

Abstract
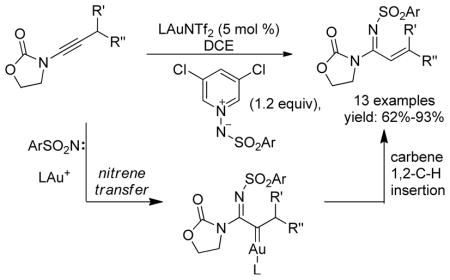
A gold-catalyzed intermolecular nitrene transfer to alkynes was developed for the first time, revealing a new mode of nitrene transfer and providing a novel access to versatile α-imino metal carbenes. Various mild nitrene-transfer reagents were examined, and iminopyridium ylides especially those based on 3,5-dichloropyridine turned out to be highly effective. With activated alkynes such as N-alkynyloxazolidinones as substrates, α, β-unsaturated amidines were formed in mostly good yields.
A rather unique and potent reactivity of gold catalysis is the capability of the metal to form gold carbenes via γ-eliminations. Although there are still some ambiguities regarding discrete reaction mechanisms,1 this strategy has been applied successfully in intramolecular transformations using tethered oxygen-delivering oxidants such as sulfoxides,2 N-oxides,1b,3 nitrones4 and epoxide5 where their oxygen-less parts (i.e., Y) behave as nucleofuges (Scheme 1A). Attempts to realized intermolecular versions of this strategy using external sulfoxides6 led to 3,3-rearrangements instead and the intermediacy of α-oxo gold carbenes was ruled out both by experiments and via calculations. We discovered, however, that pyridine N-oxides and quinoline N-oxides were suitable external oxidants for the generation of these reactive gold intermediates intermolecularly, thus offering valuable opportunities to replace hazardous and potentially explosive diazo ketones with benign alkynes in gold carbene chemistry (Scheme 1B). We have applied this strategy successfully to rather expedient syntheses of several classes of functional molecules.7
Scheme 1.

Gold-catalyzed oxygen/nitrene transfer to alkyne: concept
With all the studies so far focused on oxidation using oxygen-delivering oxidants, there is surprisingly little attention paid to nitrene-delivering oxidants. While a tethered azido group8 and a tethered sulfilimine (one example only)2a could be considered as intramolecular cases, no gold-catalyzed intermolecular nitrene transfer to alkynes has been reported (Scheme 1C). Several features of this mode of nitrene transfer are notable: 1) the nitrene moiety is delivered via an outer sphere attack, and no gold carbene complex9 is involved; this new mode of nitrene transfer is different from many known nitrene transfer reactions;10 2) it would make alkynes equivalent to α-diazo imines, which are difficult to access. It is known that α-diazo imines readily cyclize into 1,2,3-triazoles although in the presence of electron-withdrawing substituents at N-1 the reversed process could be utilized to generate α-imino rhodium carbenes.11 Herein, we disclose our effort in synthesizing and evaluating various external nitrene-transfer reagents in generating α-imino gold carbenes directly from C-C triple bonds, which led to efficient synthesis of α, β-unsaturated amidines.
At the outset, a range of nitrene-transfer reagents were synthesized for screening. As shown in Figure 1, they include sulfilimines 1 and 2 derived from dibenzothiophene,12 cyclic sulfilimine 2,13 iminoiodinane 4, which is soluble in organic solvents,14 and iminopyridinium ylides 5–8,15 and iminoquinolinium ylide 9.15
Figure 1.

Selected nitrene-transfer reagents (NTR)
After having little success with aliphatic internal alkynes, we turned our attention to activated alkynes, specifically N-alkynyloxazolidinones,7c,16 which are readily available from terminal alkynes via several convenient methods.17 Table 1 shows some of the reaction optimization results. To our delight, expected amidine 11 was indeed formed when various nitrene-transfer reagents were used. While sulfilimines 1 and 2 were mostly ineffective, cyclic sulfilimine 3 gave good yields with a range of different gold catalysts including IPrAuNTf2 (entry 5) and (F5C6)3PAuNTf2 (entry 6). While the result with IPrAuNTf2 was excellent, the fact that the propylthio group remained in the product prompted us to search further for oxidants that deliver a simple tosyl group. While soluble iminoiodinane 4 was less effective, iminopyridinium ylides offered mostly good to excellent yields (entries 8–13). Iminopyridinium ylide 8 derived from 3,5-dichloropyridine proved to be the most effective and led to the formation of 11 in excellent yields regardless the electrophilic nature of the gold catalyst (comparing entries 11, 12 and 13). Notably, steric bulk on the oxidant was detrimental, and the 2-Me group in ylide 7 decreased the reaction yield from 65% to 25% (comparing entries 8 and 10). For the same reason, iminoquinolinium ylide 9 was a poor oxidant (entry 14).
Table 1.
Reaction optimization
 | ||||||
|---|---|---|---|---|---|---|
| entrya | NTR | gold catalyst | reaction conditions | conversion | yield | E/Z |
| 1 | 1 | Ph3PAuNTf2 | rt, 12 h | 80% | 10% | 2/1 |
| 2 | 1 | IPrAuNTf2 | rt, 12 h | 20% | <5% | 3.2/1 |
| 3 | 1 | (F5C6)3PAuNTf2 | rt, 12 h | 100% | 35% | 1.5/1 |
| 4 | 2 | (F5C6)3PAuNTf2 | rt, 12 h | 100% | 30% | 1.5/1 |
| 5 | 3 | IPrAuNTf2 | rt, 3 h | 100% | 90% | - |
| 6 | 3 | (F5C6)3PAuNTf2 | rt, 12 h | 90% | 70% | 1.5/1 |
| 7 | 4 | (F5C6)3PAuNTf2 | rt, 3 h | 100% | 46% | 3/1 |
| 8 | 5 | (F5C6)3PAuNTf2 | rt, 18 h | 80% | 65% | 2.5/1 |
| 9 | 6 | (F5C6)3PAuNTf2 | rt, 48 h | 70% | 60% | 1.5/1 |
| 10 | 7 | (F5C6)3PAuNTf2 | rt, 48 h | 50% | 25% | 1.5/1 |
| 11 | 8 | (F5C6)3PAuNTf2 | rt, 12 h | 100% | 90% | 1.5/1 |
| 12 | 8 | IPrAuNTf2 | rt, 3 h | 100% | 95%b | 4/1 |
| 13 | 8 | 12c | rt, 3 h | 100% | 95%b | 4/1 |
| 14 | 9 | IPrAuNTf2 | rt, 48 h | 30% | 10% | 3/1 |
[10] = 0.05 M.
92% isolated yield.
12 = dichloro(2-picolinato)gold(III).
The amidine product 11 was formed as an inseparable mixture of two isomers. 1H NMR indicated that they were isomeric at the C-C double bond2d and the E-isomer was major but only to a small extent. Interestingly, this lack of stereoselectivity was in stark contrast to our previous result with the same substrate, where ynamide 10 was oxidized into an α, β-unsaturated imide with >50/1 E/Z ratio.7c The geometries of the C-N double bonds in both isomers were assigned as E and discussed later.
Our attempts to improve the E/Z selectivities of the C-C double bond were mostly unsuccessful. A notable feature of the optimized reaction conditions is the absence of an acid additive, which was required in our previous studies using pyridine N-oxides.7a,7b
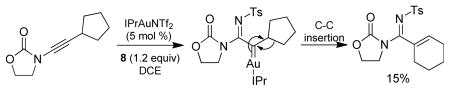
The reaction scope was then studied and the results are shown in Table 2. Good to excellent yields were obtained with substrates substituted at the alkyne terminus by a primary alkyl group (entry 1) and several functionalized ones (entries 2–6). Functional groups such as chloro (entry 2), aryl (entry 3), ester (entry 4), MsO (entry 5) and ether (entry 6) were readily tolerated; however, proximally positioned HO and azido groups appeared to interfere the desired reaction, and little or no desired product was observed. The allowance of MsO is noteworthy as it offers a reactive electrophilic site for further transformation while highlighting the mild nature of this nitrene transfer process. A cyclohexyl group was easily accommodated, leading to 14g as the only isomer in 90% yield (entry 7). Similarly, an efficient reaction was observed with a cyclopentyl group (entry 8); however, besides the expected 1,2-C-H insertion, a ring expansion occurred as a minor event, leading to a cyclohexene product18 in 15% yield. Besides tosylnitrene, other arenesulfonylnitrenes were readily transferred to alkynes using analogous iminopyridium ylides regardless of electronic properties of the arene (comparing entries 9 and 10) as well as its steric bulk (entry 11, Mts = 2,4,6-trimethylbenzenesulfonyl). While the oxazolidinone moiety appeared to be ideal for this nitrene transfer reaction, an N-phenyltoluenesulfonamido moiety, as shown in entry 12, could also play the role of activating the C-C triple bond toward this intermolecular nitrene transfer.
Table 2.
Scope studiesa
 | ||||||
|---|---|---|---|---|---|---|
1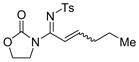 14a, 93% E/Z = 4/1 |
2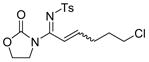 14b, 81%b E/Z = 2/1 |
3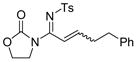 14c, 88% E/Z = 3/1 |
||||
4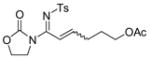 14d, 79%b E/Z = 2/1 |
5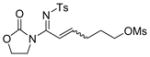 14e, 72% E/Z = 3/2 |
6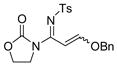 14f, 75% E/Z = 5/1 |
||||
7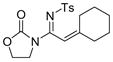 14g, 90% |
8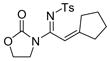 14h, 76%c |
9d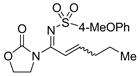 14i, 85% E/Z = 5/1 |
||||
10d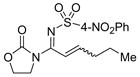 14j, 88% E/Z = 4/1 |
11d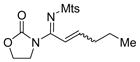 14k, 79% E/Z = 5/1 |
12 14l, 62%e E/Z = 3/2 |
||||
[13] = 0.05 M.
(F5C6)3PAuNTf2 was the catalyst.
15% of the ring-expansion product was formed.
The oxidant used was analogous to 8 with the only difference at the sulfonyl group.
dichloro(2-picolinato)gold(III).
The geometry of the C-N double bond was difficult to assign even after rather extensive NOESY studies. Eventually we were able to secure a single crystal of the major component of compound 14j, and its X-ray diffraction studies revealed that the C-N double bond has an E-configuration. Although the corresponding geometry of the minor isomer was not determined, we reasoned it should be the same as that of the major isomer as it is difficult to imagine that the geometries of both C-N and C-C double bonds would be paired in an exclusive manner (i.e., E, E and Z, Z only). The exclusive formation of the E-C-N double bond was also consistent with the fact that 14g and 14h did not have any observed geometric isomer. The reason for the selectivity, although not apparent, could be rationalized by conformational analysis. As shown in Scheme 2, assuming that gold carbene intermediate B is formed via a concerted elimination, stereoelectronic effects require bond alignments as outlined in conformation A. The preference for such a conformation can be reasoned by invoking the minimization of charge repulsion between the heteroatoms on the oxazolidinone ring and the sulfonyl oxygens. Accordingly, the geometries of the C-N double bonds in other cases were all tentatively assigned as E.
Scheme 2.

Rationale for the formation of the E-C-N double bond
In conclusion, we have developed the first examples of gold-catalyzed intermolecular nitrene transfer to alkynes and realized a new nitrene transfer strategy that provides a novel and facile access to versatile α-imino metal carbenes. Various mild nitrene-transfer reagents were examined, and iminopyridium ylides especially those based on 3,5-dichloropyridine turned out to be highly effective. With activated alkynes such as N-alkynyloxazolidinones as substrates, α-imino gold carbene intermediates were readily generated and underwent facile 1,2-C-H insertions, leading to α, β-unsaturated amidines in mostly good yields. Further studies to expand the scope to normal alkynes will be pursued in due course.
Supplementary Material
Figure 1.
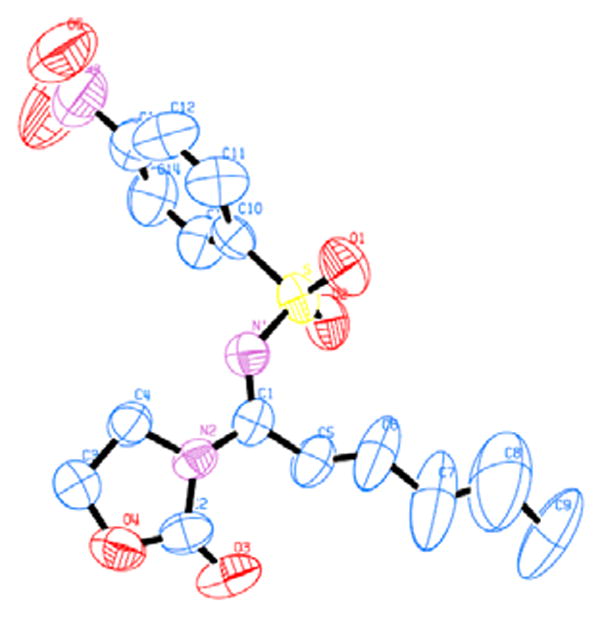
ORTEP drawing of E-14j.
Acknowledgments
The authors thank NIH and UCSB for general financial support and Dr. Guang Wu for his kind help with X-ray crystallography. LZ is an Alfred P. Sloan fellow.
Footnotes
Supporting Information Available Experimental procedures, compound characterization and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Li C-W, Pati K, Lin G-Y, Sohel SMA, Hung H-H, Liu R-S. Angew Chem, Int Ed. 2010 doi: 10.1002/anie.201004647. [DOI] [PubMed] [Google Scholar]; (b) Cui L, Zhang G, Peng Y, Zhang L. Org Lett. 2009;11:1225–1228. doi: 10.1021/ol900027h. [DOI] [PubMed] [Google Scholar]; (c) Cui L, Ye L, Zhang L. Chem Commun. 2010;46:3351–3353. doi: 10.1039/c001314e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Shapiro ND, Toste FD. J Am Chem Soc. 2007;129:4160–4161. doi: 10.1021/ja070789e. [DOI] [PubMed] [Google Scholar]; (b) Li G, Zhang L. Angew Chem, Int Ed. 2007;46:5156–5159. doi: 10.1002/anie.200701449. [DOI] [PubMed] [Google Scholar]; (c) Davies PW, Albrecht SJC. Angew Chem, Int Ed. 2009;48:8372–8375. doi: 10.1002/anie.200904309. [DOI] [PubMed] [Google Scholar]; (d) Davies PW. 2010;82:1537–1544. [Google Scholar]
- 3.Cui L, Peng Y, Zhang L. J Am Chem Soc. 2009;131:8394–8395. doi: 10.1021/ja903531g. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yeom HS, Lee Y, Lee JE, Shin S. Org Biomol Chem. 2009;7:4744–4752. doi: 10.1039/b910757f. [DOI] [PubMed] [Google Scholar]; (b) Yeom HS, Lee Y, Jeong J, So E, Hwang S, Lee JE, Lee SS, Shin S. Angew Chem, Int Ed. 2010;49:1611–1614. doi: 10.1002/anie.200906346. [DOI] [PubMed] [Google Scholar]; (c) Yeom HS, Lee JE, Shin S. Angew Chem, Int Ed. 2008;47:7040–7043. doi: 10.1002/anie.200802802. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lin GY, Li CW, Hung SH, Liu RS. Org Lett. 2008;10:5059–5062. doi: 10.1021/ol802047g. [DOI] [PubMed] [Google Scholar]; (b) Li CW, Lin GY, Liu RS. Chem Eur J. 2010;16:5803–5811. doi: 10.1002/chem.201000009. [DOI] [PubMed] [Google Scholar]; (c) Hashmi ASK, Buehrle M, Salathe R, Bats JW. Adv Synth Catal. 2008;350:2059–2064. [Google Scholar]
- 6.Cuenca AB, Montserrai S, Hossain KM, Mancha G, Lledos A, Medio-Simon M, Ujaque G, Asensio G. Org Lett. 2009;11:4906–4909. doi: 10.1021/ol9020578. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ye L, Cui L, Zhang G, Zhang L. J Am Chem Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye L, He W, Zhang L. J Am Chem Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu B, Li C, Zhang L. J Am Chem Soc. 2010;132:14070–14072. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Gorin DJ, Davis NR, Toste FD. J Am Chem Soc. 2005;127:11260–11261. doi: 10.1021/ja053804t. [DOI] [PubMed] [Google Scholar]; (b) Hiroya K, Matsumoto S, Ashikawa M, Ogiwara K, Sakamoto T. Org Lett. 2006;8:5349–5352. doi: 10.1021/ol062249c. [DOI] [PubMed] [Google Scholar]
- 9.(a) Li Z, Ding X, He C. J Org Chem. 2006;71:5876–5880. doi: 10.1021/jo060016t. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Capretto DA, Rahaman RO, He C. J Am Chem Soc. 2007;129:12058–12059. doi: 10.1021/ja0724137. [DOI] [PubMed] [Google Scholar]
- 10.(a) Fantauzzi S, Caselli A, Gallo E. Dalton Trans. 2009:5434–5443. doi: 10.1039/b902929j. [DOI] [PubMed] [Google Scholar]; (b) Diaz-Requejo MM, Perez PJ. Chem Rev. 2008;108:3379–3394. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]; (c) Mueller P, Fruit C. Chem Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 11.(a) Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J Am Chem Soc. 2008;130:14972–14974. doi: 10.1021/ja805079v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J Am Chem Soc. 2009;131:18034–18035. doi: 10.1021/ja908075u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chuprakov S, Hwang FW, Gevorgyan V. Angew Chem, Int Ed. 2007;46:4757–4759. doi: 10.1002/anie.200700804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desikan V, Liu Y, Toscano JP, Jenks WS. J Org Chem. 2008;73:4398–4414. doi: 10.1021/jo702654q. [DOI] [PubMed] [Google Scholar]
- 13.Chen CS, Tan CM, Huang CH, Chang LC, Wang JP, Cheng FC, Chern JW. Bioorg Med Chem. 2010;18:597–604. doi: 10.1016/j.bmc.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Macikenas D, Skrzypczak-Jankun E, Protasiewicz JD. J Am Chem Soc. 1999;121:7164–7165. [Google Scholar]
- 15.Gangapuram M, Redda KK. J Heterocyclic Chem. 2009;46:309–316. doi: 10.1002/jhet.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Davies PW, Cremonesi A, Martin N. Chem Commun. 2011;47:379–381. doi: 10.1039/c0cc02736g. [DOI] [PubMed] [Google Scholar]; (b) Couty S, Meyer C, Cossy J. Angew Chem, Int Ed. 2006;45:6726–6730. doi: 10.1002/anie.200602270. [DOI] [PubMed] [Google Scholar]; (c) Evano G, Coste A, Jouvin K. Angew Chem, Int Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817. [DOI] [PubMed] [Google Scholar]; (d) De KKA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem Rev. 2010;110:5064–5106. doi: 10.1021/cr100003s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Kohnen AL, Dunetz JR, Danheiser RL. Org Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]; (b) Sagamanova IK, Kurtz KCM, Hsung RP. Org Synth. 2007;84:359–367. [Google Scholar]; (c) Coste A, Couty F, Evano G. Org Synth. 2010;87:231–244. [Google Scholar]
-
18.The product was formed likely via the following sequence:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.