is necessary for development since homozygous knock-out embryos are not viable (Nasir et al. 1995; Zeitlin et al. 1995). Huntingtin also has an important role in postnatal development. Elimination of HD gene in postnatal development leads to neuronal degeneration, a motor abnormality phenotype and early mortality (Dragatsis et al. 2000). Furthermore, mutated huntingtin can rescue the knockout mice (White et al. 1997; Leavitt et al. 2001).
The first transgenic mouse model for HD was generated, carrying 5′ end of the human HD gene (CAG)115-150 (Mangiarini et al. 1996). They established three mouse lines with CAG repeat expansions that had an HD phenotype: R6/1 with 116 repeat units, R6/2 with 144 repeat units, R6/5 with wider spectrum of repeats: 128, 132, 135, 137 and 156. R6/2 mice have been studied most and show choreiform-like movements, involuntary stereotypic movements, tremor, epileptic seizures and premature death (Mangiarini et al. 1996). The age of onset is 9–11 weeks and the age of death is 10–13 weeks in R6/2 mice. R6/2 mice have huntingtin aggregates in the nucleus of neurons seen prior to developing a neurological phenotype (Davies et al. 1997). Also, the mRNA for type 1 metabotropic glutamate receptors and for D1 dopamine receptors is already reduced at the age of 4 weeks (Cha et al. 1998).
Several additional transgenic mouse models of HD have been generated with different lengths of CAG repeats and progressive HD phenotype (White et al. 1997; Reddy et al. 1998; Schilling et al. 1999; Shelbourne et al. 1999; Wheeler et al. 2000; Laforet et al. 2001; Lin et al. 2001; Menalled et al. 2002; Gray et al. 2008). A series of studies has been reported based on a yeast artificial chromosome (YAC) mouse model with full-length form of huntingtin under the endogenous promoter. These mice show CAG repeat- and age-dependent behavioral deficits which correlate to striatal atrophy and neuronal loss (Hodgson et al. 1999; Slow et al. 2003).
A conditional mouse model of HD in which mutant huntingtin gene is turned off via tetracycline-induced activation (Yamamoto et al. 2000), expresses mutated huntingtin containing 94 CAG repeats. The mice have neuronal inclusions and characteristic neuropathology when they are studied at the age of 18 weeks. Also, they show progressive motor dysfunction starting at the age of 4 weeks (Yamamoto et al. 2000). When the mutated huntingtin protein expression was blocked at the age of 18 weeks, it led to reduction of neuronal inclusions and a decrease in behavioral phenotype when mice were analyzed 16 weeks later (Yamamoto et al, 2000). The area and density of dopamine D1 receptors in caudate putamen increased significantly and striatal GFAP density decreased 16 weeks after inactivation of mutated huntingtin protein synthesis. Yamamoto (2000) thus indicate that continued synthesis of mutated huntingtin protein is needed to maintain inclusions and symptoms of HD.
A transgenic rat model of HD, with a mutated huntingtin gene containing 51 CAG repeats, expresses adult-onset neurological phenotypes, cognitive impairments, progressive motor dysfunction and neuronal nuclear inclusions in the brain (von Horsten et al. 2003). The transgenic rats have a late onset of phenotype and they die between 15 and 24 months. Transgenic HD rats have an age- and genotype-dependent deterioration of psychomotor performance and choreiform symptoms (Cao et al. 2006).
Recently, HD was modeled in rhesus macaque with a lentiviral vector (Cai et al. 2008). Yang et al. (2008) injected rhesus oocytes with lentivirus expressing exon 2 of the human huntingtin gene with 84 CAG repeats and five transgenic monkeys carrying mutant huntingtin were produced. The monkeys showed the main features of HD disease including nuclear inclusions, neuropil aggregates and a behavioral phenotype but all of them died at early stage of life.
In the HD model in Drosophila the N-terminal fragment of the human mutant protein containing 2, 75 and 120 CAG repeats were expressed in the photoreceptor neurons of the fly eye (Jackson et al. 1998). Jackson et al. found that neuronal differentiation is normal and the onset of degeneration is correlated with the length of CAG repeats. The expanded repeats are initially cytoplasmic but relocalization to the nucleus occurs before neuronal death.
C. elegans does not have huntingtin homolog, but worms are an efficient way to study cellular mechanisms. Faber et al. found CAG repeats of 2, 23, 95 and 150 in C. elegans sensory neurons and that the CAG150 repeat led to protein aggregates and degeneration of sensory neuron terminals but did not lead to cell death (Faber et al. 1999). The CAG150 repeat increased the vulnerability of sensory neurons to toxic OSM-10 green fluorescent protein. In another study, C. elegans over-expressing 128 CAG repeats led to neuronal dysfunction but did not lead to neuronal cell death (Parker et al. 2005).
Unlike the other neurodegenerative disorders described in this review, HD is linked to a single gene, huntingtin. Animal models based mutant forms of the huntingtin gene have been generated from worms to primates and have been used to study the disease mechanism and test potential therapeutics. Over 500 clinical trials are active or have been completed for HD (http://www.clinicaltrials.gov) and the animal models described above have been indispensable to the corresponding preclinical studies.
Parkinson's disease
Parkinson's disease is a slow, progressive neurodegenerative disorder that is characterized pathologically by the loss of dopaminergic neurons in the pars compacta of the substantia nigra. The cardinal symptoms of PD are resting tremor, rigidity, bradykinesia and occasionally dementia. PD is primarily idiopathic resulting from a complex interaction of genetics and environment. A subset (<15% of cases) of patients have a family history of PD and pedigree analyses have identified 13 PARK loci to date (OMIM 168600). Molecular genetics studies have identified genes associated with 11 of 16 PARK loci and transgenic animal models for six of these genes (i.e. α-synuclein, parkin, DJ-1, PINK1, UCH-L1 and LRRK2) will be discussed below.
α-Synuclein (PARK1 and PARK4)
The first gene linked to PD was the α-synuclein gene and the first mutation, A53T, was found in a large family with an autosomal dominant pattern of inherited PD (Polymeropoulos et al. 1997). Two additional mutants A30P and E46K were subsequently identified in independent linkage studies (Ueda et al. 1993; Zarranz et al. 2004). Alpha-synuclein is found in presynaptic nerve terminals and originally was identified as a non-β amyloid component of Alzheimer's disease-related amyloid plaques (Ueda et al. 1993). In post-mortem brains of PD patients, α-synuclein was detected in Lewy bodies which are pathological hallmarks of PD (Spillantini et al. 1997). Normal α-synuclein is important for physiologic function of dopamine neurons as deletion of α-synuclein leads to reduction in striatal dopamine and reduction in TH-positive fibers but does not affect number of TH-positive neurons in aging mice (Abeliovich et al. 2000; Al-Wandi et al. 2008).
Transgenic mice over-expressing human α-synuclein showed an accumulation of α-synuclein in neuronal inclusion bodies found in several brain regions including the substantia nigra (Masliah et al. 2000). The formation of inclusion bodies correlated with loss of striatal dopaminergic terminals and motor impairment. Transgenic mice expressing human α-synuclein with the A30P mutation exhibited increased accumulation of the mutant α-synuclein in the neuronal cell bodies and neurites (Kahle et al. 2000). In a recent study the A30P mutation in mice led to development of motor performance deficits at the age of 13 months and reduced levels of dopamine in striatum at the age of 15 months (Plaas et al. 2008). Similarly, transgenic mice expressing human α-synuclein with both the A53T mutation or “wild-type” human α-synuclein exhibited Lewy body-like pathology, evidence of neurodegeneration and motor deficits; however, the transgenic protein was not expressed in nigral neurons (Kahle et al. 2000). Using a different promoter, transgenic mice expressing the A53T mutant, but not the human “wild type” α-synuclein, developed severe motor deficits that led to death, paralleled by the accumulation of cytoplasmic filamentous inclusions in neurons (Giasson et al. 2002). In the Giasson et al. (2002) study, the mouse prion protein promoter was used, compared to Thy-1 promoter used in the studies by Kahle et al. (2000) and van der Putten et al. (2000) indicating that the temporal and spatial expression of the human synuclein forms is important for the manifestation of neurodegenerative effects (Kahle et al. 2000; van der Putten et al. 2000). A study comparing transgenic mice expressing human α -synuclein, A53T mutant and A30P mutant from the mouse prion promoter, found that the A53T mutant caused more neurodegeneration associated with abnormal accumulation of detergent-insoluble aggregates of α-synuclein (Giasson et al. 2002). Mice overexpressing human α-synuclein with both A53T and A30P mutations had accumulation of α-synuclein in neurons, decreased dopamine concentrations in striatum and motor impairment (Ikeda et al. 2009). Although the aforementioned studies are models of α-synucleopathies, the lack of selective expression in nigral neurons or nigral degeneration thus fails to generate a full phenocopy of PD.
Selective expression of the wild-type, A53T or A30P mutants in dopaminergic neurons using the tyrosine hydroxylase promoter did not cause selective loss of nigral neurons or striatal dopamine, or the development of inclusions in dopaminergic neurons (Matsuoka et al. 2001). A separate study by Rathke-Hartlieb et al. (2001) showed similar findings and reported no change in sensitivity to the dopaminergic toxin, MPTP. Richfield et al. (2002) used the rat tyrosine hydroxylase 9.0 kb promoter to express a transgenic double mutant (A53T and A30P) form of α-synuclein in dopaminergic neurons. Expression of the double mutant led to age-related declines in motor coordination, striatal dopamine and integrity of dopaminergic terminals but no inclusion bodies were reported (Richfield et al. 2002). In contrast to a previous study (Rathke-Hartlieb et al. 2001), mice expressing wild-type or double-mutant human α-synuclein were more susceptible to MPTP toxicity (Richfield et al. 2002). A communication by Tofaris et al. (2006) used the rat TH promoter to express a truncated form of human α-synuclein in a mouse α-synuclein null background. Unlike previous reports, inclusion bodies were observed in the substantia nigra. An age-related decline in striatal dopamine and locomotor activity was also observed in these animals (Tofaris et al. 2006). This study indicated that the C-terminal portion of α-synuclein is important for the formation of inclusions. To summarize, overexpressing human α-synuclein or its mutated forms in transgenic mice is not sufficient to cause a complete Parkinsonian phenotype. However, this approach is valuable for understanding synucleopathies and for studying vulnerability to environmental toxins and possible therapies aimed at preventing synucleopathies. Despite numerous studies indicating that α-synuclein is involved in synaptic turnover/function and synaptic trafficking, its endogenous function and pathological role as a component of Lewy bodies remains to be determined.
Recent studies indicate transgenic mice with both the double-mutant human form of α-synuclein and the exon 3 knockout of parkin have combined age- and tissue-related effects on mitochondrial morphology and integrity (Stichel et al. 2007). Further studies on the relationship of α-synuclein and parkin may provide insights into the molecular mechanisms of mitochondrial dysfunction in PD.
Parkin (parkin; PARK2)
Studies of autosomal recessive inheritance pattern of early-onset PD in a group of Japanese families led to the identification of the parkin gene at the PARK2 locus (Kitada et al. 1998). Since the study by Kitada and colleagues, multiple studies have shown that mutations in Parkin are linked to autosomal recessively inherited PD. Unlike α-synuclein that has few identified mutations, more than a 100 mutations have been identified in the parkin gene (Abbas et al. 1999; Lucking et al. 2000; Hedrich et al. 2001, 2004; Itier et al. 2003; Oliveira et al. 2003; Klein et al. 2007). Parkin has an E3 ubiquitin-protein ligase activity (Shimura et al. 2000) and targets proteins for degradation by the proteosome (Zhang et al. 2000; Chung et al. 2001; Imai et al. 2001).
Several laboratories have generated parkin knockout mice by targeting different exons of the parkin gene (Bonifati et al. 2003; Goldberg et al. 2003; Palacino et al. 2004; Von Coelln et al. 2004; Perez et al. 2005; Sato et al. 2006). The phenotype of the mice varies and overall they fail to develop a Parkinsonian phenotype, but the different knockout models may provide a means to examine the role of parkin in protein turnover, oxidative stress and mitochondrial dysfunction.
DJ-1 (PARK7)
The DJ-1 gene was identified at the PARK7 locus (van Duijn et al. 2001) with a point mutation (L166P) that cosegregated with the disease allele in an Italian family (Bonifati et al. 2003). Mutations in DJ-1 are autosomal recessive and are associated with early onset of PD (Abou-Sleiman et al. 2003; Bonifati et al. 2003; Hague et al. 2003; Hedrich et al. 2004; Annesi et al. 2005). DJ-1 is a member of the THiJ/pfpI family and exhibits a redox-dependent chaperone function which may be important for combating oxidative-stress (Shenk et al. 2009).
Studies of DJ-1 knockout animals demonstrated that absence of DJ-1 expression (1) decreases motor function and (2) alters dopamine function in the nigrostriatal pathway (Chen et al. 2005; Goldberg et al. 2005; Kim et al. 2005). A recent study demonstrated that upregulation of DJ-1 can compensate for defects in pink1 mutant flies (Hao et al. 2010). Development of transgenic animals based on other mutations in DJ-1 as well as further studies on DJ-1 knockout mice may provide a useful platform for testing gene therapeutic strategies for patients carrying DJ-1 mutations and offer opportunities to further understand the role of DJ-1 in oxidative stress.
PTEN-induced kinase 1 (PINK1; PARK6)
Point mutations in the PTEN-induced kinase or PINK1 gene, a putative mitochondrial kinase at the PARK6 locus, are the second most frequently occurring cause of autosomal recessively inherited early-onset PD (Valente et al. 2001,2004; Hatano et al. 2004; Rogaeva et al. 2004; Rohe et al. 2004; Bonifati et al. 2005; Bender et al. 2006; Ibanez et al. 2006). PINK1 is present throughout the brain and localizes to mitochondria where it is thought to protect against mitochondrial dysfunction (Gandhi et al. 2006).
Studies examining knockdown or knockout of the PINK1 gene failed to produce a PD phenotype (Kitada et al. 2007; Zhou et al. 2007). However, a transgenic mouse created by targeting the human G309D-PINK1 mutation into mice created a knockout mouse that exhibited mitochondrial dysfunction in brain, impaired spontaneous locomotor activity, weight loss and dopaminergic synapse dysfunction in aged mice (Gispert et al. 2009). Although there was no signs of neurodegeneration in the mice described by Gispert et al., the observed changes in dopamine, α-synuclein and locomotion in older animals suggests that this mouse model may recapitulate certain features of early PD. Consistent with this model, mutations in both DJ-1 and PINK1 genes have been identified in subset of patients with early onset PD. Biochemical studies suggest DJ-1 stabilizes PINK1 and works cooperatively with it to protect cells against oxidative stress (Gandhi et al. 2006). Studies in Drosophilia have found that PINK1 and Parkin are involved in a common pathway important for mitochondrial function and morphology (Clark et al. 2006; Park et al. 2006, 2009; Deng et al. 2008; Kim et al. 2008; Yang et al. 2008). Future studies examining the role of PINK1 and parkin in mitochondrial function in mammalian models may lead to the development of a mouse model that more closely resembles the pathology of PD.
Ubiquitin carboxy-terminal hydrolase-L1 (UCH-L1; PARK5)
A missense mutation (I93M) in the ubiquitin carboxy-terminal hydrolase-L1 (UCH-L1) gene was identified in a sibling pair affected by PD (Leroy et al. 1998). Although no additional mutations in UCH-L1 have been linked to PD, a variant (S18Y) has been identified that correlates with lowered risk of developing PD (Lincoln et al. 1999; Maraganore et al. 1999, 2004). The gracile axonal dystrophy (gad) mouse has a deletion in the UCH-L1 gene, but the mice do not exhibit behavioral and pathological characteristics of PD (Saigoh et al. 1999). Recently, Setsuie et al. (2007) generated a transgenic mouse expressing the I93M mutant form of UCH-L1 under expression of the PDGF-beta promoter. Mice over-expressing UCH-L1 (I93M) had fewer dopaminergic neurons in the substantia nigra and decreased striatal dopamine levels compared to wild-type mice. The transgenic mice also exhibited increased levels of insoluble UCH-L1 in the midbrain, increased sensitivity to MPTP (Setsuie et al. 2007) and over-expression of α-synuclein (Yasuda et al. 2009). Overall, the occurrence of the UCH-L1 mutation in PD patients is rare, but a transgenic mouse study using the I93M mutations, suggests that UCH-L1 function may contribute to dopaminergic degeneration. Additional genetic analysis studies in PD patients or studies on the effects of expressing the S18Y mutation in mice are needed to explore the relevance of UCH-L1 in PD.
Leucine-rich repeat kinase 2 (LRRK2; PARK8)
The leucine-rich repeat kinase 2 (LRRK2) gene at the PARK8 locus (Funayama et al. 2002) was initially identified from a pedigree of autosomal dominantly inherited PD (Paisan-Ruiz et al. 2004; Zimprich et al. 2004). The LRRK2 protein, approximately 286 kD, has multiple predicted domains. In rodent basal ganglia LRRK2 is found in lysosomes, endosomes, transport vesicles and mitochondria (Biskup et al. 2006) and overexpression or knockdown of LRRK2 has been shown to impair synaptic vesicle endocytosis (Shin et al. 2008). Nearly 50 mutations in LRRK2 have been identified and currently constitute the most common cause of inherited PD. The common G2019S mutation is found in idiopathic cases of PD (Lesage et al. 2007) and has been shown to increase kinase activity in vitro (Chen et al. 2005). West and colleagues also described altered kinase activity of the R1441C/G mutation in the GTPase domain of LRRK2 (West et al. 2005).
Recently, a transgenic mouse was generated by introducing a BAC containing a FLAG-tagged LRRK2 expressed from 35 kb of the LRRK2 promoter and 60 kb of the LRRK2 3′ non-coding region (Li et al. 2007). The FLAG-LRRK2 was expressed in several brain regions including the dopaminergic neurons of the substantia nigra. Affinity-purified FLAG-LRRK2 from brains of transgenic mice had higher kinase and GTPase activity compared to FLAG-LRRK2 from lung or from FLAG-LRRK2 transfected into HEK-293T cells. HEK-293T cells transfected with the R1441C/G mutant of LRRK2 had decreased GTPase activity compared to non-mutant LRRK2 (Ekstrand et al. 2007). Although these in vivo results support previous in vitro reports of kinase and GTPase activity of LRRK2 and its possible role in PD, the effects of overexpressing the FLAG-LRRK2 on the dopaminergic nigrostriatal neurons or locomotor activity was not described.
The loss of LRRK2 expression in Drosophila leads to dopaminergic neuron degeneration and altered locomotor activity (Lee et al. 2007). Recent studies on mice containing LRRK2 WT, LRRK2 G2019S or LRRK2 R1441G mutations do not result in a PD phenotype (Li et al. 2009, 2010; Lin et al. 2009; Tong et al. 2009). Given there is no obvious neurodegeneration, these models do not currently provide an opportunity to evaluate therapeutic strategies. Further studies examining how environmental stressors combined with LRRK2 mutations may lead to PD-like phenotype may provide a better phenocopy of PD.
Although the loss of dopaminergic neurons in the SN is the primary “mechanism” that causes PD, the molecular changes that lead to the loss of this specific cell type is unknown. Several genes described above are linked to PD but the majority of patients (80–85%) do not appear to have an inherited genetic link. The disease likely results from the complex interplay of genetic predisposition seeded in the genes described above and related genes in common cellular pathways, the chemical properties of dopamine and its metabolism, and environmental factors known to affect the dopaminergic system. The use of transgenic models for PD has been valuable in our understanding of disease mechanism. Future studies utilizing these models to understand the complex interplay of genes, dopamine metabolism and the environment may lead to therapies tailored to halt progression of disease or prevent the disease from occurring.
Concluding remarks
Transgenic animal models based on mutations in human genes enable researchers to dissect the function of a mutant gene or combination of mutant genes in the pathophysiology of the disease. More importantly, once a transgenic model has been established to have characteristics of a disease, it can be used as a platform for evaluating potential genetic and pharmacological therapies. As we gain more understanding of how the genes identified in human pedigree analyses and gene linkage studies fit into cellular pathways that contribute to disease, we can use transgenic animals to examine how the combination of multiple “mutations” leads to neurodegeneration.
Acknowledgments
The authors thank Dr. Marc Halterman for his recommendations on the table. This work was supported by the Intramural Research Program at the National Institute on Drug Abuse.
References
- Abbas N, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol Aging. 2008;31:796–804. doi: 10.1016/j.neurobiolaging.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families Alzheimer's Disease Collaborative Group. Nat Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- Anagnostou G, Akbar MT, Paul P, Angelinetta C, Steiner TJ, de Belleroche J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol Aging. 2008;31:969–985. doi: 10.1016/j.neurobiolaging.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Annesi G, Savettieri G, Pugliese P, D'Amelio M, Tarantino P, Ragonese P, La Bella V, Piccoli T, Civitelli D, Annesi F, Fierro B, Piccoli F, Arabia G, Caracciolo M, Ciro Candiano IC, Quattrone A. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58:803–807. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bennett SP, Boyd TD, Norden M, Padmanabhan J, Neame P, Wefes I, Potter H. A novel technique for simultaneous bilateral brain infusions in a mouse model of neurodegenerative disease. J Neurosci Methods. 2009;184:320–326. doi: 10.1016/j.jneumeth.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–1457. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TD. Genetic aspects of Alzheimer disease. Genet Med. 2008;10:231–239. doi: 10.1097/GIM.0b013e31816b64dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bonifati V, et al. Early-onset Parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283:28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer's disease: an update. Ann Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Brunden KR, Trojanowski JQ, Lee VM. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Lin X, Xie C, Laird FM, Lai C, Wen H, Chiang HC, Shim H, Farah MH, Hoke A, Price DL, Wong PC. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J Neurosci. 2005;25:7567–7574. doi: 10.1523/JNEUROSCI.1645-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Shim H, Lai C, Xie C, Lin X, Yang WJ, Chandran J. ALS2/alsin knockout mice and motor neuron diseases. Neurodegener Dis. 2008;5:359–366. doi: 10.1159/000151295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Temel Y, Blokland A, Ozen H, Steinbusch HW, Vlamings R, Nguyen HP, von Horsten S, Schmitz C, Visser-Vandewalle V. Progressive deterioration of reaction time performance and choreiform symptoms in a new Huntington's disease transgenic rat model. Behav Brain Res. 2006;170:257–261. doi: 10.1016/j.bbr.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai A, Withers J, Koh YH, Parry K, Bao H, Zhang B, Budnik V, Pennetta G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum Mol Genet. 2008;17:266–280. doi: 10.1093/hmg/ddm303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, Griffin JW, Cornblath DR. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet. 1998;62:633–640. doi: 10.1086/301769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Hong R, Wada M, Koyama S, Kimura H, Arawaka S, Kawanami T, Kurita K, Kadoya T, Aoki M, Itoyama Y, Kato T. Neuroprotective effect of oxidized galectin-1 in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;194:203–211. doi: 10.1016/j.expneurol.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH. Deleterious variants of FIG 4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Conforti FL, Sprovieri T, Mazzei R, Ungaro C, La Bella V, Tessitore A, Patitucci A, Magariello A, Gabriele AL, Tedeschi G, Simone IL, Majorana G, Valentino P, Condino F, Bono F, Monsurro MR, Muglia M, Quattrone A. A novel angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul Disord. 2008;18:68–70. doi: 10.1016/j.nmd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D'Alfonso S. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009;30:688–694. doi: 10.1002/humu.20950. [DOI] [PubMed] [Google Scholar]
- Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupre N, Camu W, Meininger V, Rouleau GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46:112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O'Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Zhai H, Fu R, Shi Y, Gorrie GH, Yang Y, Liu E, Dal Canto MC, Mugnaini E, Siddique T. Distal axonopathy in an alsin-deficient mouse model. Hum Mol Genet. 2007;16:2911–2920. doi: 10.1093/hmg/ddm251. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devon RS, Orban PC, Gerrow K, Barbieri MA, Schwab C, Cao LP, Helm JR, Bissada N, Cruz-Aguado R, Davidson TL, Witmer J, Metzler M, Lam CK, Tetzlaff W, Simpson EM, McCaffery JM, El-Husseini AE, Leavitt BR, Hayden MR. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci USA. 2006;103:9595–9600. doi: 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, May P. Overview on rodent models of Alzheimer's disease. Curr Protoc Neurosci. 2005 doi: 10.1002/0471142301.ns0922s33. Chap. 9:Unit 9.22. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive Parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder GA, Gama Sosa MA, De Gasperi R, Dickstein DL, Hof PR. Presenilin transgenic mice as models of Alzheimer's disease. Brain Struct Funct. 2010;214:127–143. doi: 10.1007/s00429-009-0227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci USA. 1999;96:179–184. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Baudry M, Diaz Brinton R, Thompson RF. Estrogen and hippocampal plasticity in rodent models. J Alzheimers Dis. 2008;15:589–603. doi: 10.3233/jad-2008-15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, Revesz T. PINK1 protein in normal human brain and Parkinson's disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Garcia-Sierra F, Mondragon-Rodriguez S, Basurto-Islas G. Truncation of tau protein and its pathological significance in Alzheimer's disease. J Alzheimers Dis. 2008;14:401–409. doi: 10.3233/jad-2008-14407. [DOI] [PubMed] [Google Scholar]
- Gellera C, Colombrita C, Ticozzi N, Castellotti B, Bragato C, Ratti A, Taroni F, Silani V. Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics. 2008;9:33–40. doi: 10.1007/s10048-007-0111-3. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Gimenez-Llort L, Blazquez G, Canete T, Johansson B, Oddo S, Tobena A, LaFerla FM, Fernandez-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31:125–147. doi: 10.1016/j.neubiorev.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Gispert S, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway MJ, Alexander MD, Ennis S, Traynor BJ, Corr B, Frost E, Green A, Hardiman O. A novel candidate region for ALS on chromosome 14q11.2. Neurology. 2004;63:1936–1938. doi: 10.1212/01.wnl.0000144344.39103.f6. [DOI] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Jr, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306:234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- Hadano S, Benn SC, Kakuta S, Otomo A, Sudo K, Kunita R, Suzuki-Utsunomiya K, Mizumura H, Shefner JM, Cox GA, Iwakura Y, Brown RH, Jr, Ikeda JE. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet. 2006;15:233–250. doi: 10.1093/hmg/ddi440. [DOI] [PubMed] [Google Scholar]
- Hague S, Rogaeva E, Hernandez D, Gulick C, Singleton A, Hanson M, Johnson J, Weiser R, Gallardo M, Ravina B, Gwinn-Hardy K, Crawley A, St George-Hyslop PH, Lang AE, Heutink P, Bonifati V, Hardy J, Singleton A. Early-onset Parkinson's disease caused by a compound heterozygous DJ-1 mutation. Ann Neurol. 2003;54:271–274. doi: 10.1002/ana.10663. [DOI] [PubMed] [Google Scholar]
- Haltia M, Viitanen M, Sulkava R, Ala-Hurula V, Poyhonen M, Goldfarb L, Brown P, Levy E, Houlden H, Crook R, et al. Chromosome 14-encoded Alzheimer's disease: genetic and clinicopathological description. Ann Neurol. 1994;36:362–367. doi: 10.1002/ana.410360307. [DOI] [PubMed] [Google Scholar]
- Hao LY, Giasson BI, Bonini NM. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci USA. 2010;107:9747–9752. doi: 10.1073/pnas.0911175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset Parkinsonism. Ann Neurol. 2004;56:424–427. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- HDCRG. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O, Schwinger E, Vieregge P, Lang AE, Breakefield XO, Ozelius LJ, Pramstaller PP, Klein C. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset Parkinsonism. Hum Mol Genet. 2001;10:1649–1656. doi: 10.1093/hmg/10.16.1649. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004;62:389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Honson NS, Kuret J. Tau aggregation and toxicity in tauopathic neurodegenerative diseases. J Alzheimers Dis. 2008;14:417–422. doi: 10.3233/jad-2008-14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett DR, Richardson JC. The pathology of APP transgenic mice: a model of Alzheimer's disease or simply overexpression of APP? Histol Histopathol. 2009;24:83–100. doi: 10.14670/HH-24.83. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Huntington G. On chorea. Med Surg Rep. 1872;26:317–321. [Google Scholar]
- Ibanez P, Lesage S, Lohmann E, Thobois S, De Michele G, Borg M, Agid Y, Durr A, Brice A. Mutational analysis of the PINK1 gene in early-onset Parkinsonism in Europe and North Africa. Brain. 2006;129:686–694. doi: 10.1093/brain/awl005. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Kawarabayashi T, Harigaya Y, Sasaki A, Yamada S, Matsubara E, Murakami T, Tanaka Y, Kurata T, Wuhua X, Ueda K, Kuribara H, Ikarashi Y, Nakazato Y, Okamoto K, Abe K, Shoji M. Motor impairment and aberrant production of neurochemicals in human alpha-synuclein A30P + A53T transgenic mice with alpha-synuclein pathology. Brain Res. 2009;1250:232–241. doi: 10.1016/j.brainres.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Itier JM, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Jacquier A, Buhler E, Schafer MK, Bohl D, Blanchard S, Beclin C, Haase G. Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann Neurol. 2006;60:105–117. doi: 10.1002/ana.20886. [DOI] [PubMed] [Google Scholar]
- Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- Jonsson PA, Graffmo KS, Brannstrom T, Nilsson P, Andersen PM, Marklund SL. Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol. 2006;65:1126–1136. doi: 10.1097/01.jnen.0000248545.36046.3c. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, Fenner B, Hardiman O, Prehn JH. Control of motoneuron survival by angiogenin. J Neurosci. 2008;28:14056–14061. doi: 10.1523/JNEUROSCI.3399-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with Parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, Schellenberg GD. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnlein P, Sperfeld AD, Vanmassenhove B, Van Deerlin V, Lee VM, Trojanowski JQ, Kretzschmar HA, Ludolph AC, Neumann M. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008;65:1185–1189. doi: 10.1001/archneur.65.9.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, Chandran J, Shim H, Shimoji M, Cookson MR, Huganir RL, Rothstein JD, Price DL, Wong PC, Martin LJ, Zhu JJ, Cai H. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J Neurosci. 2006;26:11798–11806. doi: 10.1523/JNEUROSCI.2084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Amouyel P. Genetic heterogeneity of Alzheimer's disease: complexity and advances. Psychoneuroendocrinology. 2007;32(Suppl 1):S62–S70. doi: 10.1016/j.psyneuen.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–324. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesage S, et al. LRRK2 exon 41 mutations in sporadic Parkinson disease in Europeans. Arch Neurol. 2007;64:425–430. doi: 10.1001/archneur.64.3.425. [DOI] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer's disease locus on chromosome 1. Science. 1995a;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995b;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Lin X, Shim H, Cai H. Deficiency in the ALS2 gene does not affect the motor neuron degeneration in SOD1(G93A) transgenic mice. Neurobiol Aging. 2007;28:1628–1630. doi: 10.1016/j.neurobiolaging.2006.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln S, Vaughan J, Wood N, Baker M, Adamson J, Gwinn-Hardy K, Lynch T, Hardy J, Farrer M. Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson's disease. Neuroreport. 1999;10:427–429. doi: 10.1097/00001756-199902050-00040. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, Farrer MJ, Hardy JA, Lincoln SJ, McDonnell SK, Rocca WA. Case-control study of the ubiquitin carboxy-terminal hydrolase L1 gene in Parkinson's disease. Neurology. 1999;53:1858–1860. doi: 10.1212/wnl.53.8.1858. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, de Andrade M, Rocca WA. UCHL1 is a Parkinson's disease susceptibility gene. Ann Neurol. 2004;55:512–521. doi: 10.1002/ana.20017. [DOI] [PubMed] [Google Scholar]
- Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Wu Y, Olivieri M, Li XJ, Li H, Zeitlin S, Chesselet MF. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science (New York, NY. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Al-Chalabi A, Zatz M. A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum Genet. 2005;118:499–500. doi: 10.1007/s00439-005-0031-y. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oliveira SA, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann Neurol. 2003;53:624–629. doi: 10.1002/ana.10524. [DOI] [PubMed] [Google Scholar]
- Otomo A, Kunita R, Suzuki-Utsunomiya K, Mizumura H, Onoe K, Osuga H, Hadano S, Ikeda JE. ALS2/alsin deficiency in neurons leads to mild defects in macropinocytosis and axonal growth. Biochem Biophys Res Commun. 2008;370:87–92. doi: 10.1016/j.bbrc.2008.01.177. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in Parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- Paubel A, Violette J, Amy M, Praline J, Meininger V, Camu W, Corcia P, Andres CR, Vourc'h P. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. 2008;65:1333–1336. doi: 10.1001/archneur.65.10.1333. [DOI] [PubMed] [Google Scholar]
- Perez FA, Curtis WR, Palmiter RD. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 2005;6:71. doi: 10.1186/1471-2202-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaas M, Karis A, Innos J, Rebane E, Baekelandt V, Vaarmann A, Luuk H, Vasar E, Koks S. Alpha-synuclein A30P point-mutation generates age-dependent nigrostriatal deficiency in mice. J Physiol Pharmacol. 2008;59:205–216. [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haass C, Schulz JB. Sensitivity to MPTP is not increased in Parkinson's disease-associated mutant alpha-synuclein transgenic mice. J Neurochem. 2001;77:1181–1184. doi: 10.1046/j.1471-4159.2001.00366.x. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- Rohe CF, Montagna P, Breedveld G, Cortelli P, Oostra BA, Bonifati V. Homozygous PINK1 C-terminus mutation causing early-onset Parkinsonism. Ann Neurol. 2004;56:427–431. doi: 10.1002/ana.20247. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- Sato S, Chiba T, Nishiyama S, Kakiuchi T, Tsukada H, Hatano T, Fukuda T, Yasoshima Y, Kai N, Kobayashi K, Mizuno Y, Tanaka K, Hattori N. Decline of striatal dopamine release in Parkin-deficient mice shown by ex vivo autoradiography. J Neurosci Res. 2006;84:1350–1357. doi: 10.1002/jnr.21032. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Sawamura N, Morishima-Kawashima M, Waki H, Kobayashi K, Kuramochi T, Frosch MP, Ding K, Ito M, Kim TW, Tanzi RE, Oyama F, Tabira T, Ando S, Ihara Y. Mutant presenilin 2 transgenic mice. A large increase in the levels of Abeta 42 is presumably associated with the low density membrane domain that contains decreased levels of glycerophospholipids and sphingomyelin. J Biol Chem. 2000;275:27901–27908. doi: 10.1074/jbc.M004308200. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Setsuie R, Wang YL, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun YJ, Kwon J, Kabuta T, Yoshimi K, Aoki S, Mizuno Y, Noda M, Wada K. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson's disease-associated UCH-L1 I93M mutant. Neurochem Int. 2007;50:119–129. doi: 10.1016/j.neuint.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RM. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8:763–774. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- Shenk JC, Liu J, Fischbach K, Xu K, Puchowicz M, Obrenovich ME, Gasimov E, Alvarez LM, Ames BN, Lamanna JC, Aliev G. The effect of acetyl-L-carnitine and R-alpha-lipoic acid treatment in ApoE4 mouse as a model of human Alzheimer's disease. J Neurol Sci. 2009;283:199–206. doi: 10.1016/j.jns.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian V, Crabtree B, Acharya KR. Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum Mol Genet. 2008;17:130–149. doi: 10.1093/hmg/ddm290. [DOI] [PubMed] [Google Scholar]
- Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, Gatei M, Criscuolo C, Filla A, Chessa L, Fusser M, Epe B, Gueven N, Lavin MF. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kanekura K, Levine TP, Kohno K, Olkkonen VM, Aiso S, Matsuoka M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J Neurochem. 2009;108:973–985. doi: 10.1111/j.1471-4159.2008.05857.x. [DOI] [PubMed] [Google Scholar]
- Takashima A. Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J Alzheimers Dis. 2008;14:371–375. doi: 10.3233/jad-2008-14403. [DOI] [PubMed] [Google Scholar]
- Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303:496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor EL, Perkinton MS, Schmidt A, Ackerley S, Brownlees J, Jacobsen NJ, Byers HL, Ward M, Hall A, Leigh PN, Shaw CE, McLoughlin DM, Miller CC. ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J Biol Chem. 2005;280:34735–34740. doi: 10.1074/jbc.M506216200. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW. Localization of a novel locus for autosomal recessive early-onset Parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset Parkinsonism. Ann Neurol. 2004;56:336–341. doi: 10.1002/ana.20256. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P. Park7, a novel locus for autosomal recessive early-onset Parkinsonism, on chromosome lp36. Am J Hum Genet. 2001;69:629–634. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA. 2004;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Horsten S, et al. Transgenic rat model of Huntington's disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, Sullivan PM. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiology of Disease. 2005a;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C, Copeland NG, Jenkins NA, Borchelt DR. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: alphaB-crystallin modulates aggregation. Hum Mol Genet. 2005b;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Yasui K, Nakano T, Doi K, Fukada Y, Kitayama M, Ishimoto M, Kurihara S, Kawashima M, Fukuda H, Adachi Y, Inoue T, Nakashima K. Mouse motor neuron disease caused by truncated SOD1 with or without C-terminal modification. Brain Res Mol Brain Res. 2005;135:12–20. doi: 10.1016/j.molbrainres.2004.11.019. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS. Animal models of Alzheimer's disease: therapeutic implications. J Alzheimers Dis. 2008;15:507–521. doi: 10.3233/jad-2008-15401. [DOI] [PubMed] [Google Scholar]
- Wu D, Yu W, Kishikawa H, Folkerth RD, Iafrate AJ, Shen Y, Xin W, Sims K, Hu GF. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann Neurol. 2007;62:609–617. doi: 10.1002/ana.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Miller TM, McAlonis-Downes M, Chun SJ, Cleveland DW. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann Neurol. 2006;60:95–104. doi: 10.1002/ana.20888. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Nihira T, Ren YR, Cao XQ, Wada K, Setsuie R, Kabuta T, Hattori N, Mizuno Y, Mochizuki H. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson's disease. J Neurochem. 2009;108:932–944. doi: 10.1111/j.1471-4159.2008.05827.x. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46 K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3:242–250. doi: 10.7150/ijbs.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant Parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]