

Abstract
In the last two decades several significant changes have been proposed in the receptor theory that describes how ligands can interact with G protein-coupled receptors (GPCRs). Here we briefly summarize the evolution of receptor theory and detail recent prominent advances. These include: (i) the existence of spontaneously active GPCRs that are capable of signalling even though they are unoccupied by any ligand; (ii) the discovery of ligands that can inactivate these spontaneously active receptors; (iii) the notion that a ligand may simultaneously activate more than one GPCR signalling pathway; and (iv) the notion that certain ligands may be able to preferentially direct receptor signalling to a specific pathway. Because the data supporting these receptor theory ideas are derived primarily from studies using artificial expression systems, the physiological relevance of these new paradigms remains in question. As a potential example of how these new perspectives in receptor theory relate to drug actions and clinical outcomes, we discuss their relevance to the recent controversy regarding the chronic use of β2-adrenoceptor agonists in the treatment of asthma.
LINKED ARTICLES
This article is part of a themed issue on Respiratory Pharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-1
Keywords: asthma, β2-adrenoceptor, agonist, inverse agonist, constitutive activity, biased agonism, efficacy
Introduction
G protein-coupled receptors (GPCRs) are the largest family of mammalian cell-surface receptors, representing more than 1% of human genes. These receptors transduce a wide variety of extracellular signals into intracellular events allowing an organism to both sense the external environment and communicate inter- and intracellular messages. From a drug discovery perspective, GPCRs account for the largest number of therapeutic targets, and somewhere between one-half and two-thirds of all drugs on the market produce their effect through GPCRs.
The last quarter century has brought about the first meaningful changes to classical receptor theory since 1966. These changes include: (i) the incorporation of multiple receptor states for GPCRs, including the existence of spontaneously active unoccupied GPCRs; and ligands termed ‘inverse agonists’ capable of turning off the spontaneous activity; (ii) the notion of a single GPCR activating more than one G protein, and signalling via non-G protein pathways; and (iii) the concept that one ligand may be able to stimulate a specific intracellular pathway while another may cause the same receptor to preferentially activate a second pathway, a concept now known as ‘biased agonism’. Despite a great deal of evidence to support these observations based on in vitro studies in artificial expression systems, there is still a question as to their relevance clinically and significance in drug discovery. Here we briefly review the evolution of receptor theory and discuss whether some of the recent modifications to this theory explain the effects of drugs used in the management of asthma and possibly other airway diseases.
Understanding receptor agonism
A.J. Clarke began the formalization of receptor theory when he proposed the occupation-response theory of receptor activation, which assumed that the greater the number of receptors occupied by an agonist, the larger the resulting response (Clark, 1933; 1937;). It was later discovered that different agonists tested in a given system expressing the same number of receptors could produce different maximal responses (Ariens, 1954). Ariens proposed the term ‘intrinsic activity’ and suggested that the agonist producing the largest response (full agonist) be assigned an intrinsic activity value of one (1.0), and fractions of this value be assigned to agonists that stimulated comparatively lesser responses (partial agonists). Stephenson later coined the term ‘ligand efficacy’ to describe the activation function of ligands with affinity for a given receptor (Stephenson, 1956). Stephenson's contribution allowed a conceptual framework for defining agonists – ligands with affinity (the ability to bind to the receptor) and efficacy (the ability to activate the receptor); and antagonists, ligands with affinity but no efficacy for the receptor. This concept of efficacy was associated with Ariens' intrinsic activity in that ligands with the highest efficacy are generally regarded to be full agonists in most systems. Like Ariens' intrinsic activity, the efficacy function described by Stephenson was also subject to ‘system-dependence’ in that agonists would appear more ‘efficacious’ in a system with greater capacity (i.e. a greater density of receptors), than in systems with lower receptor numbers where fewer agonist-receptor complexes would be formed. Antagonist efficacy was thought to equal zero in all systems and thus, would not be affected by receptor density. Then, in an attempt to make agonist efficacy a completely ligand-dependent parameter, Furchgott proposed dividing Stephenson's efficacy term by the number of total receptors in the system (Furchgott, 1966). The resulting value was termed ‘intrinsic efficacy’ (activated response per receptor) and was thought to be a unique constant for any given agonist-receptor interaction. We now know that ligand efficacy, and thus the biological response, is determined by a ligand's effect on the structure and biophysical properties of the receptor.
This concept, that intrinsic efficacy is a constant for any given drug-receptor complex, remained essentially unaltered and unchallenged for the next quarter century. The concept worked in explaining most data describing obvious agonist responses. For example, changes in heart rate, or relaxation of airway smooth muscle to β-adrenoceptor agonists, when using native tissues or systems (i.e. not transfected or over-expressed systems). However, more recent findings that demonstrate the complexity of GPCR signalling require a revision of these hypotheses (see following discussion).
The model system – the prototypical GPCR
The β2-adrenoceptor (β2AR) is one of the most studied proteins in biology and considered by many the prototypical GPCR. In its canonical signalling pathway, agonist binding couples the β2AR to the Gs subtype of G proteins. Gs activation leads to stimulation of adenylyl cyclase, production of cyclic adenosine monophosphate (cAMP) and activation of the cAMP-dependent protein kinase [protein kinase A (PKA)], which mediates most of the functional consequences of Gs-coupled receptor activation. In airway smooth muscle, β2AR-stimulated PKA activity mediates relaxation through phosphorylation of multiple proteins involved in regulating intracellular calcium levels, calcium sensitivity, and cross-bridge cycling. Other GPCRs follow a similar receptor-G protein-effector transmembrane signalling paradigm, often resulting in the activation of an intracellular kinase and signalling pathway (Penn, 2008).
Among the many early findings of studies of βAR activation was the observation that both the signalling and functional effects of receptor activation waned with time. Chronic exposure to βAR-agonist resulted in loss of effect on functional (‘tachyphylaxis’) or signalling (‘desensitization’) events mediated by the receptor. Similar tachyphylaxis/desensitization of other GPCRs was also observed (Bristow et al., 1982; Terwilliger et al., 1994; Iaccarino et al., 1998; Bohn et al., 2000). The mechanistic basis of this effect was unclear, although loss of cellular receptor density was shown to be associated with decreased catecholamine sensitivity in human heart failure (in which chronic elevation of circulating catecholamines occurs) (Bristow et al., 1982). Ultimately, the role of receptor phosphorylation by intracellular kinases was discovered to be critical to the desensitization of GPCRs (Pitcher et al., 1998). PKA was the first kinase shown capable of phosphorylating the mammalian β2AR (Benovic et al., 1985). Whether activated as a consequence of β2AR stimulation (homologous desensitization) or via some other means such as another Gs-coupled receptor (heterologous desensitization), PKA was shown to phosphorylate β2ARs causing a conformational change in the receptor and its consequent reduced coupling to G proteins. Additionally, the β2AR can also be phosphorylated by members of a kinase family known as G protein receptor kinases (GRKs). GRK-mediated phosphorylation of the β2AR, and numerous other GPCRs, serves to diminish receptor-G protein coupling and is specific for the agonist-occupied, or spontaneously active form of the receptor, whereas PKA and other second messenger kinases can phosphorylate receptors independent of their occupancy or activity status. Importantly, GRK-mediated phosphorylation of GPCRs also promotes the binding of β-arrestin (βarrestin) proteins to the receptor. βarrestin-1 and βarrestin-2 are expressed ubiquitously and constitute, with the two visual arrestins, the arrestin family of proteins (Yamaki et al., 1987; Lohse et al., 1990; Attramadal et al., 1992). βarrestin binds the intracellular tail of the receptor and sterically hinders coupling between the receptor and the G protein.
Arrestins act as scaffold proteins that link desensitized receptors to the endocytic machinery that internalizes the receptor (Laporte et al., 2000). Internalized receptors directed to recycling endosomes are dephosphorylated and returned to the plasma membrane where they are ready to signal again (resensitized). In cases of prolonged agonist exposure, receptors are more likely to traffic to lysosomes where they are degraded and thus not available for further signalling (down-regulation). More recently it has been shown that βarrestins bound to the endocytic machinery utilize additional docking sites to link various mitogen-activated protein kinases (MAPKs) to receptors. The receptor/βarrestin/MAPK scaffolds form internalized signalling complexes, or signalosomes, which can initiate a variety of cellular responses (Lefkowitz and Whalen, 2004; Shenoy and Lefkowitz, 2005). Thus, the βarrestin-dependent signalling pathway can be independent of the classical G protein-dependent signalling pathway and its existence has forced a major paradigm shift in classical GPCR theory. Consistent with its recognition as the prototypical GPCR, the β2AR was the first receptor for which βarrestin-dependent signal transduction was suggested (Luttrell et al., 1999). Several years later, with the aid of small interfering RNA and other technologies, the β2AR-mediated βarrestin-dependent signalling pathway was more fully described (Shenoy et al., 2006).
Receptor trafficking (recycling endosome, lysosome, signalosome) may explain the sometimes paradoxical effects observed when using β2AR drugs in the chronic management of certain diseases such as asthma and heart failure. As discussed in the following, GRKs, βarrestins and the nature of ligands are important determinants of receptor fate, and thus, physiological function.
Receptor theory modification #1: Can ligands ‘tell’ a receptor which way to go?
Although classic receptor theory could account for an agonist-receptor complex activating more than a single signalling pathway, it assumed that this capacity extended to all agonists of a given GPCR and was a function of agonist efficacy. With very little experimental data, Terry Kenakin proposed in 1995 that agonists might cause a receptor to activate a second pathway via an alternative mechanism (Kenakin, 1995a,b;). Kenakin termed his proposed potential dual signalling mechanism ‘ligand-directed trafficking of receptor “signalling” or “stimulus” ’ in an attempt to differentiate it from the intracellular desensitization/internalization mechanism used by a receptor (however, the phrase involving trafficking was still confusing and has now been largely replaced by the term ‘biased agonism’). Kenakin suggested the simplest way an agonist could activate multiple pathways is via the ‘strength of signal’ generated by different drugs, or via ligands selectively activating a specific pathway. The Ockham's razor explanation would be the ‘strength of signal’ argument because this was consistent with classic receptor theory and relied simply on agonists of differing efficacies. In this case, agonists of low to intermediate efficacy could activate a certain number of receptors and the receptors could activate their preferred G protein or pathway. However, ligands of very high efficacy capable of activating a greater number of receptors could saturate the receptor's preferred pathway, and ‘overflow’ active receptors could then activate a second pathway. An alternative theory was that one agonist could induce a receptor conformation that had preference for one signalling pathway, whereas a second agonist could give rise to a different receptor conformation that preferentially stimulated an alternative signalling pathway, the concept now often termed ‘biased agonism’. Kenakin argued that the best proof in support of biased agonism was if in the same system, two agonists could display reversed orders of potency for the two different signalling pathways.
The evidence for agonist reversal began to show up in the literature in the next few years with perhaps the most prominent example being a study using 5-hydroxytryptamine (5-HT)2C receptors (Berg et al., 1998). In this study the authors simultaneously measured the phospholipase C-inositol phosphate (PLC-IP) pathway and the phospholipase A2-arachadonic acid (PLA2-AA) pathway and discovered some 5-HT2C agonists (e.g. 3-trifluoromethylphenyl-piperazine) preferentially activated the PLC-IP pathway, whereas others (e.g. lysergic acid diethylamide) favoured the PLA2-AA pathway. Another study used a native system, rat cardiac myocytes, and provided evidence that some β2AR ligands could produce β2AR-coupling to both Gs and Gi, while another ligand only activated the Gs pathway (Xiao et al., 2003). Other examples of biased agonism, in which ligands exhibit different capacities to stimulate different signalling pathways (e.g. G protein- or βarrestin-dependent), exist for several ligands acting at a variety of GPCRs, including the β2AR (Drake et al., 2008; Rajagopal et al., 2010). Using a set of fluorescence resonance energy transfer (FRET)-based live-cell biosensors, Drake et al. measured both the kinetics and amplitude of β2AR ligand-induced cAMP generation as an indicator of G protein-dependent signalling activation (Drake et al., 2008). The rate of ligand-induced βarrestin translocation to the β2AR, which is an event proximal to βarrestin-dependent signalling, was also measured. For more than a dozen β2AR agonist compounds, equal relative efficacies for G protein-dependent and βarrestin-associated activities were observed. However, three ligands (ethyl-substituted catecholamines) were identified that demonstrated marked bias toward βarrestin-dependent signalling at the β2AR (Drake et al., 2008). In addition to the discovery of β2AR biased agonists, this paper demonstrated that β2AR agonists such as salbutamol and formoterol, commonly prescribed for their G protein-mediated relaxation of airway smooth muscle, also activate the βarrestin-dependent signalling pathway for which the physiological effect is virtually unknown.
The existence of biased ligands has forced the historical receptor model, which proposed that receptors could exist in an inactive (R) or active (R*) receptor conformation, to expand to include at least four receptor conformations. The inactive conformation, plus active conformations that stimulate βarrestin-dependent signalling (R*βarr), G protein-dependent signalling (R*G) or both (R*dual)(Figure 1). For example, an imperfect βarrestin-biased ligand would preferentially stabilize the receptor in the R*βarr, versus the R*dual, conformation.
Figure 1.
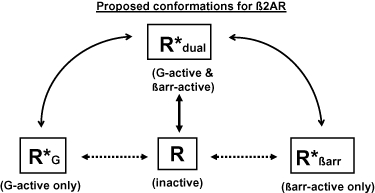
Aside from the inactive conformation (R), all receptor conformations would be capable of constitutive activity according to their respective equilibrium constants. Straight solid, and dotted, arrows represent the equilibriums for ligands that are unbiased or perfectly biased, respectively. Curved arrows represent the spectrum of possible equilibriums for ligands that demonstrate imperfect bias.
Presumably, the endogenous agonist(s) for a receptor would posses the capacity to stimulate all of the receptor's possible signalling pathways, under physiologic, and/or pathologic conditions. The determinants of which possible pathway(s) are activated by a given ligand are likely the result of the ligand's ability to stabilize or induce different conformations of the receptor capable of enriching or producing a receptor conformation with higher affinity and/or efficacy for a given pathway. As such, biased agonism has important implications for the use of existing, and design of new, therapeutic GPCR ligands. For example, because the differing pathways stimulated by a given receptor can lead to distinct functional outcomes, drug discovery must now consider not only the effect of a drug on the classical G protein-dependent pathway (as has been the practice to date), but also on the βarrestin-dependent and other signalling pathways. Accordingly, both drug development screens and basic science research outcomes need to consider all signalling events that a given receptor-ligand interaction can produce.
Receptor theory modification #2: Spontaneous GPCR signalling – not all antagonists are equal
As briefly described earlier, there were only two classes of ligands for GPCRs: agonists and, antagonists. Agonists possessed affinity and efficacy, while antagonists had affinity but zero efficacy. Indeed, it was because they had zero efficacy, therefore leaving a single variable, that it was possible to get an affinity measurement for antagonists. The most famous method for measuring antagonist affinity was described by Schild and formed the foundation for many drug discovery projects seeking to discover GPCR antagonists (Arunlakshana and Schild, 1959). In classical receptor theory all unoccupied GPCRs were thought to be in a quiescent state until activated by an agonist. In other words, ‘empty’ receptors did not signal or activate pathways, only receptors activated by agonists were capable of generating a signal.
However, beginning in the late 1980s and through the 1990s it became evident that spontaneously or constitutively active GPCRs existed. Although no agonist was present, these empty GPCRs were capable of spontaneously assuming a conformation that allowed G protein binding and thus, were able to activate the same pathway(s) as receptors occupied by an agonist (Lee et al., 1997) (reviewed in Bond and Ijzerman, 2006).
One of the earliest examples of spontaneously active GPCRs was provided by Costa and Herz in 1989, when they showed that in a neuroblastoma-glioma cell line (NG-108–15), delta opioid receptors could exhibit spontaneous (ligand-independent) activation of G proteins (Costa and Herz, 1989). Simultaneous with this discovery was the fact that a subset of antagonists could inactivate these constitutively active receptors. To differentiate those antagonists that could ‘turn off’ spontaneously active receptors from those that bind receptors but did not turn them off, Costa and Herz termed the former ‘negative antagonists’ and left the latter as simply ‘antagonists’ (note that the term ‘negative antagonists’ was subsequently replaced with the term, ‘inverse agonists’). Thus we now had three classes of compounds for GPCRs: agonists, antagonists, and inverse agonists. Ariens' ‘intrinsic activity’ had now expanded to a range from −1 to 1; antagonists had 0 intrinsic activity, but inverse agonists had intrinsic activities ranging from −1 (for ‘full’ inverse agonists) to approaching 0 for those weak, ‘partial’ inverse agonists; just as agonist intrinsic activities ranged from 1 (for ‘full’ agonists) to approaching 0 for those weak, ‘partial’ agonists. (Figure 2)
Figure 2.
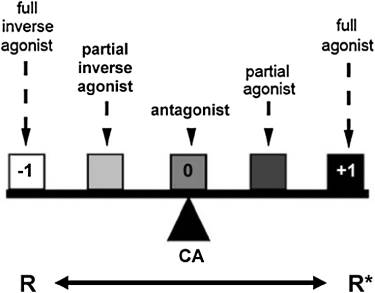
Model of G protein-coupled receptor activation. Full agonists maximally stabilize receptors in an active conformation (R*), whereas full inverse agonists stabilize the inactive receptor conformation (R). Neutral antagonists, simply referred to as antagonists, have no effect on the R/R* equilibrium, but allow constitutive activity (CA) and block the effects of agonists and inverse agonists. Intrinsic activity is shown as −1, 0 and +1. Figure modified from Seifert and Wenzel-Seifert (Seifert and Wenzel-Seifert, 2002) and reproduced with permission of Naunyn–Schmiedeberg's Archives of Pharmacology.
Initially, experimental approaches for revealing receptor constitutive activity were difficult. In most physiologic systems the number of spontaneously active receptors (R*) is a small percentage of the total receptors as most are in the inactive conformation (R). Work from the Lefkowitz laboratory with adrenoceptors showed that a much more efficient means of achieving constitutive receptor activity was to produce mutations in the third intracellular loop of the receptor (Kjelsberg et al., 1992). These mutations elicited a conformational change in the receptor that favoured (ligand-independent) G protein binding and thus caused a shift in the R/R* equilibrium in favour of more R*. The other manner of demonstrating spontaneous receptor activity was to over-express the receptors (Chidiac et al., 1994) where, although only a small percentage of total receptors were constitutively active, the spontaneous signal was greater due to the increased absolute number of receptors in the R* conformation.
Despite the constitutive receptor signal often being of relatively low magnitude, it was soon learned that some GPCR-related diseases result from ‘gain’ or ‘loss’ of receptor constitutive activity. For example, a variety of thyroid diseases result from elevated thyroid-stimulating hormone receptor (TSHR) constitutive activity induced by a variety of TSHR mutations (Parma et al., 1993). Conversely, it has been suggested that the melanocortin 4 receptor constitutively transmits a satiety signal to the brain and that loss of this tonic message contributes to obesity (Vaisse et al., 2000; Srinivasan et al., 2004). Additionally, studies from transgenic mice that overexpress wild-type receptors or express highly constitutively active mutant receptors support the finding that alterations in basal GPCR signalling can lead to altered physiology or pathophysiology (reviewed in Milligan, 2003; Tao, 2008). For example, transgenic overexpression (∼75-fold increase over endogenous expression levels) of the β2AR in airway smooth muscle cells resulted in a significant increase in constitutive G protein-dependent cAMP signalling sufficient to markedly reduce MCh-induced airway smooth muscle tone (McGraw et al., 1999). Studies that describe pathologies resulting from GPCR mutations that cause increased receptor constitutive activity have only measured (canonical) G protein-dependent signalling. Thus, it remains to be determined what role, if any, constitutive βarrestin-dependent signalling might play in these diseases. Although classical receptor theory has evolved to include a description of ‘intrinsic activity’ for inverse agonists, this characterization must now diverge to provide separate descriptions for G- and βarrestin-dependent signalling pathways. Indeed, there exist several examples in which inverse agonists that block G protein-dependent signalling activate or permit βarrestin-dependent signalling (Gesty-Palmer et al., 2006; Wisler et al., 2007). One explanation for these findings is that constitutively active receptors signal via both the G protein- and βarrestin-dependent pathways (Rdual), and that binding of some inverse agonists stabilizes these receptors in the Rbarr conformation. Drug development strategies, as well as clinical and basic science study designs, need to accommodate the implication of a modern receptor theory based on constitutive activity of the receptor for multiple possible signalling pathways, and the potential biased agonism of ligands, in order to fully appreciate the properties of a given GPCR ligand.
Therapeutic effects of ligands: what dictates their benefit and their harm?
With the evolution of GPCR theory, there are numerous factors that influence the choice/utility of ligands. Important considerations include whether the ligand is an agonist, antagonist or inverse agonist; the pleiotropic signalling nature (G protein-dependent, βarrestin-dependent) of the ligand; the chronicity of ligand use; and the identity of the receptor and cellular targets.
These recent advances in receptor theory provide a framework with which to examine the confounding observation that acute use of a particular agonist leads to beneficial effects, whereas chronic administration of the same agonist may lead to harmful effects. Receptor desensitization and associated waning of beneficial effects is well chronicled and perhaps most easily conceptualized as a result of reduced cell surface expression of receptors. Additionally, receptor uncoupling from the signalling mechanism or modification of downstream signalling proteins/events may desensitize receptor signalling independent of any loss of receptor density. However, it is doubtful that functional tachyphylaxis of beneficial signalling pathways accounts for all adverse effects associated with chronic agonist administration. Alternatively, it appears that activation of pro-inflammatory or other adverse signalling pathways may contribute to deleterious effects. For example, adverse signalling may result when a receptor couples to a different G protein, activates the G protein-independent (βarrestin-dependent) signalling pathway, or activates an untargeted cell type. How chronic agonist use may shift the balance of signalling to a deleterious pathway is currently unknown, but receptor trafficking is likely involved.
In contrast to agonist use, antagonist/inverse agonist administration can elicit harmful effects acutely, but have beneficial effects after continued chronic use. One mechanism by which this may occur is through inhibition of receptor down-regulation and restoration of receptor responsiveness to agonists. These actions are now readily appreciated for inverse agonists in the treatment of heart failure. Multiple in vitro and in vivo experiments show that loss of βAR signalling sensitivity, part of the pathogenic mechanism of heart failure, is a result of chronic agonist-induced up-regulation of the GRK/arrestin classical desensitization pathway that ultimately contributes to βAR down-regulation (Petrofski and Koch, 2003). Certain previously contraindicated ‘β-blockers’ are now the standard of care to treat heart failure and are associated with reduced morbidity and mortality. These ligands inhibit excessive receptor signalling, that would otherwise occur in the face of chronically elevated endogenous catecholamines characteristic of heart failure, and thereby prevent enhanced GRK2 expression and cardiomyocyte βAR down-regulation (Petrofski and Koch, 2003). However, not all β-blockers are equally effective. Recent findings from the COMET study showed that heart failure patients prescribed carvedilol gain a significant mortality advantage over those who use metoprolol (Poole-Wilson et al., 2003), and some ‘β-blockers’ do not produce any reduction in mortality (Investigators, 2001). While the COMET study has been criticized as an unfair test due to the use of the short acting metoprolol tartrate as opposed to the longer acting succinate salt currently used in heart failure, there is also speculation that this advantage results from carvedilol's unique property as a weak agonist for βarrestin-dependent signalling (Wisler et al., 2007). Several mouse studies have now demonstrated a cardioprotective effect of βarrestin-dependent signalling induced by β1-adrenoceptor (β1AR) and angiotensin II type 1a receptor (AT1aR) activation (reviewed in Patel et al., 2009).
Biased agonism is another example of a potential therapeutic approach that could be utilized to emphasize the beneficial, and reduce the harmful, effects of a ligand. For example, Gesty-Palmer et al. showed in mice that activation of the parathyroid hormone-related protein receptor (PTH1R) by (D-Trp12,Tyr34)-PTH(7–34), a biased agonist for the βarrestin-dependent signalling pathway, leads to anabolic bone formation without stimulating G protein-mediated bone resorption. If reproducible in humans, these findings hold promise for the treatment of osteoporosis and related diseases (Gesty-Palmer et al., 2009). Interestingly, although chronic activation of both β1AR- and PTH1R-mediated βarrestin-dependent signalling appears to promote beneficial effects in heart failure and bone homeostasis (respectively), activation of the βarrestin-dependent signalling pathway downstream of β2AR appears to be detrimental in asthma (discussed in the following).
The β2AR response in asthma reflects the complexity of receptor agonism
Asthma represents restricted airflow as a result of airway smooth muscle pro-contractile agents, together with lumenal occlusion by mucus and plasma, and airway wall thickening. The principal means of reversing acute bronchoconstriction is via exogenous inhaled β2AR-agonists, which act on airway smooth muscle cell β2ARs to counteract the pro-contractile agents and relax airways via the intracellular mechanisms noted earlier. However, the therapeutic response to beta-agonists can vary among patients, the response to different beta-agonists can vary, and recent studies suggest that β2ARs on cell types other than airway smooth muscle play an important role in both asthma pathogenesis and the therapeutic efficacy of inhaled beta-agonists (Penn and Benovic, 2008).
Although generally highly effective in reversing acute bronchoconstriction, a number of studies link chronic β-agonist use with adverse patient outcomes such as functional β2-adrenergic receptor (β2AR) tachyphylaxis (Newnham et al., 1994; 1995; Grove and Lipworth, 1995), deterioration of asthma control (Sears, 2002; Salpeter et al., 2006) and death (Stolley and Schinnar, 1978; Spitzer et al., 1992; Pearce et al., 1995). Although chronic beta-agonism is not always associated with adverse events (Drazen et al., 1996; Dennis et al., 2000; Bateman et al., 2008), there is clearly a dearth of mechanistic understanding of the physiological effects of chronic β2AR stimulation.
Much attention has been focused recently on the limitations of β2AR agonists in chronic asthma treatment. With respect to therapeutic efficacy, it has been long appreciated that the ability of β2AR agonists to control the bronchoconstrictive state can wane over time. This is best appreciated as a loss of bronchoprotective effect (BPE), whereby with continued use the ability of inhaled beta-agonist to prevent a drop in forced expired volume in 1 s (FEV1) upon challenge with (bronchoconstricting) methacholine diminishes (Deshpande and Penn, 2006; Penn, 2008).
Of greater concern has been the speculated role of β2AR-agonist treatment on asthma mortality. Concerns initially arose from an unusually high incidence of asthma mortality that occurred from the 1960s through the 1980s, in several countries but particularly in the United Kingdom and New Zealand, that were associated with the use of high-dose preparations of the high intrinsic efficacy, short-acting beta-agonists isoproterenol and fenoterol. Use of these drugs in the treatment of asthma was ultimately discontinued. However, additional safety concerns, this time associated with use of the long-acting, low intrinsic efficacy, beta-agonist salmeterol, arose as a result of mortality data from the 1993 Serevent Nationwide Surveillance (SNS) Study in the United Kingdom (Castle et al., 1993) and Salmeterol Multicenter Asthma Research Trial (SMART) (Nelson et al., 2006) conducted in the United States that was terminated in 2003. The prospective, randomized SNS Study reported a small but non-significant (P = 0.105) increase in mortality in those subjects taking salmeterol versus those talking salbutamol (with mortality rates of 0.07 and 0.02%, respectively). The SMART study reported small but significant increases in respiratory-related deaths, asthma-related deaths, and in combined asthma-related deaths or life-threatening experiences in subjects receiving salmeterol versus placebo, with salmeterol-associated deaths occurring more frequently in the African American subpopulation. A subsequent meta-analysis (Salpeter et al., 2006) of 19 randomized placebo-control trials (weighted heavily by SMART study results) reported an increase in life-threatening exacerbations and asthma-related deaths associated with LABA therapy when compared with placebo, Results from these three studies revived the debate over beta-agonist safety that had abated somewhat. And although to date these three studies are frequently cited as evidence justifying safety concerns over the use of LABAs, critics have noted several study design flaws. Two major flaws relate to underpowering of the studies (due to asthma-related death being such a rare event) and the failure for control for either concomitant steroid or (other) beta-agonist use (see Ortega and Peters, 2010) for related discussion). Despite their design flaws that render interpretation problematic, these studies undoubtedly influenced the subsequent Food and Drug Administration (FDA) decision to require a black box warning for treatments including long-acting beta-agonists (reviewed in Ortega and Peters, 2010). Due to disagreement over the meaning of LABA clinical safety trials and those meta-analyses based on these studies, the FDA decision and the issue of beta-agonist safety remains a controversial and hotly debated topic (see Taylor, 2009) and the review by Cazzola and colleagues in this issue). Interestingly, this debate has progressed despite little if any mechanistic basis for an increase in asthma morbidity and mortality conferred by β2AR agonist use. However, recent studies suggest that adverse effects associated with chronic β2AR agonism may result not only from loss of beneficial signalling, but activation of deleterious signalling pathways.
β2AR desensitization contributing to loss of BPE
Because the loss of BPE occurs with chronic β-agonist (maintenance or daily) use, a widely held assumption has been that β2AR desensitization underlies this functional tachyphylaxis. This assumption has been supported by recent studies that demonstrate rodent and human airway smooth muscle β2ARs are subject to GRK- and arrestin- mediated desensitization (Finney et al., 2000; Penn et al., 2001; Deshpande et al., 2008; Kong et al., 2008). Molecular strategies that inhibit GRKs or arrestins improve the signalling of β2ARs in both human and murine airway smooth muscle cells (Deshpande et al., 2008; Penn and Benovic, 2008), and mice lacking the βarrestin-2 gene have a greater airway smooth muscle relaxant response to beta-agonists both ex vivo and in vivo (Deshpande et al., 2008). In addition to this classical mechanism of β2AR desensitization (GRK/βarrestin-mediated receptor uncoupling from Gs) there are other mechanisms that may contribute to a loss of β2AR responsiveness. For example, a reduction in β-agonist stimulated airway smooth muscle cAMP accumulation and thus, reduced bronchodilation, may occur as a result of increased expression or activity of phosphodiesterases (Hansen et al., 2000), reduced expression or activity of either Gs or adenylyl cyclase (reviewed in Billington and Penn, 2003; Guo et al., 2005), or a switch in coupling of the β2AR from Gs to (adenylyl cyclase-inhibiting) Gi (Baillie et al., 2003; McGraw et al., 2007). Thus, the capacity of current therapeutic beta-agonists to induce mechanisms of β2AR desensitization at the receptor locus, or reduce β2AR signalling via effects on proximal downstream events, appears to play an important role in limiting their clinical efficacy.
β2AR signalling promoting up-regulation of pro-contractile signalling
To model chronic stimulation of the β2AR, McGraw et al. (2003) generated transgenic mice that overexpress airway smooth muscle β2ARs. Interestingly, these mice demonstrated increased airway constrictive responses that were associated with induced expression of phospholipase C (PLC)-β1, the effector of ASM Gq-coupled contraction that stimulates phosphoinositide and subsequent calcium-mediated cellular contraction. This up-regulation of PLC-β1 was also observed in an allergen-driven murine asthma model (Lin et al., 2008). Similarly, prolonged β2AR agonism in human airway smooth muscle cultures increased histamine-induced production of phosphoinositides (Sayers et al., 2006). This ‘antithetical’ effect of beta-agonism, along with β2AR desensitization, renders beta-agonists self-limiting, and could conceivably render the airway hyper-responsive to pro-contractile stimuli.
β2AR agonism promoting or permitting inflammation
The effect of beta-agonist therapy on airway inflammation in asthma has been debated for years. Whereas some studies looking at clinical indices of airway inflammation have suggested some anti-inflammatory properties of beta-agonists (reviewed by Remington and Digiovine, 2005), several studies have reported various in vivo indices of inflammation associated with asthma to be unaffected or increased by inhaled beta-agonist therapy (reviewed in Loza et al., 2008). A widely-articulated but yet-to-be-proven belief is that β-agonists can provide symptomatic relief via their capacity to directly relax ASM, but this effect serves to mask an increasing level of airway inflammation (Nelson et al., 2006), and contribute to asthma exacerbations and subtle deterioration of asthma control.
Perhaps the most compelling evidence for a problematic role of β2AR activity in airway inflammation and asthma pathogenesis comes from several recent studies demonstrating the effect of manipulating β2AR activation via either pharmacological or genetic means on indices of allergic airway inflammation. Using a mouse model of asthma, Bond and colleagues demonstrated that whereas chronic β-agonist (salbutamol) administration exacerbated the lung eosinophil phenotype relative to allergen-treated control mice, chronic inverse agonist treatment (nadolol or ICI-118,551) significantly reduced it (Callaerts-Vegh et al., 2004; Nguyen et al., 2008). Furthermore, airway epithelial cell mucin production was significantly inhibited by chronic treatment with the β2AR inverse agonists nadolol and ICI-118,551. However, chronic salbutamol administration did not enhance this index of mucous metaplasia (Nguyen et al., 2008). Building on this work, Nguyen et al. (Nguyen et al., 2009) showed that the significant reduction in asthma phenotype associated with inverse agonist treatment is copied in mice lacking the β2AR. Collectively, these studies demonstrate that β2AR signalling is required for the full asthma phenotype development in mice. Thus, chronic treatment with therapeutic beta-agonists could, through activation of a β2AR-mediated ‘adverse’ signalling pathway, exacerbate the asthma phenotype or accelerate asthma severity. The relationship between agonists and inverse agonists has been reciprocal in nature with regards to effects on cell signalling pathways. This gives rise to the concept that if chronic treatment with an agonist may be detrimental, then chronic treatment with the inverse agonist may be beneficial.
Recent studies showing that β2AR agonism affects both T cell and airway epithelium inflammatory function provide additional mechanistic insight into how β2AR agonism might adversely affect asthma. Loza et al. (Loza et al., 2007; 2008;) recently reported that β2AR-agonists, at physiologically and clinically relevant concentrations, stimulate increased antigen-independent and cytokine-stimulated accumulation of type 2 T cells by increasing cell survival. Futamura and colleagues (Futamura et al., 2010) showed, using cultured normal human bronchial epithelial cells and bronchial smooth muscle cells, that cytokine-induced up-regulation of thymic stromal lymphopoietin (TSLP) is significantly enhanced by both long- and short-acting β2AR-agonists. Mouse lung overexpression of TSLP, an indispensable cytokine in the Th2-mediated development of allergic diseases, results in goblet cell metaplasia as well as severe airway inflammation and hyper-responsiveness (Zhou et al., 2005). Thus, β2AR-agonists have the potential to regulate T-cell development and cytokine expression to affect disease pathogenesis or the efficacy of therapies.
β2AR agonism promoting mucous metaplasia
A second target cell through which β2AR agonism may exacerbate inflammation is the airway epithelial cell. Besides the evidence earlier with pharmacological or genetic inhibition of β2AR signalling, a recent study in allergen-sensitized and -challenged mice showed that daily dosing with β2AR agonists worsened airway hyper-responsiveness and that this was associated with physiologically relevant increases in airway inflammation and mucous cell metaplasia (Riesenfeld et al., 2010). These conclusions are supported by studies that implicate β2AR-agonists in mucin production in rats (Kamachi et al., 2001) and airway epithelial cell proliferation and airway wall thickening in mice (Tamaoki et al., 2004). β2ARs on human airway epithelial cells have been shown to regulate mucin secretion (Leikauf et al., 1984) and, through regulation of cystic fibrosis transmembrane conductance regulator (CFTR), mucus viscosity (Leikauf et al., 1984; Delavoie et al., 2009). Additionally, epithelial cell proliferation is stimulated by β-agonist-mediated activation of β2ARs (Nishimura et al., 2002; Tamaoki et al., 2004). Collectively, these studies are consistent with the putative role of mucus plugging in fatal human asthma (Kamachi et al., 2001).
So what do we target and how?
How do we take advantage of our current understanding of β2AR signalling and its consequences in the airway to improve on asthma management? We might consider the following possibilities.
Maintenance therapy with inverse agonists of the β2AR to minimize β2AR desensitization in ASM, mucin production in epithelial cells and inflammation induced by allergen
Based on murine studies a predicted effect of therapy with β2AR inverse agonists would be to suppress the inflammatory response, as well as the associated AHR, that is triggered by allergen exposure in most asthmatics. Thus, the fundamental problem (bronchoconstriction) might be largely avoided. Even in the event of acute bronchospastic exacerbations, rescue beta-agonists (in doses capable of competing with the inverse agonist) could conceivably be more effective given the ‘sensitizing’ effect of inverse agonists on β2AR responsiveness. Interestingly, a recent study by Hanania et al. (Hanania et al., 2010) demonstrated that after 13 weeks of nadolol treatment in 10 mild asthmatics, the dose of albuterol required to reverse the loss of FEV1 after methacholine challenge was similar to the dose required at baseline (pre-nadolol treatment).
In addition, excessive mucin production promoted by either endogenous or exogenous beta-agonist in the asthmatic lung could be greatly reduced by inverse agonist treatment. The critical question that remains is whether safety concerns would override the benefit of these prophylactic effects of inverse agonist therapy. Although a large clinical trial will be required to resolve this question, two preliminary studies examining the effects of nadolol in 20 (10 per trial) mild asthmatics reported that while some subjects experienced a moderate, non-symptomatic drop in expired airflow after the initial dose of nadolol, after 9 or 13 weeks of treatment sensitivity to methacholine was reduced as evidenced by significant increase in PC20 values (Hanania et al., 2008; 2010;).
Biased beta-agonists that promote the good β2AR signalling and forego the bad
β2AR receptor expression, and thus β2AR-mediated signalling, is required for the full development of the asthma phenotype in mice. The lack of asthma phenotype observed in βarrestin-2-KO mice suggests that β2AR-mediated βarrestin-dependent signalling may be the culprit transducing the negative consequences of beta-agonists. Conceivably, βarrestin-dependent signalling may mediate increased survival in T cells, increase other inflammatory cell numbers and activity, and increase epithelial cell mucin production. Indeed, sorting out the signalling determinants that mediate these responses in both cell-based and integrative models represents an exciting new area in airway biology and pharmacology. A possible solution to the mixed bag of signalling and consequences of current beta-agonists used in asthma therapy would be a biased beta-agonist that preferentially activates G protein-dependent signalling and functions as an antagonist or inverse agonist for the βarrestin-dependent signalling pathway. Although most β2AR ligands tested demonstrate proportional effects on both signalling pathways, some ligands are biased and preferentially activate the G protein-dependent pathway relative to the βarrestin-dependent signalling pathway or vice-versa (Violin and Lefkowitz, 2007; Drake et al., 2008; Violin et al., 2010). Future drug development strategies that consider biased agonism properties appear required for the discovery of new beta-agonists that preferentially activate G protein-dependent signalling (Evans et al., 2010).
Adjunct therapies that target the negative consequences of β2AR agonism
For addressing the increased Gq-coupled receptor signalling that occurs with chronic β2AR agonism, general approaches might include yet-to-be developed drugs or gene therapy that successfully antagonize Gq (functioning similar to RGS proteins or regulators of G protein GAP activity (Penn, 2008; Druey, 2009) or phospholipase C (or Gq activation of PLC analogous to the effect of the N-terminal domain of GRK2 (Carman et al., 1999). Currently available adjuncts that target specific Gq-coupled receptors include m3 muscarinic acetylcholine receptor (m3mAChR) antagonists (e.g. tiotropium) and cysteinyl leukotriene-1 receptor (CysLT1R) antagonists (e.g. monteleukast).
To target the inflammation for which β2AR agonism appears required, steroids are an obvious adjunct. Given the most common asthma therapy is in fact an inhaler of a long-acting beta-agonist plus corticosteroid, it is of course possible that at least one of the negative consequences of β2AR agonism is already addressed. However, this remains to be fully investigated, and the possibility remains that more selective drugs (e.g. inhibitors of nuclear factor kappa-light-chain enhancer of activated B cells (NF-kB) signalling (e.g. IκB kinase inhibitors) or regulators of specific glucocorticoid-regulated genes) could better complement β2AR ligands avoiding some of the negative consequences of steroid therapy.
Conclusion
Advances in our understanding of GPCR signalling, which will undoubtedly continue to evolve, may address the unwanted clinical results elicited by various β2AR ligands and guide future development of more effective and safer drugs for the treatment of asthma and other respiratory diseases.
Acknowledgments
Some of the ideas contained in this paper were the result of work supported by grants from the NIH (HL084123, HL093103, JKLW, HL585506, HL093103, RBP, NAH, R01AI079236, BFD and R01AI079236, RAB) and the American Asthma Foundation (RAB).
Glossary
Abbreviations
- AT1aR
angiotensin II type 1a receptor
- β1AR
β1-adrenoceptor
- β2AR
β2-adrenoceptor
- β-arrestin
βarrestin
- BPE
bronchoprotective effect
- cAMP
cyclic adenosine monophosphate
- CFTR
cystic fibrosis transmembrane conductance regulator
- CysLT1R
cysteinyl leukotriene-1 receptor
- FEV1
forced expired volume in 1 s
- FRET
fluorescence resonance energy transfer
- GPCR
G protein-coupled receptor
- GRK
G protein receptor kinase
- m3mAChR
m3 muscarinic acetylcholine receptor
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PKA
protein kinase A
- PLA2-AA
phospholipase A2-arachadonic acid
- PLC-IP
phospholipase C-inositol phosphate
- TSHR
thyroid-stimulating hormone receptor
- TSLP
thymic stromal lymphopoietin
- 5-HT
5-hydroxytryptamine
Conflicts of interest
RBP is the recipient of an independent investigator grant from GSK. RAB owns shares in Inverseon.
Supporting Information
Teaching Materials; Figs 1–2 as PowerPoint slide.
References
- Ariens E. Affinity and intrinsic activity in the theory of competitive inhibition. I. Problems and theory. Arch Int Pharmacodyn Ther. 1954;99:32–49. [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, et al. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA. 2003;100:940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bateman E, Nelson H, Bousquet J, Kral K, Sutton L, Ortega H, et al. Meta-analysis: effects of adding salmeterol to inhaled corticosteroids on serious asthma-related events. Ann Intern Med. 2008;149:33–42. doi: 10.7326/0003-4819-149-1-200807010-00229. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Pike LJ, Cerione RA, Staniszewski C, Yoshimasa T, Codina J, et al. Phosphorylation of the mammalian beta-adrenergic receptor by cyclic AMP-dependent protein kinase. Regulation of the rate of receptor phosphorylation and dephosphorylation by agonist occupancy and effects on coupling of the receptor to the stimulatory guanine nucleotide regulatory protein. J Biol Chem. 1985;260:7094–7101. [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin Type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Billington C, Penn R. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res. 2003;4:2. [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin F-T, Lefkowitz RJ, Caron MG. [mu]-Opioid receptor desensitization by [beta]-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bond RA, Ijzerman AP. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci. 2006;27:92–96. doi: 10.1016/j.tips.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, et al. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- Callaerts-Vegh Z, Evans KLJ, Dudekula N, Cuba D, Knoll BJ, Callaerts PFK, et al. Effects of acute and chronic administration of β-adrenoceptor ligands on airway function in a murine model of asthma. Proc Natl Acad Sci USA. 2004;101:4948–4953. doi: 10.1073/pnas.0400452101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Parent J-L, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, et al. Selective regulation of Gαq/11 by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem. 1999;274:34483–34492. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- Castle W, Fuller R, Hall J, Palmer J. Serevent nationwide surveillance study: comparison of salmeterol with salbutamol in asthmatic patients who require regular bronchodilator treatment. Br Med J. 1993;306:1034–1037. doi: 10.1136/bmj.306.6884.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidiac P, Hebert TE, Valiquette M, Dennis M, Bouvier M. Inverse agonist activity of beta-adrenergic antagonists. Mol Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- Clark AJ. The Mode of Action of Drugs on Cells. London: E Arnold & Company; 1933. [Google Scholar]
- Clark AJ. General Pharmacology. 4th edn. Supplement. Berlin: Julius Springer; 1937. [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delavoie F, Molinari M, Milliot M, Zahm J-M, Coraux C, Michel J, et al. Salmeterol restores secretory functions in cystic fibrosis airway submucosal gland serous cells. Am J Respir Cell Mol Biol. 2009;40:388–397. doi: 10.1165/rcmb.2008-0037OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis SM, Sharp SJ, Vickers MR, Frost CD, Crompton GK, Barnes PJ, et al. Regular inhaled salbutamol and asthma control: the TRUST randomised trial. Lancet. 2000;355:1675–1679. doi: 10.1016/s0140-6736(00)02238-8. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 2006;18:2105–2120. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JKL. {beta}-Arrestins specifically constrain {beta}2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J. 2008;22:2134–2141. doi: 10.1096/fj.07-102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. β-Arrestin-biased Agonism at the β2-Adrenergic Receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- Drazen JM, Israel E, Boushey HA, Chinchilli VM, Fahy JV, Fish JE, et al. Comparison of regularly scheduled with as-needed use of albuterol in mild asthma. N Engl J Med. 1996;335:841–847. doi: 10.1056/NEJM199609193351202. [DOI] [PubMed] [Google Scholar]
- Druey K. Regulation of G-protein-coupled signaling pathways in allergic inflammation. Immunol Res. 2009;43:62–76. doi: 10.1007/s12026-008-8050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BA, Sato M, Sarwar M, Hutchinson DS, Summers RJ. Ligand-directed signalling at β-adrenoceptors. Br J Pharmacol. 2010;159:1022–1038. doi: 10.1111/j.1476-5381.2009.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney PA, Belvisi MG, Donnelly LE, Chuang T-T, Mak JCW, Scorer C, et al. Albuterol-induced downregulation of Gsα accounts for pulmonary β2-adrenoceptor desensitization in vivo. J Clin Invest. 2000;106:125–135. doi: 10.1172/JCI8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF. The use of beta-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor agonist complexes. Adv Drug Res. 1966;3:21–56. [Google Scholar]
- Futamura K, Orihara K, Hashimoto N, Morita H, Fukuda S, Sagara H, et al. beta2-Adrenoceptor agonists enhance cytokine-induced release of thymic stromal lymphopoietin by lung tissue cells. Int Arch Allergy Immunol. 2010;152:353–361. doi: 10.1159/000288288. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, et al. A β-arrestin–biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1–9. doi: 10.1126/scitranslmed.3000071. 1 1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove A, Lipworth BJ. Bronchodilator subsensitivity to salbutamol after twice daily salmeterol in asthmatic patients. Lancet. 1995;346:201–206. doi: 10.1016/s0140-6736(95)91265-7. [DOI] [PubMed] [Google Scholar]
- Guo M, Pascual RM, Wang S, Fontana MF, Valancius CA, Panettieri RA, et al. Cytokines regulate beta-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA- and EP2 receptor-dependent mechanisms. Biochemistry. 2005;44:13771–13782. doi: 10.1021/bi051255y. [DOI] [PubMed] [Google Scholar]
- Hanania NA, Singh S, El-Wali R, Flashner M, Franklin AE, Garner WJ, et al. The safety and effects of the beta-blocker, nadolol, in mild asthma: an open-label pilot study. Pulm Pharmacol Ther. 2008;21:134–141. doi: 10.1016/j.pupt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanania NA, Mannava B, Franklin AE, Lipworth BJ, Williamson PA, Garner WJ, et al. Response to salbutamol in patients with mild asthma treated with nadolol. Eur Respir J. 2010;36:963–965. doi: 10.1183/09031936.00003210. [DOI] [PubMed] [Google Scholar]
- Hansen G, Jin S-LC, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci USA. 2000;97:6751–6756. doi: 10.1073/pnas.97.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ. Reciprocal in vivo regulation of myocardial G protein–coupled receptor kinase expression by ?-Adrenergic receptor stimulation and blockade. Circulation. 1998;98:1783–1789. doi: 10.1161/01.cir.98.17.1783. [DOI] [PubMed] [Google Scholar]
- Investigators B-BEoST. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659–1667. doi: 10.1056/NEJM200105313442202. [DOI] [PubMed] [Google Scholar]
- Kamachi A, Munakata M, Nasuhara Y, Nishimura M, Ohtsuka Y, Amishima M, et al. Enhancement of goblet cell hyperplasia and airway hyperresponsiveness by salbutamol in a rat model of atopic asthma. Thorax. 2001;56:19–24. doi: 10.1136/thorax.56.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy I: mechanisms of efficacy and receptor promiscuity. Trends Pharmacol Sci. 1995a;16:188–192. doi: 10.1016/s0165-6147(00)89020-3. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy II: agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995b;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kjelsberg MA, Cotecchia S, Ostrowski J, Caron MG, Lefkowitz RJ. Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J Biol Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- Kong KC, Gandhi U, Martin TJ, Anz CB, Yan H, Misior AM, et al. Endogenous Gs-coupled receptors in smooth muscle exhibit differential susceptibility to GRK2/3-mediated desensitization†. Biochemistry. 2008;47:9279–9288. doi: 10.1021/bi801056w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated Pits. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- Lee TW, Cotecchia S, Milligan G. Up-regulation of the levels of expression and function of a constitutively active mutant of the hamster alpha1B-adrenoceptor by ligands that act as inverse agonists. Biochem J. 1997;325:733–739. doi: 10.1042/bj3250733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Whalen EJ. [beta]-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Leikauf GD, Ueki IF, Nadel JA. Autonomic regulation of viscoelasticity of cat tracheal gland secretions. J Appl Physiol. 1984;56:426–430. doi: 10.1152/jappl.1984.56.2.426. [DOI] [PubMed] [Google Scholar]
- Lin R, Peng H, Nguyen LP, Dudekula NB, Shardonofsky F, Knoll BJ, et al. Changes in [beta]2-adrenoceptor and other signaling proteins produced by chronic administration of [‘][beta]-blockers’ in a murine asthma model. Pulm Pharmacol Ther. 2008;21:115–124. doi: 10.1016/j.pupt.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. β-arrestin: a protein that regulates β-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Loza MJ, Peters SP, Foster S, Khan IU, Penn RB. [beta]-Agonist enhances type 2 T-cell survival and accumulation. J Allergy Clin Immunol. 2007;119:235–244. doi: 10.1016/j.jaci.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Loza MJ, Foster S, Peters SP, Penn RB. Interactive effects of steroids and beta-agonists on accumulation of type 2 T cells. J Allergy Clin Immunol. 2008;121:750 e751–755 e753. doi: 10.1016/j.jaci.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, nbsp G, Daaka Y, Miller WE, et al. Beta-arrestin-dependent formation of 2 adrenergic receptor-src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Forbes SL, Kramer LA, Witte DP, Fortner CN, Paul RJ, et al. Transgenic overexpression of β2-adrenergic receptors in airway smooth muscle alters myocyte function and ablates bronchial hyperreactivity. J Biol Chem. 1999;274:32241–32247. doi: 10.1074/jbc.274.45.32241. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Almoosa KF, Paul RJ, Kobilka BK, Liggett SB. Antithetic regulation by β-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway β-agonist paradox. J Clin Invest. 2003;112:619–626. doi: 10.1172/JCI18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw DW, Elwing JM, Fogel KM, Wang WCH, Glinka CB, Mihlbachler KA, et al. Crosstalk between Gi and Gq/Gs pathways in airway smooth muscle regulates bronchial contractility and relaxation. J Clin Invest. 2007;117:1391–1398. doi: 10.1172/JCI30489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol. 2003;64:1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM. The salmeterol multicenter asthma research trial*. Chest. 2006;129:15–26. doi: 10.1378/chest.129.1.15. [DOI] [PubMed] [Google Scholar]
- Newnham DM, McDevitt DG, Lipworth BJ. Bronchodilator subsensitivity after chronic dosing with eformoterol in patients with asthma. Am J Med. 1994;97:29–37. doi: 10.1016/0002-9343(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Newnham DM, Grove A, McDevitt DG, Lipworth BJ. Subsensitivity of bronchodilator and systemic beta 2 adrenoceptor responses after regular twice daily treatment with eformoterol dry powder in asthmatic patients. Thorax. 1995;50:497–504. doi: 10.1136/thx.50.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Omoluabi O, Parra S, Frieske JM, Clement C, Ammar-Aouchiche Z, et al. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. Am J Respir Cell Mol Biol. 2008;38:256–262. doi: 10.1165/rcmb.2007-0279RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ, et al. β2-Adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci USA. 2009;106:2435–2440. doi: 10.1073/pnas.0810902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Tamaoki J, Isono K, Aoshiba K, Nagai A. β-adrenergic receptor-mediated growth of human airway epithelial cell lines. Eur Respir J. 2002;20:353–358. doi: 10.1183/09031936.02.01352001. [DOI] [PubMed] [Google Scholar]
- Ortega VE, Peters SP. Beta-2 adrenergic agonists: focus on safety and benefits versus risks. Curr Opin Pharmacol. 2010;10:246–253. doi: 10.1016/j.coph.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Parma J, Duprez L, Sande JV, Cochaux P, Gervy C, Mockel J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- Patel PA, Tilley DG, Rockman HA. Physiologic and cardiac roles of [beta]-arrestins. J Mol Cell Cardiol. 2009;46:300–308. doi: 10.1016/j.yjmcc.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Pearce N, Beasley R, Crane J, Burgess C, Jackson R. End of the New Zealand asthma mortality epidemic. Lancet. 1995;345:41–44. doi: 10.1016/s0140-6736(95)91159-6. [DOI] [PubMed] [Google Scholar]
- Penn R. Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:149–169. doi: 10.1007/s00210-008-0263-1. [DOI] [PubMed] [Google Scholar]
- Penn RB, Benovic JL. Regulation of heterotrimeric G protein signaling in airway smooth muscle. Proc Am Thorac Soc. 2008;5:47–57. doi: 10.1513/pats.200705-054VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn RB, Pascual RM, Kim Y-M, Mundell SJ, Krymskaya VP, Panettieri RA, et al. Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J Biol Chem. 2001;276:32648–32656. doi: 10.1074/jbc.M104143200. [DOI] [PubMed] [Google Scholar]
- Petrofski JA, Koch WJ. The [beta]-adrenergic receptor kinase in heart failure. J Mol Cell Cardiol. 2003;35:1167–1174. doi: 10.1016/s0022-2828(03)00243-8. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Poole-Wilson PA, Swedberg K, Cleland JGF, Di Lenarda A, Hanrath P, Komajda M, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington TL, Digiovine B. Long-acting beta-agonists: anti-inflammatory properties and synergy with corticosteroids in asthma. Curr Opin Pulm Med. 2005;11:74–78. doi: 10.1097/01.mcp.0000146784.56834.ff. [DOI] [PubMed] [Google Scholar]
- Riesenfeld E, Sullivan M, Thompson-Figueroa J, Haverkamp H, Lundblad L, Bates J, et al. Inhaled salmeterol and/or fluticasone alters structure/function in a murine model of allergic airways disease. Respir Res. 2010;11:22. doi: 10.1186/1465-9921-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter S, Buckley N, Ormiston T, Salpeter E. Meta-analysis: effect of long-acting {beta}-agonists on severe asthma exacerbations and asthma-related deaths. Ann Intern Med. 2006;144:904–912. doi: 10.7326/0003-4819-144-12-200606200-00126. [DOI] [PubMed] [Google Scholar]
- Sayers I, Swan C, Hall IP. The effect of [beta]2-adrenoceptor agonists on phospholipase C (beta1) signalling in human airway smooth muscle cells. Eur J Pharmacol. 2006;531:9–12. doi: 10.1016/j.ejphar.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Sears MR. Adverse effects of [beta]-agonists. J Allergy Clin Immunol. 2002;110(Part 2):S322–S328. doi: 10.1067/mai.2002.129966. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J Biol Chem. 2005;280:15315–15324. doi: 10.1074/jbc.M412418200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Spitzer WO, Suissa S, Ernst P, Horwitz RI, Habbick B, Cockcroft D, et al. The use of β-agonists and the risk of death and near death from asthma. N Engl J Med. 1992;326:501–506. doi: 10.1056/NEJM199202203260801. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR, et al. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson RP. A modification of receptor theory. Br J Pharmacol Chemother. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolley PD, Schinnar R. Association between asthma mortality and isoproterenol aerosols: a review. Prev Med. 1978;7:519–538. doi: 10.1016/0091-7435(78)90265-7. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Tagaya E, Kawatani K, Nakata J, Endo Y, Nagai A. Airway mucosal thickening and bronchial hyperresponsiveness induced by inhaled β2-agonist in mice*. Chest. 2004;126:205–212. doi: 10.1378/chest.126.1.205. [DOI] [PubMed] [Google Scholar]
- Tao Y-X. Constitutive activation of G protein-coupled receptors and diseases: insights into mechanisms of activation and therapeutics. Pharmacol Ther. 2008;120:129–148. doi: 10.1016/j.pharmthera.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR. The {beta}-agonist saga and its clinical relevance: on and on it goes. Am J Respir Crit Care Med. 2009;179:976–978. doi: 10.1164/rccm.200901-0055CC. [DOI] [PubMed] [Google Scholar]
- Terwilliger RZ, Ortiz J, Guitart X, Nestler EJ. Chronic morphine administration increases β-adrenergic receptor kinase (βARK) Levels in the rat locus coeruleus. J Neurochem. 1994;63:1983–1986. doi: 10.1046/j.1471-4159.1994.63051983.x. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Lefkowitz RJ. [beta]-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging β-arrestins at the AT1R reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R-P, Zhang S-J, Chakir K, Avdonin P, Zhu W, Bond RA, et al. Enhanced Gi signaling selectively negates {beta}2-adrenergic receptor (AR)- but not {beta}1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–1639. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
- Yamaki K, Takahashi Y, Sakuragi S, Matsubara K. Molecular cloning of the S-antigen cDNA from bovine retina. Biochem Biophys Res Commun. 1987;142:904–910. doi: 10.1016/0006-291x(87)91499-9. [DOI] [PubMed] [Google Scholar]
- Zhou B, Comeau MR, Smedt TD, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.