

Abstract
Sulfur mustard (HD) is a vesicating agent that has been used as a chemical warfare agent in a number of conflicts, posing a major threat in both military conflict and chemical terrorism situations. Currently, we lack effective therapies to rescue skin injuries by HD, in part, due to the lack of appropriate animal models, which are required for conducting laboratory studies to evaluate the therapeutic efficacy of promising agents that could potentially be translated in to real HD-caused skin injury. To address this challenge, the present study was designed to assess whether microvesication could be achieved in mouse skin by an HD analog 2-chloroethyl ethyl sulfide (CEES) exposure; notably, microvesication is a key component of HD skin injury in humans. We found that skin exposure of male SKH-1 hairless mice to CEES caused epidermal-dermal separation indicating microvesication. In other studies, CEES exposure also caused an increase in skin bi-fold thickness, wet/dry weight ratio, epidermal thickness, apoptotic cell death, cell proliferation, and infiltration of macrophages, mast cells and neutrophils in male SKH-1 hairless mouse skin. Taken together, these results establish CEES-induced microvesication and inflammation-related histopathological changes in mouse skin, providing a potentially relevant laboratory model for developing effective countermeasures against HD skin injury in humans.
Keywords: Sulfur mustard, 2-chloroethyl ethyl sulfide (CEES), male SKH-1 hairless mouse, inflammatory biomarkers, blisters, epidermal-dermal separation
1. INTRODUCTION
Sulfur mustard (bis (2-chloroethyl) sulfide, HD) is a powerful bifunctional alkylating and vesicating agent that has been used as a major chemical warfare agent in a number of conflicts (Noort et al., 2002; Saladi et al., 2006). For the development of useful prophylactic and/or therapeutic interventions to counter HD’s toxic actions, understanding its mechanism of action is essential. Although primary targets of HD are skin, eyes and respiratory organs, it also causes injuries to other organs such as respiratory tract, reproductive system, central nervous system and gastrointestinal tract (Graham et al., 2005). Upon exposure, HD causes severe cutaneous injury that occurs hours after its exposure and is associated with erythema, inflammation, blister formation and cell death mainly of basal epidermal keratinocytes (Dacre and Goldman, 1996; Graham et al., 2005; Wormser, 1991). HD-related skin toxicity is a complex process where inflammation plays an important role in the pathological changes including vesication (Shakarjian et al., 2010). Histopathological analysis of HD-exposed skin reveals apoptosis in basal keratinocytes leading to a severe inflammatory response including vasodilatation and infiltration of various inflammatory cells at the site of injury (Smith et al., 1997; Smith et al., 1998).
A large number of animal models including the weanling pig, hairless guinea pig, hairless mouse and rabbit have been utilized to study HD-induced vesication and the associated toxicity in the skin (Blaha et al., 2000; Brodsky et al., 2006; Smith et al., 1997). Several in vitro models such as epidermal keratinocyte cell culture and bioengineered human skin have also been employed to understand the mechanisms of HD-induced skin toxicity (Greenberg et al., 2006; Hayden et al., 2009; Smith et al., 1990; Tewari-Singh et al., 2010). Although these efforts have helped gain an insight into the pathogenesis and mechanism of action of HD, an animal skin toxicity model that mimics the vesicating effect of HD in humans is needed in a laboratory setting to accelerate the development of efficacious countermeasures. In this regard, the commercially available monofunctional analog of HD, 2-chloroethyl ethyl sulfide (CEES), has been used in cell culture studies, and in animal studies at a concentration of 0.5–6 mg/kg/animal causing skin injuries similar to HD (Black et al., 2010; Chatterjee et al., 2004; Han et al., 2004; Tewari-Singh et al., 2010; Tewari-Singh et al., 2009). In recent studies, we reported that CEES-induced inflammatory and pathogenic alterations in female SKH-1 hairless mouse skin and uncovered molecular mechanisms associated with such skin injury end points (Pal et al., 2009; Tewari-Singh et al., 2009). However, microvesication that is a key component of HD skin injury in humans, was not observed in mice exposed to the 2 mg CEES dose that was used in our studies.
Taken together, the present study was designed to address whether microvesication could be achieved in mouse skin by CEES exposure, as well as whether there are sex-related variations in CEES-induced inflammatory responses in mouse skin. Because 2 mg CEES-related histopathological changes observed in our studies were similar in both the male and female hairless mice, we further exposed only male mice to a higher dose of 4 mg CEES that caused microvesication and could be used as a key biomarker in efficacy studies. Our results herein also demonstrate that CEES causes inflammation-related histopathological alterations in male hairless mouse skin, which are comparable to those in female mice of the same strain. This newly developed mouse CEES-vesicant laboratory model provides an opportunity for accelerated agent efficacy studies to develop counteragents against HD skin injury in humans without having to use the highly toxic HD compound itself.
2. Materials and Methods
2.1. Materials
CEES was purchased from Sigma-Aldrich Chemicals Co. (St. Louis, MO). Apoptosis detection kit (DeadEnd Colorimetric Terminal deoxynucleotidyl transferase dUTP nick end labeling {TUNEL} System) was purchased from Promega (Madison, WI). Anti proliferative cell nuclear antigen (PCNA) antibody was purchased from DAKO (Carpinteria) and BM8 monoclonal F4/80 rat anti-mouse IgG2a antibody was obtained from Caltag, (Invitrogen, Carlsbad, CA). Other chemicals used were purchased from Sigma-Aldrich Co. (St. Louis, MO) unless otherwise specified.
2.2. Animals
Male SKH-1 hairless mice (4–5 weeks of age) were purchased from Charles River Laboratories (Wilmington, MA) and housed under standard condition at the University of Colorado centre of Laboratory Animal Care. The animals were acclimatized for one week before their use in our experimental studies following the specified protocol approved by the IACUC of the University of Colorado Denver, CO.
2.3. CEES exposure
CEES was dissolved in acetone and applied topically to the dorsal skin of the male SKH-1 hairless mice according to the protocol published earlier by Tewari-Singh et al., (2009). Acetone, a well established vehicle for skin topical treatments, was used as a vehicle for CEES application due to its ability to enhance the skin permeability to hydrophilic and amphipathic compounds (Tewari-Singh et al., 2009; Tsai et al., 2001). To assess CEES-induced toxic effects on the skin of male SKH-1 hairless mice, 200 μl acetone alone or 1 or 2 mg CEES/mouse in the same acetone volume was applied for 9, 12, 24 and 48 h; an untreated mouse group was included as a further control. The selection of CEES doses and time of exposure were based on our earlier studies in female SKH-1 hairless mice (Tewari-Singh et al., 2009). To study the toxic effect of a higher dose of CEES for its ability to induce blistering, the above detailed treatment protocol was repeated in male mice but with a 4 mg CEES dose/mouse. At the end of each experiment, mice were euthanized and dorsal skin was collected as described earlier (Saladi et al., 2006; Tewari-Singh et al., 2009). Importantly, enough tissue slices were harvested from each animal to allow processing in all of the following manners: 1) snap freezing using liquid nitrogen, 2) fixation in 10% phosphate-buffered formalin, and 3) fixation in Bouin’s solution.
2.4. Measurement of skin bi-fold thickness and wet/dry weight ratio
Following topical application of acetone or 2 mg CEES in acetone, skin bi-fold thickness was measured (in mm) at 9, 12, 24 and 48 h using an electronic digital caliper (Marathon, Inc. Belleville, ON, Canada). For wet/dry weight ratio measurements, 10 mm skin tissue biopsies were collected, weighed before and after drying in an oven at 37°C for 24 h, and the wet/dry weight ratios were calculated.
2.5. Histopathological evaluation of skin sections and measurement of epidermal thickness
The fixed skin tissue samples were dehydrated in ascending concentrations of ethanol, cleared with xylene and embedded in paraffin (Triangle Biomedical Sciences, Durham, NC). Five-micron sections were cut and after haematoxylin and eosin (H&E) staining, tissue was microscopically evaluated for epidermal thickness, epidermal-dermal separation, apoptotic cell death, epidermal necrosis, and infiltration of inflammatory cells. The epidermal thickness (μm) was measured randomly in at least five fields per tissue sample from two sets of H&E stained slides using Axiovision Rel 4.5 software (Carl Zeiss, Inc. Germany; ×400 magnification).
2.6. Apoptotic cell detection via TUNEL staining
Apoptotic cells were detected using the DeadEnd Colorimetric TUNEL system according to the manufacturer’s protocol with some modifications as described earlier (Tewari-Singh et al., 2009). The brown colored TUNEL positive cells were quantified in 10 randomly selected fields at 400× magnification, and an apoptotic cell index was calculated as the number of apoptotic cells ×100 divided by total number of cells.
2.7. Immunohistochemical staining for the detection of proliferating cells and macrophages
Paraffin embedded sections were deparaffinized, rehydrated and treated for antigen retrieval using 10 mM sodium citrate (pH 6.0) for 30 min at 70°C. The endogenous peroxide activity was blocked using 3% hydrogen peroxide in methanol (v/v) and the sections were incubated with either mouse monoclonal PCNA antibody or with BM8 monoclonal F4/80 rat anti-mouse IgG2a antibody for the detection of macrophages in PBS for 2 h at 37°C in a humidity chamber. The N-Universal negative control rabbit IgG antibody (DAKO) was used as a negative control. After washings in PBS, the sections were incubated with the appropriate biotinylated secondary antibody for 1 h followed by incubation with HRP conjugated streptavidin (DAKO) in PBS for 30 min at RT in a humidity chamber. The sections were then incubated in DAB working solution for 10 min at RT and counterstained with diluted hematoxylin for 2 min followed by dehydration and mounting for microscopic observation. The DAB positive nuclei were counted in 10 randomly selected fields (×400 magnification), and the proliferation index was determined as number of PCNA positive cells ×100/total number of cells. The macrophages were counted per cm2 field in five randomly selected fields per section (×400 magnification).
2.8. Toluidine blue staining for mast cells
The toluidine blue staining for mast cells in the skin sections was carried out as reported earlier (Tewari-Singh et al., 2009). Briefly, the sections were treated with toluidine blue (Sigma-Aldrich Chemical Co. St. Louis., MO) working solution (5 ml toluidine blue + 45 ml of 1% sodium chloride, pH 2.0–2.5) for 2–3 min, washed in distilled water, dehydrated, cleared in xylene and mounted for microscopic observation. The mast cells were quantified by counting their number per cm2 field in five randomly selected fields (×400 magnification).
2.9. Measurement of myeloperoxidase (MPO) activity
The MPO activity was measured in skin tissue samples by using a fluorescent MPO detection kit from Cell Technology (Mountain View, CA) as detailed in our previous report (Tewari-Singh et al., 2009). In brief, about 100 mg skin tissue samples without the red blood cells were homogenized in 1X homogenization buffer with N-ethylmaleimide using a polytron PT 10–35 (Kinematica, AG, Bohemia, NY). After homogenization, 1 ml of solubilization buffer was added to the pellet and samples were further homogenized and sonicated for 30 sec. The protein was isolated via freeze thaw cycles and estimated by Lowry’s method (Bio-Rad DC protein assay kit, Bio-Rad laboratories, Hercules, CA). For MPO assay, 50 μl reaction mixture (50 μM of detection reagent, 20 mM hydrogen peroxide, 1X buffer, 0.1 μM eosinophil peroxidase inhibitor and 20 mM catalase inhibitor (3-amino-1,2,4-trizole)) and 50 μl of prepared sample or serially diluted MPO (for the standard curve) were added in a 96-well opaque black plate for reaction. The plate was incubated in the dark for 60 min at room temperature and the fluorescence was measured using a fluorescence reader at 530 nm excitation and 590 nm emission. The blank reading was subtracted from the entire sample reading and the MPO activity was expressed as a mU/μg protein using the MPO standard curve.
2.10. Immunohistochemical and statistical analyses
All the immunohistochemical and H&E stained slides were observed under Zeiss Axioscop 2 microscope (Carl Zeiss, Inc., Germany), and image analyses were done by using Carl Zeiss Axiovision Rel 4.5 software. The data were analyzed and all statistical calculations were done using SigmaStat software version 2.03 (Jandal Scientific Corp., San Raphael, CA). Data are expressed as mean ± SEM and were analyzed via one way ANOVA followed by the Bonferroni t-test for multiple comparisons. P< 0.05 was considered statistically significant.
3. Results
3.1. CEES topical exposure causes inflammation and microvesication in male SKH-1 hairless mouse skin
Recently, we reported that topical application of up to 2 mg CEES, causes strong skin toxicity, including edema and inflammation but not vesication in female SKH-1 hairless mice. Accordingly, to assess whether CEES could induce microvesication, male SKH-1 hairless mice were exposed to a higher dose of 4 mg CEES, and skin samples were temporally examined for inflammation-related histopathological alterations and microvesication. This higher dose of CEES caused a substantial increase in skin bi-fold thickness that peaked between 9 (2.0 mm) and 12 (2.04 mm) h post-CEES exposure and declined thereafter, compared to 1.06 and 1.16 mm for untreated and vehicle control groups, respectively (Fig. 1A). Similarly, CEES (4 mg) also caused a significant (p<0.01) increase in the skin wet/dry weight ratio at 9 h (5.50 mm) post-CEES exposure (Fig. 1B).
Fig. 1.
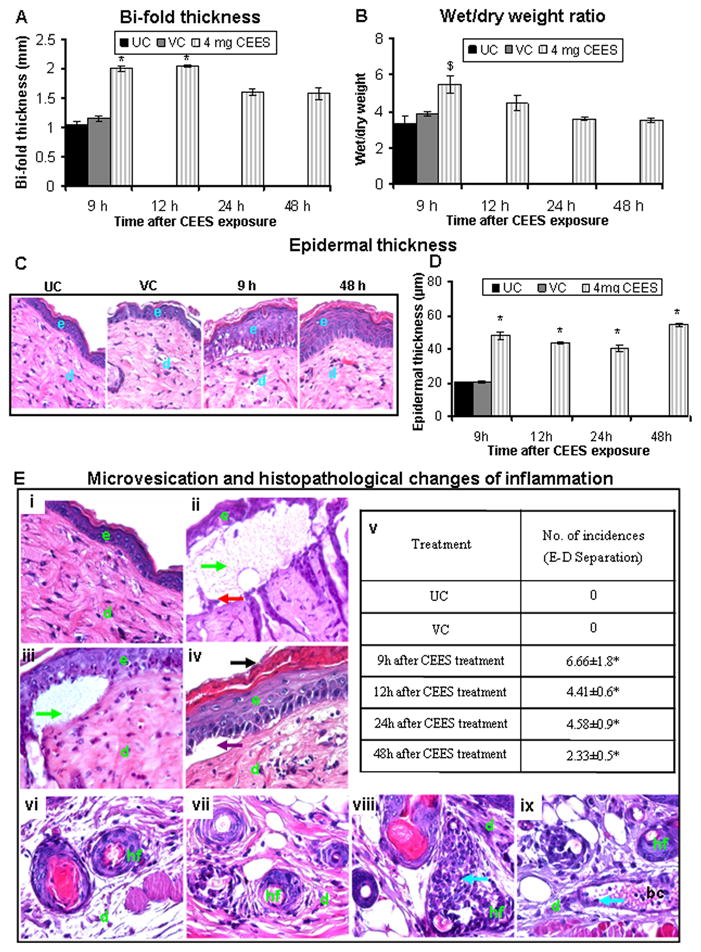
CEES topical exposure at a 4 mg dose causes inflammation-related histopathological alterations and microvesication in male SKH-1 hairless mouse skin. Following 4 mg CEES and control exposures, skin bi-fold thickness (A) and wet/dry weight ratio (B) were recorded as detailed under Materials and Methods. Representative H&E stained skin tissue sections from untreated control, vehicle control, and 4 mg CEES exposed mice at 9 to 48 h post exposure showing changes in epidermal thickness (×400 magnification; C) and quantitated epidermal thickness (D). Representative H&E stained skin sections from untreated (i and vi) and vehicle (vii) controls, 4 mg CEES treatment for 9 h (ii and viii), 12 h (iii and ix), and number of epidermal-dermal separations incidences (v) per 6 cm2 field, ×400 magnification (E); (iv) shows the artifact of microvesication because it is not filled with protein exudate and not counted as true microvesication. Data presented are mean ± SEM of four-five animals in each treatment group. Statistical significance of difference between CEES and control groups were determined by one way ANOVA followed by Bonferroni t-test for pair wise multiple comparisons. *, p<0.001; $, p<0.01 and #, p<0.05 as compared to untreated control. UC, untreated control; VC, vehicle control; e, epidermis; d, dermis; hf, hair follicle; bc, blood capillary; green arrows, epidermal-dermal separations (microvesication); black arrow, parakeratosis; violet arrow, artifact (not microvesication); red arrow, hemidesmosomal components; and blue arrows, neutrophils.
Microscopic examination of H&E stained skin tissue sections following the 4 mg dose of CEES also revealed an increased epidermal thickness (Fig. 1C). Compared to the untreated control (epidermal thickness of 20.04 μm), 4 mg CEES exposure significantly (p<0.001) increased epidermal thickness to 48.14, 43.68, 40.59 and 54.41 μm at 9, 12, 24 and 48 h, respectively (Fig. 1D). In addition to increasing the epidermal thickness, higher dose of 4 mg CEES also caused microvesication, which was observed histologically as an epidermal-dermal separation with attendant dermal edema and inflammation (Fig. 1E, ii and iii, green arrows); violet arrow (Fig. 1E, iv) shows the artifact of microvesication because it is not filled with protein exudate and is not counted as true microvesication. This effect was most severe at 9 h post-CEES exposure (Fig. 1E, ii, green arrows). The observed microvesication was quantified as the number of incidences per 6 cm2 field (×400 magnification). Compared to the untreated and vehicle controls which showed no incidences of microvesication, we observed 6.66, 4.41, 4.58 and 2.33 average incidences of this event at 9, 12, 24 and 48 h post-CEES exposure of skin, respectively (Fig. 1E, v). Detachment of hemidesmosomes at the epidermal-dermal junction is known to play an important role in microvesication (Shakarjian et al., 2010). Consistent with this, hemidesmosomal components were also observed in the CEES-induced microblisters (Fig. 1E, ii, red arrow). Bouin’s fixative is known to reduce tissue stretching to allow better confirmatory observation of any changes in epidermis and microblister formation. Accordingly, in addition to formalin fixation, skin samples from control and 4 mg CEES exposed mice were also fixed in Bouin’s fixative and then processed similarly. This approach helped in identifying the induction of parakeratosis (retention of nuclei in the stratum corneum layer), also present in many skin diseases including psoriasis and exfoliative dermatitis (Brady, 2004). This process was more severe at 24 and 48 h post-CEES exposure (Fig. 1E, iv, black arrow). Other observed change with CEES (4 mg) exposure was an increase in the inflammatory cells such as mast cells and neutrophils in the dermis of the skin tissue (Fig. 1E, viii and ix, blue arrows).
Most of our previous studies have employed female SKH-1 hairless mice, however, the major portion of the human population expected to be exposed to HD is male including battlefield casualties or chemical terrorism-associated first responders. This rationale related to our second question addressed in this study, namely whether there are any sex-related variations in CEES-induced inflammatory responses in mouse skin. To address this issue, male SKH-1 hairless mice were exposed to similar treatments and assessed for similar inflammatory responses to what was observed in female mice. In this study, compared to untreated controls, CEES exposure caused a significant (p<0.001–p<0.05) increase in skin bi-fold thickness that peaked at 24 h (Fig. 2A). Next, we assessed skin tissue wet/dry weight ratio following CEES exposure and observed a significant (p<0.001) increase at 24 h (4.68) in CEES-exposed compared to untreated (2.75 ) and vehicle (3.32) control groups (Fig. 2B).
Fig. 2.
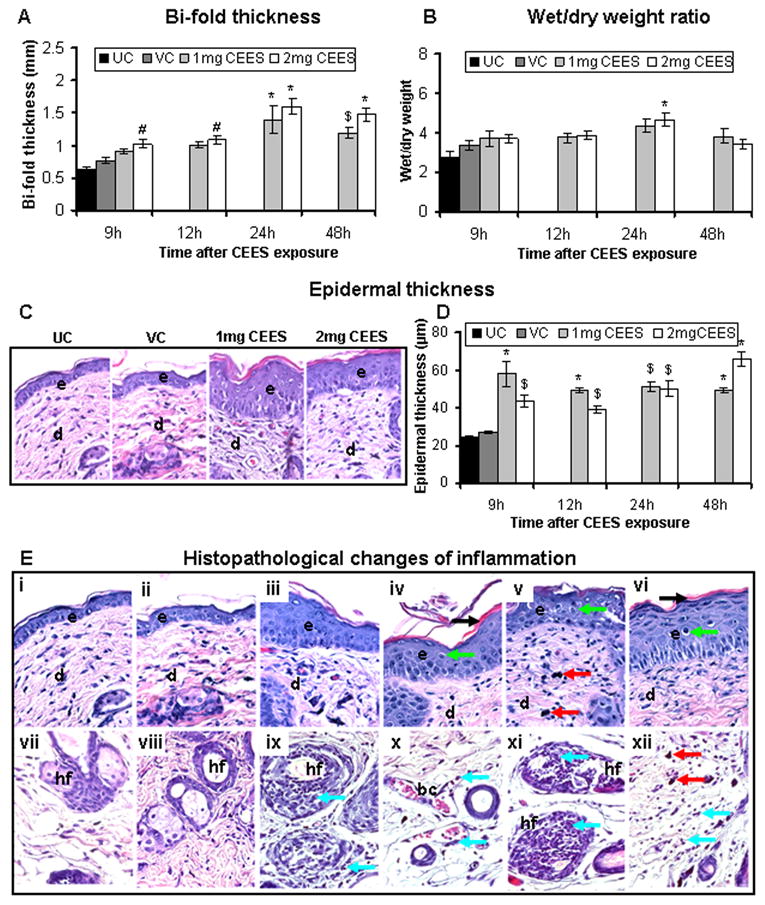
CEES topical exposure at 1 or 2 mg dose causes inflammation-related histopathological alterations in male SKH-1 hairless mouse skin. Following 1 or 2 mg CEES and control exposures, skin bi-fold thickness (A) and wet/dry weight ratio (B) were recorded as detailed in Materials and Methods. Representative H&E stained skin tissue sections from untreated control, vehicle control, and 1 or 2 mg CEES exposed mice at 9 h post exposure, showing changes in epidermal thickness (×400 magnification; C) and its measurement (D) Representative H&E stained skin sections from untreated (i and vii) and vehicle (ii and viii) controls, 2 mg CEES for 9 (iii, ix and x), 12 (iv, xi and xii), 24 (v) and 48 (vi) h, ×400 magnification (E). Data presented are mean ± SEM of five animals in each treatment group. Statistical significance of difference between the CEES exposed and untreated control group was determined by one way ANOVA followed by Bonferroni t-test for pair wise multiple comparisons. *, p<0.001; $, p<0.01 and #, p<0.05 as compared to untreated control group. UC, untreated control; VC, vehicle control; e, epidermis; d, dermis; hf, hair follicle; bc, blood capillary; black arrow, epidermal necrosis; red arrows, mast cells; blue arrows, neutrophils; green arrows, langerhans cells.
Since an increase in skin bi-fold thickness following CEES exposure was associated with inflammation in our previous study in female SKH-1 hairless mice, we next examined CEES-caused histopathological changes in male SKH-1 hairless mouse skin, which could possibly be associated with the observed increase in skin bi-fold thickness and edema. Quantitative analysis of H&E stained sections showed a significant (p<0.001–p<0.01) increase in epidermal thickness following CEES exposure compared to untreated and vehicle controls (Fig. 2C and D). In addition, CEES caused an increase in upper epidermal cell necrosis and desquamation (Fig. 2E, iv, black arrow). There also appeared to be an increase in Langerhans cells (Fig. 2E, iv to vi, green arrows). In the dermal region, increased numbers of mast cells (Fig. 2E, v and xii, red arrows) and neutrophils (Fig. 2E, ix to xii, blue arrows) were observed after CEES exposure. These inflammation-related histopathological alterations in male SKH-1 hairless mouse skin following CEES exposure were further analyzed and quantified; in particular, mast cells (toluidine blue staining), macrophages (F4/80 immunohistochemical staining), neutrophil infiltration (MPO activity), apoptotic cell death (TUNEL), and cell proliferation (PCNA) all were quantitated.
3.2. CEES topical exposure causes increase in the mast cells, macrophages and MPO activity in male SKH-1 hairless mouse skin
Inflammatory cells are reported to play an important role in HD-caused skin toxicity and vesication (Cowan and Broomfield, 1993). Consistent with this idea, earlier, we too observed an increase in epidermal inflammatory cell types post CEES exposure of female SKH-1 mice (Tewari-Singh et al., 2009). In the present study, H&E staining of the CEES-treated skin sections showed an increase in mast cells as assessed by toluidine blue staining (Fig. 3A). Quantification of the toluidine blue-stained skin sections showed that all 1, 2, and 4 mg CEES exposures resulted in significant (p<0.001) increases in number of mast cells in the dermis as compared to respective controls (Fig. 3B & C). Similarly, immunohistochemical staining for F4/80 antigen demonstrated an increase in the total numbers of macrophages in CEES-exposed skin from male SKH-1 hairless mice (Fig. 3D). Quantification of the F4/80 staining clearly showed that 1, 2, and 4 mg CEES exposures caused a significant (p<0.001) increase in the number of macrophages in the dermis compared to controls (Fig. 3E & F). However, there was no significant difference found either between exposures with 1 or 2 mg of CEES doses or between untreated and vehicle control groups at any studies time point (Fig. 3E).
Fig. 3.
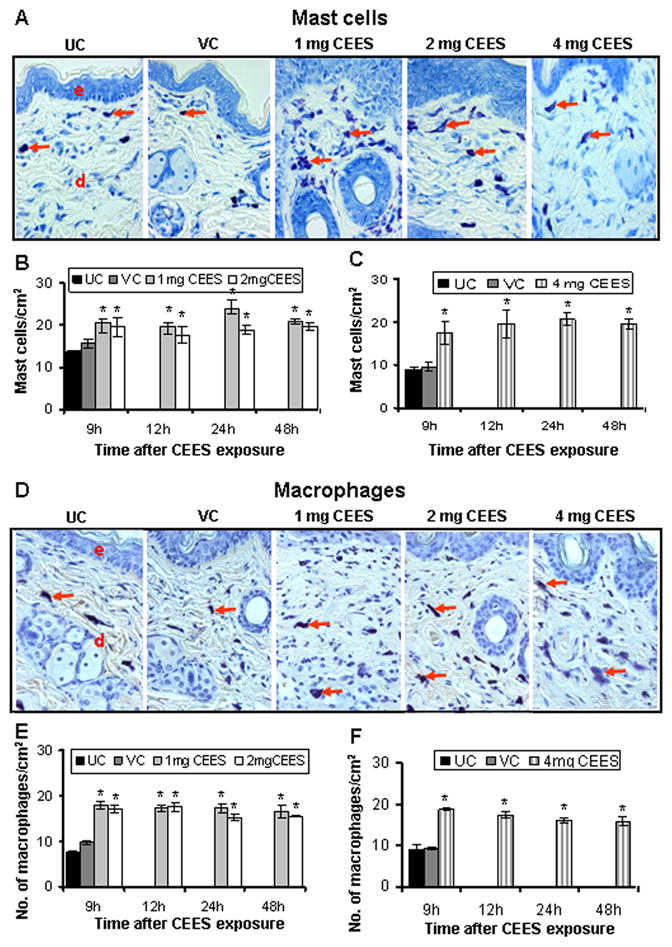
CEES topical exposure causes an increase in the number of mast cells and macrophages in male SKH-1 hairless mouse skin. Following 1, 2 or 4 mg CEES and control exposures, skin samples were collected as a function of time, and processed and stained as detailed under Materials and Methods. Representative toluidine blue stained mast cells (A) and F4/80 stained macrophages (D) in the skin sections from untreated and vehicle controls, and 1, 2 or 4 mg CEES exposed groups (×400 magnification). Mast cells/cm2 (B and C) and macrophages/cm2 (E and F) were calculated from various treatment groups by counting positive stained cells in randomly selected five fields per sample (×400 magnification) as detailed under Materials and Methods. Data presented are mean ± SEM of five animals of each group. Statistical significance of difference between the CEES exposed and control groups were determined by one way ANOVA followed by Bonferroni t-test for pair wise multiple comparisons. *, p<0.001; $, p<0.01 as compared to untreated control group. UC, untreated control; VC, vehicle control; e, epidermis; d, dermis; red arrows, toluidine blue or F4/80 positive cells.
Tissue MPO activity, an indicator of neutrophil infiltration, is another established biomarker of HD/CEES-induced skin toxicity in the hairless guinea pig and mouse models (Bradley et al., 1982; Tewari-Singh et al., 2009). Indeed, H&E stained skin sections of male SKH-1 hairless mice showed neutrophil infiltration following CEES exposure and skin injury (Fig. 2E). We measured MPO activity in order to biochemically quantitate this observed neutrophil infiltration. As shown in Fig. 4A, 2 mg CEES exposure caused a significant (p<0.01) increase in MPO activity at 9 h (3.14 mU/μg protein) and 12 h (2.77 mU/μg protein) compared with their controls (1.64 mU/μg protein). However, 1 mg CEES dose demonstrated a statistically significant (p<0.01) increase in MPO activity only after 12 h of exposure. Comparable observations were made following the exposure to 4 mg CEES dose (Fig. 4B); however the maximum increase in MPO activity was observed at 48 h (2.72 mU/μg protein) compared to untreated controls (1.06 mU/μg protein, Fig. 4B).
Fig. 4.
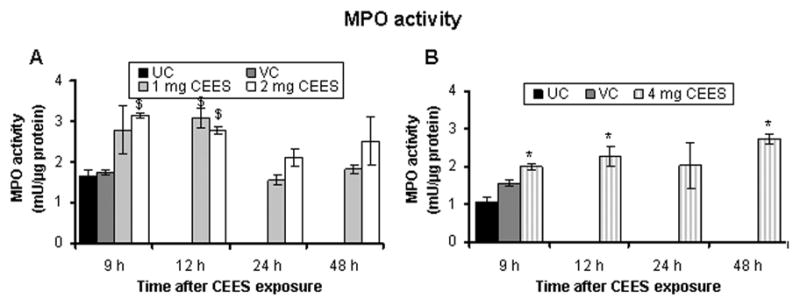
CEES topical exposure causes an increase in MPO activity, indicating neutrophil infiltration, in male SKH-1 hairless mouse skin. Following 1, 2 (A) or 4 (B) mg CEES and control exposures, skin samples were collected as a function of time, and MPO activity was determined using a fluorescence assay kit as described under Materials and Methods. Data presented are mean ± SEM of three-five animals in each treatment group and samples were taken in duplicate for the MPO assay. Statistical significance of difference between the CEES exposed and control groups were determined by one way ANOVA followed by Bonferroni t-test for pair wise multiple comparisons. *, p<0.001; $, p<0.01 as compared to untreated control group. UC, untreated control; VC, vehicle control.
3.3. CEES topical exposure causes increase in the apoptotic and proliferating cells in male SKH-1 hairless mouse skin
Based on our observation of pyknotic keratinocytes in H&E stained skin tissue sections from CEES exposed male SKH-1 hairless mice, we estimated an apoptotic index using TUNEL staining. As seen in the representative TUNEL-stained skin sections from various treatment groups, a substantial increase in the number of TUNEL- positive cells compared to controls was observed in CEES-exposed mice (Fig. 5A). The percent of TUNEL- positive cells was significantly (p<0.001) increased following 1, 2 or 4 mg CEES exposure for 9–48 h (Fig. 5B & C). Our previous study also reported PCNA as a biomarker of CEES- induced skin toxicity in female SKH-1 hairless mice (Tewari-Singh et al., 2009), therefore we also examined that marker in the present study. Quantification of PCNA-stained skin sections showed a substantial increase in PCNA positive cells the basal epidermal layer of the CEES-exposed skin tissues (Fig. 5D), and compared to control tissues, CEES exposure caused a significant (p<0.001 – 0.01) increase in cell proliferation at all doses (Fig. 5E & F).
Fig. 5.
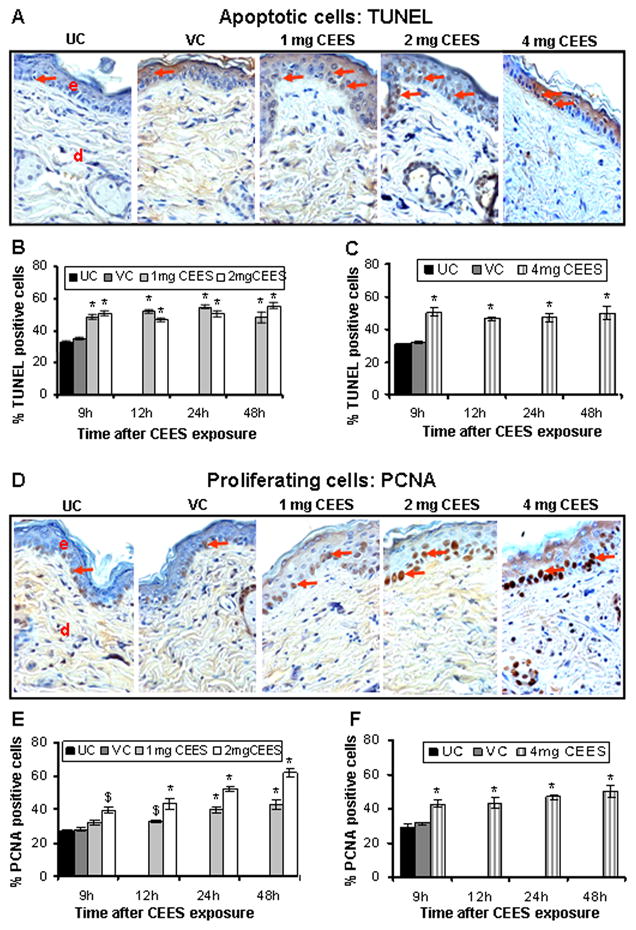
CEES topical exposure causes an increase in both apoptotic and proliferating cells in male SKH-1 hairless mouse skin. Following 1, 2 or 4 mg CEES and control exposures, skin samples were collected as a function of time, and processed and stained for apoptotic cell death (TUNEL) and cell proliferation (PCNA) as detailed in Materials and Methods. Representative TUNEL (A) and PCNA (D) stained skin sections from untreated and vehicle controls, and 1, 2 or 4 mg CEES exposed groups (×400 magnification). Percent TUNEL positive cells (B and C) and percent PCNA positive cells (E and F) were calculated from various treatment groups by counting positive stained cells in randomly selected five fields per sample (×400 magnification) as detailed in Materials and Methods. Data presented are mean ± SEM of five animals of each group. Statistical significance of difference between the CEES exposed and control groups were determined by one way ANOVA followed by Bonferroni t-test for pair wise multiple comparisons. *, p<0.001; $, p<0.01 as compared to untreated control group. UC, untreated control; VC, vehicle control; e, epidermis; d, dermis; red arrows, TUNEL or PCNA positive cells.
4. Discussion
HD, a potent vesicating chemical warfare agent, poses an imminent threat both in military conflicts and against civilians (Noort et al., 2002; Saladi et al., 2006). Earlier studies have demonstrated that high doses of HD induce severe skin lesions that include erythema, edema, inflammation and formation of blisters (Balali-Mood and Hefazi, 2005; Greenberg et al., 2006; Shakarjian et al., 2006; Wormser et al., 2005). The results of the present study using a male SKH-1 hairless mouse model demonstrate similar findings to those with HD but through use of the less toxic HD-analog, CEES. A dose of 2 mg CEES in this study demonstrates inflammation-related histopathological changes in male SKH-1 hairless mice, which are comparable to our earlier reported study in female mice of this strain (Tewari-Singh et al., 2009). In our previous studies of CEES-induced mouse skin alterations (Tewari-Singh et al., 2009), we employed female mice because most previous skin toxicity studies, including chemical carcinogen-caused skin tumorigenesis and/or tumor promotion events have employed female rather than male mice (Gu et al., 2005a; Gu et al., 2007). However, due to the higher probability of males being exposed to HD in the battlefield and as first responders during any attack, we compared our 2 mg CEES studies in female mice with the male mice and conducted further studies in male mouse skin injury model. In this report, exposure of male mice to a higher dose of 4 mg CEES induced microvesication (epidermal-dermal separation), an important consequence similar to the vesication inflicted following HD exposure, which was not achieved in our studies with 2 mg CEES in this rodent model. These toxic effects are induced in humans with a much lower exposure of 10–40 μg/cm2 of HD (Kehe et al., 2008), and CEES being less toxic requires a higher concentration for this effect. The concentrations of CEES used here that mimics the inflammation and microvesication effects of HD in this model, overcome the drawback of non-occurrence of vesication in animal models with CEES, and could be used appropriately for the efficacy studies to identify effective agents that can counter these toxic effects of HD in humans.
HD/CEES-induced skin toxicity is mainly characterized by inflammation in the dermis and apoptosis or necrosis of basal keratinocytes. These changes may be due to a direct DNA damaging effect, alkylation or an oxidative stress (Kehe and Szinicz, 2005; Paromov et al., 2007). The HD and CEES-induced cell death in the epidermis may also lead to an inflammatory response (Kan et al., 2003; Rosenthal et al., 2001). Following apoptosis, cell proliferation allows replacement of dead cells, and PCNA plays an important role in DNA replication and damage repair, serving as a biomarker of proliferation (Gu et al., 2005b; Tewari-Singh et al., 2009). Consistent with this and other reports (Graham et al., 2005; Kan et al., 2003; Kehe and Szinicz, 2005), we observed apoptotic death of epidermal keratinocytes and an increase in the PCNA -positive cells following mouse skin exposure to CEES. Indeed, these results suggest that CEES mediated DNA damage could play an important role in the apoptotic cell death of keratinocytes as observed in epidermal keratinocytes after CEES exposure (Tewari-Singh et al., 2010).
The CEES-induced inflammatory response including recruitment of mast cells, neutrophils, macrophages and their mediators is thought to be important for vesication (Cowan and Broomfield, 1993). HD exposure has been documented to result in the release of several inflammatory cytokines including TNFα and multiple interleukins (ILs), together with activation of the transcription factor NF-κB and increased expression of MMP-9 (Paromov et al., 2007; Wormser et al., 2005). Both TNFα and ILs have strong chemotactic activity for neutrophils and macrophages (Arroyo et al., 2001; Sabourin et al., 2002; Sabourin et al., 2000). The CEES-induced increase in the numbers of mast cells observed in our present study may be involved in the production and delivery of inflammatory mediators such as histamine, proteases, chemotactic factors and cytokines (Theoharides and Cochrane, 2004). Additionally, the observed increase in mast cells herein may also be associated with neutrophil infiltration and the increased number of macrophages in the male SKH-1 mouse skin. Importantly, our results are similar to the documented HD-induced inflammatory cell response reported using the mouse ear vesicant model (MEVM) model (Wormser et al., 2005).
Vesication is an important sequel of human HD exposure; however, most animal models do not mimic this effect and only show microvesication in response to HD (Black et al., 2010; Casillas et al., 2000; Hayden et al., 2009). In the present study, CEES induced microvesication in SKH-1 mouse model. Other mouse models such as the liquid HD MEVM and the HD vapor hairless mouse vesicant model have been widely used to access the HD-induced skin toxicity since they show microblister (microvesication) formation with dermal edema (Casillas et al., 2000; Ricketts et al., 2000). For instance, in the MEVM model, microblistering occurs without a marked inflammatory infiltrate; this is helpful to study the relative role of proteases in microblister formation (Casillas et al., 2000; Powers et al., 2000). The hairless guinea pig also has been widely used to study HD-induced skin toxicity since it possesses a multilayer epidermis similar to human skin. In the guinea pig model, microblisters, necrosis in epidermis, degenerative changes in the stroma with vascular necrosis, and hemorrhage have all been reported following HD exposure (Smith et al., 1997; Smith et al., 1995). The weanling pig is another model used for wound healing studies because of its similarity with human skin. This model shows microblister formation and necrosis following exposure to HD at higher doses (King and Monteiro-Riviere, 1990; Reid et al., 2000). From an experimental standpoint it is important to mention that most of the skin vesicant and toxicity models developed utilizing exposure to HD require special containment facilities and involve multiple procedural limitations. In contrast, the SKH-1 hairless mouse vesicant model reported here utilizing CEES can be used in a less restrictive laboratory setting.
It has been reported that vesication occurs via apoptosis in basal keratinocytes and degradation of anchoring filaments/hemidesmosomes after HD exposure (Greenberg et al., 2006; Kan et al., 2003; Mol et al., 2009). Furthermore, detachment of hemidesmosomes at the epidermal-dermal junction plays an important role in HD-induced microvesication (Shakarjian et al., 2010). Consistent with these studies, our CEES-treated animals also showed microvesication with detachment of hemidesmosomes at the epidermal-dermal junction and damage to the keratinocytes; both possibly leading to microvesication. More studies are needed to further elucidate the involvement of hemidesmosomes, type VIII collagen and MMPs that have been reported to play a role in HD-induced vesication (Hayden et al., 2009; Mol et al., 2009; Shakarjian et al., 2006). Future studies are also needed to define the mechanisms involved in the observed CEES-induced microvesication and to identify whether these are the same as reported following HD treatment.
Lastly, in our hairless mouse skin injury model with 4 mg CEES, we observed induction of parakeratosis (retention of nuclei in the stratum corneum layer). Parakeratosis is present in many skin diseases including psoriasis and exfoliative dermatitis and may be due to a number of inciting agents (Brady, 2004). Many of the reported causes of parakeratosis involve large increases in the number of dermal neutrophils. We too observed a large neutrophil influx into and around the hair follicles following CEES application. The significance of this finding will require further investigation, which could facilitate our mechanistic studies of CEES-induced cell death and vesication in future.
In conclusion, the new information in this study and associated significance are that for the first time we found that skin exposure of male SKH-1 hairless mice to HD analog CEES causes epidermal-dermal separation indicating microvesication, which is a key component of HD skin injury in humans. Notably, HD poses major threat in both military conflict and chemical terrorism situations and currently we lack effective therapies to rescue skin injuries by HD in part due to the lack of appropriate models. Accordingly the major significance of the findings, namely CEES-induced microvesication and inflammation-related histopathological changes in mouse skin, is that they provide potentially useful relevant laboratory model for developing effective countermeasures against HD skin injury in humans.
Acknowledgments
Funding
The research is supported by the CounterACT Program, National Institute of Health (NIH) Office of the Director, and National Institute of Environmental Health Sciences (NIEHS), Grant Number (U54 ES015678). The study sponsor (NIH) had no involvement in the study design; collection, analysis and interpretation of data; the writing of the manuscript; and the decision to submit the manuscript for publication.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Anil K. Jain, Email: Anil.Jain@ucdenver.edu.
Neera Tewari-Singh, Email: Neera.Tewari-Singh@ucdenver.edu.
David J. Orlicky, Email: David.Orlicky@ucdenver.edu.
Carl W White, Email: whitec@njc.org.
References
- Arroyo CM, Broomfield CA, Hackley BE., Jr The role of interleukin-6 (IL-6) in human sulfur mustard (HD) toxicology. Int J Toxicol. 2001;20:281–296. doi: 10.1080/109158101753253027. [DOI] [PubMed] [Google Scholar]
- Balali-Mood M, Hefazi M. The pharmacology, toxicology, and medical treatment of sulphur mustard poisoning. Fundam Clin Pharmacol. 2005;19:297–315. doi: 10.1111/j.1472-8206.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- Black AT, Joseph LB, Casillas RP, Heck DE, Gerecke DR, Sinko PJ, Laskin DL, Laskin JD. Role of MAP kinases in regulating expression of antioxidants and inflammatory mediators in mouse keratinocytes following exposure to the half mustard, 2-chloroethyl ethyl sulfide. Toxicol Appl Pharmacol. 2010;245:352–360. doi: 10.1016/j.taap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha M, Bowers W, Jr, Kohl J, DuBose D, Walker J, Alkhyyat A, Wong G. Effects of CEES on inflammatory mediators, heat shock protein 70A, histology and ultrastructure in two skin models. J Appl Toxicol. 2000;20(Suppl 1):S101–108. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat672>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Brady SP. Parakeratosis. J Am Acad Dermatol. 2004;50:77–84. doi: 10.1016/s0190-9622(03)02801-9. [DOI] [PubMed] [Google Scholar]
- Brodsky B, Trivedi S, Peddada S, Flagler N, Wormser U, Nyska A. Early effects of iodine on DNA synthesis in sulfur mustard-induced skin lesions. Arch Toxicol. 2006;80:212–216. doi: 10.1007/s00204-005-0032-6. [DOI] [PubMed] [Google Scholar]
- Casillas RP, Kiser RC, Truxall JA, Singer AW, Shumaker SM, Niemuth NA, Ricketts KM, Mitcheltree LW, Castrejon LR, Blank JA. Therapeutic approaches to dermatotoxicity by sulfur mustard. I. Modulaton of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. 2000;20(Suppl 1):S145–151. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat665>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Chatterjee D, Mukherjee S, Smith MG, Das SK. Evidence of hair loss after subacute exposure to 2-chloroethyl ethyl sulfide, a mustard analog, and beneficial effects of N-acetyl cysteine. J Biochem Mol Toxicol. 2004;18:150–153. doi: 10.1002/jbt.20020. [DOI] [PubMed] [Google Scholar]
- Cowan FM, Broomfield CA. Putative roles of inflammation in the dermatopathology of sulfur mustard. Cell Biol Toxicol. 1993;9:201–213. doi: 10.1007/BF00755599. [DOI] [PubMed] [Google Scholar]
- Dacre JC, Goldman M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol Rev. 1996;48:289–326. [PubMed] [Google Scholar]
- Graham JS, Chilcott RP, Rice P, Milner SM, Hurst CG, Maliner BI. Wound healing of cutaneous sulfur mustard injuries: strategies for the development of improved therapies. J Burns Wounds. 2005;4:e1. [PMC free article] [PubMed] [Google Scholar]
- Greenberg S, Kamath P, Petrali J, Hamilton T, Garfield J, Garlick JA. Characterization of the initial response of engineered human skin to sulfur mustard. Toxicol Sci. 2006;90:549–557. doi: 10.1093/toxsci/kfi306. [DOI] [PubMed] [Google Scholar]
- Gu M, Dhanalakshmi S, Mohan S, Singh RP, Agarwal R. Silibinin inhibits ultraviolet B radiation-induced mitogenic and survival signaling, and associated biological responses in SKH-1 mouse skin. Carcinogenesis. 2005a;26:1404–1413. doi: 10.1093/carcin/bgi096. [DOI] [PubMed] [Google Scholar]
- Gu M, Dhanalakshmi S, Singh RP, Agarwal R. Dietary feeding of silibinin prevents early biomarkers of UVB radiation-induced carcinogenesis in SKH-1 hairless mouse epidermis. Cancer Epidemiol Biomarkers Prev. 2005b;14:1344–1349. doi: 10.1158/1055-9965.EPI-04-0664. [DOI] [PubMed] [Google Scholar]
- Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67:3483–3491. doi: 10.1158/0008-5472.CAN-06-3955. [DOI] [PubMed] [Google Scholar]
- Han S, Espinoza LA, Liao H, Boulares AH, Smulson ME. Protection by antioxidants against toxicity and apoptosis induced by the sulphur mustard analog 2-chloroethylethyl sulphide (CEES) in Jurkat T cells and normal human lymphocytes. Br J Pharmacol. 2004;141:795–802. doi: 10.1038/sj.bjp.0705591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden PJ, Petrali JP, Stolper G, Hamilton TA, Jackson GR, Jr, Wertz PW, Ito S, Smith WJ, Klausner M. Microvesicating effects of sulfur mustard on an in vitro human skin model. Toxicol. 2009 doi: 10.1016/j.tiv.2009.07.021. In Vitro. [DOI] [PubMed] [Google Scholar]
- Kan RK, Pleva CM, Hamilton TA, Anderson DR, Petrali JP. Sulfur mustard-induced apoptosis in hairless guinea pig skin. Toxicol Pathol. 2003;31:185–190. doi: 10.1080/01926230390183661. [DOI] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Emmler J, Kreppel H, Jochum M, Thiermann H. Sulfur mustard research-strategies for the development of improved medical therapy. Eplasty. 2008;8:e32. [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Szinicz L. Medical aspects of sulphur mustard poisoning. Toxicology. 2005;214:198–209. doi: 10.1016/j.tox.2005.06.014. [DOI] [PubMed] [Google Scholar]
- King JR, Monteiro-Riviere NA. Cutaneous toxicity of 2-chloroethyl methyl sulfide in isolated perfused porcine skin. Toxicol Appl Pharmacol. 1990;104:167–179. doi: 10.1016/0041-008x(90)90292-3. [DOI] [PubMed] [Google Scholar]
- Mol MA, van den Berg RM, Benschop HP. Involvement of caspases and transmembrane metalloproteases in sulphur mustard-induced microvesication in adult human skin in organ culture: directions for therapy. Toxicology. 2009;258:39–46. doi: 10.1016/j.tox.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Noort D, Benschop HP, Black RM. Biomonitoring of exposure to chemical warfare agents: a review. Toxicol Appl Pharmacol. 2002;184:116–126. [PubMed] [Google Scholar]
- Pal A, Tewari-Singh N, Gu M, Agarwal C, Huang J, Day BJ, White CW, Agarwal R. Sulfur mustard analog induces oxidative stress and activates signaling cascades in the skin of SKH-1 hairless mice. Free Radic Biol Med. 2009;47:1640–1651. doi: 10.1016/j.freeradbiomed.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paromov V, Suntres Z, Smith M, Stone WL. Sulfur mustard toxicity following dermal exposure: role of oxidative stress, and antioxidant therapy. J Burns Wounds. 2007;7:e7. [PMC free article] [PubMed] [Google Scholar]
- Powers JC, Kam CM, Ricketts KM, Casillas RP. Cutaneous protease activity in the mouse ear vesicant model. J Appl Toxicol. 2000;20(Suppl 1):S177–182. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat678>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Reid FM, Graham J, Niemuth NA, Singer AW, Janny SJ, Johnson JB. Sulfur mustard-induced skin burns in weanling swine evaluated clinically and histopathologically. J Appl Toxicol. 2000;20(Suppl 1):S153–160. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat673>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Ricketts KM, Santai CT, France JA, Graziosi AM, Doyel TD, Gazaway MY, Casillas RP. Inflammatory cytokine response in sulfur mustard-exposed mouse skin. J Appl Toxicol. 2000;20(Suppl 1):S73–76. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat685>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Rosenthal DS, Simbulan-Rosenthal CM, Liu WF, Velena A, Anderson D, Benton B, Wang ZQ, Smith W, Ray R, Smulson ME. PARP determines the mode of cell death in skin fibroblasts, but not keratinocytes, exposed to sulfur mustard. J Invest Dermatol. 2001;117:1566–1573. doi: 10.1046/j.0022-202x.2001.01578.x. [DOI] [PubMed] [Google Scholar]
- Sabourin CL, Danne MM, Buxton KL, Casillas RP, Schlager JJ. Cytokine, chemokine, and matrix metalloproteinase response after sulfur mustard injury to weanling pig skin. J Biochem Mol Toxicol. 2002;16:263–272. doi: 10.1002/jbt.10050. [DOI] [PubMed] [Google Scholar]
- Sabourin CL, Petrali JP, Casillas RP. Alterations in inflammatory cytokine gene expression in sulfur mustard-exposed mouse skin. J Biochem Mol Toxicol. 2000;14:291–302. doi: 10.1002/1099-0461(2000)14:6<291::AID-JBT1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Saladi RN, Smith E, Persaud AN. Mustard: a potential agent of chemical warfare and terrorism. Clin Exp Dermatol. 2006;31:1–5. doi: 10.1111/j.1365-2230.2005.01945.x. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Bhatt P, Gordon MK, Chang YC, Casbohm SL, Rudge TL, Kiser RC, Sabourin CL, Casillas RP, Ohman-Strickland P, Riley DJ, Gerecke DR. Preferential expression of matrix metalloproteinase-9 in mouse skin after sulfur mustard exposure. J Appl Toxicol. 2006;26:239–246. doi: 10.1002/jat.1134. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Casillas R, Graham J, Skelton HG, Stemler F, Hackley BE., Jr Histopathologic features seen with different animal models following cutaneous sulfur mustard exposure. J Dermatol Sci. 1997;14:126–135. doi: 10.1016/s0923-1811(96)00560-9. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Graham JS, Moeller RB, Okerberg CV, Skelton H, Hurst CG. Histopathologic features seen in sulfur mustard induced cutaneous lesions in hairless guinea pigs. J Cutan Pathol. 1995;22:260–268. doi: 10.1111/j.1600-0560.1995.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Smith WJ, Hamilton T, Skelton HG, Graham JS, Okerberg C, Moeller R, Hackley BE., Jr Histopathologic and immunohistochemical features in human skin after exposure to nitrogen and sulfur mustard. Am J Dermatopathol. 1998;20:22–28. doi: 10.1097/00000372-199802000-00005. [DOI] [PubMed] [Google Scholar]
- Smith WJ, Gross CL, Chan P, Meier HL. The use of human epidermal keratinocytes in culture as a model for studying the biochemical mechanisms of sulfur mustard toxicity. Cell Biol Toxicol. 1990;6:285–291. doi: 10.1007/BF02443803. [DOI] [PubMed] [Google Scholar]
- Tewari-Singh N, Gu M, Agarwal C, White CW, Agarwal R. Biological and molecular mechanisms of sulfur mustard analogue-induced toxicity in JB6 and HaCaT cells: possible role of ataxia telangiectasia-mutated/ataxia telangiectasia-Rad3-related cell cycle checkpoint pathway. Chem Res Toxicol. 2010;23:1034–1044. doi: 10.1021/tx100038b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari-Singh N, Rana S, Gu M, Pal A, Orlicky DJ, White CW, Agarwal R. Inflammatory biomarkers of sulfur mustard analog 2-chloroethyl ethyl sulfide-induced skin injury in SKH-1 hairless mice. Toxicol Sci. 2009;108:194–206. doi: 10.1093/toxsci/kfn261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoharides TC, Cochrane DE. Critical role of mast cells in inflammatory diseases and the effect of acute stress. J Neuroimmunol. 2004;146:1–12. doi: 10.1016/j.jneuroim.2003.10.041. [DOI] [PubMed] [Google Scholar]
- Tsai JC, Sheu HM, Hung PL, Cheng CL. Effect of barrier disruption by acetone treatment on the permeability of compounds with various lipophilicities: implications for the permeability of compromised skin. J Pharm Sci. 2001;90:1242–1254. doi: 10.1002/jps.1077. [DOI] [PubMed] [Google Scholar]
- Wormser U. Toxicology of mustard gas. Trends Pharmacol Sci. 1991;12:164–167. doi: 10.1016/0165-6147(91)90534-y. [DOI] [PubMed] [Google Scholar]
- Wormser U, Brodsky B, Proscura E, Foley JF, Jones T, Nyska A. Involvement of tumor necrosis factor-alpha in sulfur mustard-induced skin lesion; effect of topical iodine. Arch Toxicol. 2005;79:660–670. doi: 10.1007/s00204-005-0681-5. [DOI] [PubMed] [Google Scholar]