

Abstract
A series of α-ketooxazoles containing conformational constraints in the C2 acyl side chain of 2 (OL-135) were examined as inhibitors of fatty acid amide hydrolase (FAAH). Only one of the two possible enantiomers displayed potent FAAH inhibition (S vs R enantiomer), and their potency is comparable or improved relative to 2, indicating that the conformational restriction in the C2 acyl side chain is achievable. A cocrystal X-ray structure of the -ketoheterocycle 12 bound to a humanized variant of rat FAAH revealed its binding details, confirmed that the (S)-enantiomer is the bound active inhibitor, shed light on the origin of the enantiomeric selectivity, and confirmed that the catalytic Ser241 is covalently bound to the electrophilic carbonyl as a deprotonated hemiketal. Preliminary in vivo characterization of the inhibitors 12 and 14 is reported demonstrating that they raise brain anandamide levels following either intraperitoneal (i.p.) or oral (p.o.) administration indicative of effective in vivo FAAH inhibition. Significantly, the oral administration of 12 caused dramatic accumulation of anandamide in the brain, with peak levels achieved between 1.5–3 h and these elevations were maintained over 9 h. Additional studies of these two representative members of the series (12 and 14) in models of thermal hyperalgesia and neuropathic pain are reported, including the demonstration that 12 administered orally significantly attenuated mechanical (>6 h) and cold (>9 h) allodynia for sustained periods consistent with its long acting effects in raising the endogenous concentration of anandamide.
Introduction
Fatty acid amide hydrolase (FAAH)1,2 serves as the catabolic regulator of several endogenous lipid amides3–6 including anandamide (1a)7–10 and oleamide (1b),11–13 Figure 1. Its distribution is consistent with its role in hydrolyzing and regulating such signaling fatty acid amides6 at their sites of action.3 Although it is a member of the amidase signature family of serine hydrolases for which there are a number of prokaryotic enzymes, it is the only well characterized mammalian enzyme bearing the family’s unusual Ser–Ser–Lys catalytic triad.14–17
Figure 1.

Substrates of FAAH.
Due to the therapeutic potential of inhibiting FAAH18–20 for the treatment of pain,21–23 inflammation,24 or sleep disorders,13,25 there has been growing interest in the development of selective inhibitors of the enzyme.26 Early studies following the initial characterization of the enzyme led to the discovery that the endogenous sleep-inducing molecule 2-octyl α-bromoacetoacetate is an effective FAAH inhibitor,27 and the disclosure of a series of nonselective reversible inhibitors bearing an electrophilic ketone (e.g., trifluoromethyl ketone-based inhibitors)28–31 and irreversible inhibitors32–37 (e.g., fluorophosphonates and sulfonyl fluorides) that were used to characterize the enzyme as a serine hydrolase. Subsequent studies have defined two major classes of inhibitors that provide opportunities for the development of inhibitors with greater therapeutic potential. One class is the reactive aryl carbamates and ureas38–50 that irreversibly acylate a FAAH active site serine49 and that have been shown to exhibit anxiolytic activity38 and produce antinociceptive effects.39 A second class is the -ketoheterocycle-based inhibitors51–60 that bind to FAAH by reversible hemiketal formation with an active site serine. Many of these latter reversible competitive inhibitors have been shown to be potent and selective for FAAH versus other mammalian serine hydrolases, and members of this class have been shown to be efficacious in preclinical animal models of pain.61
In these latter studies, 2 (OL-135)53 emerged as an important lead inhibitor for further study. It is a potent (Ki = 4.7 nM)53 and selective (>60–300 fold)31 FAAH inhibitor that induces analgesia and increases endogenous anandamide levels.61 It exhibits antinociceptive and anti-inflammatory activity in a range of preclinical animal models that include the tail flick assay, hot plate assay, formalin test of noxious chemical pain (1st and 2nd phase), the mild thermal injury (MTI) model of peripheral pain, the spinal nerve ligation (SNL) and chronic constriction injury (CCI) models of neuropathic pain, and an inflammatory model of pruritus with efficacies that match or exceed those of morphine (@1–3 mg/kg in MTI/SNL), ibuprofen (@100 mg/kg in MTI), or gabapentin (@500 mg/kg in SNL) and at administered doses (10–20 mg/kg, i.p.) that approach or are below those of such common pain or anti-inflammatory medications.61 The compound lacks significant offsite target activity (Cerep assay profiling), does not bind cannabinoid (CB1 or CB2) or vanilloid receptors, and does not significantly inhibit common P450 metabolism enzymes or the human ether-a-go-go related gene (hERG). The observed antinociceptive effects are not associated with concomitant respiratory depression or tolerance/dependence characteristic of opioid administration, nor with increased feeding or decreased locomotor coordination frequently observed with CB agonists.61 Significantly, 2 does not produce antinociception in FAAH knockout mice61 establishing that FAAH is the relevant target responsible for the observed in vivo effects.
Consequently, we have conducted a series of systematic structure–activity relationship (SAR) studies on 2 exploring the 4- and 5-position of the central oxazole,53,55,56,60 the C2 acyl side chain,53,57,58 and the central heterocycle,54,59 each of which was found to independently impact inhibitor potency or selectivity, Figure 2.62 Herein, we report results of studies examining candidate inhibitors containing further conformational constraints in the C2 acyl side chain of 2 and related inhibitors, the X-ray crystal structure characterization of a prototypical inhibitor in the series bound to the enzyme, and preliminary in vivo characterization of two representative inhibitors in this series.
Figure 2.

Progression of the inhibitor series.
Chemistry
The general method for the synthesis of the oxazole-based inhibitors bearing a C5 aryl substituent and containing the additional conformational restriction in the C2 acyl side chain is shown in Scheme 1. The inhibitors contain a tetrahydronaphthalene or indane central to the C2 acyl chain, a newly introduced chiral center adjacent to the electrophilic carbonyl, and pendant terminal aryl groups that were varied in our studies. Expectations are that the introduction of further conformational restraints in the C2 acyl chain would improve oral bioavailability of the candidate inhibitors, potentially maintain or enhance FAAH inhibitory activity and selectivity, and sterically further reduce potential competitive metabolic or off target reactivity of the electrophilic carbonyl.
Scheme 1.

Introduction of a methyl ester α to the ketone of the commercially available 6-methoxytetralone or 6-methoxyindanone proceeded as reported using dimethylcarbonate and NaH, and was followed by hydrogenation removal of the cyclic ketone using H2 and Pd/C.63 Simultaneous deprotection of the aryl methyl ether and the methyl ester with aqueous HBr in HOAc yielded the phenolic carboxylic acid. Esterification of the carboxylic acid using H2SO4 and MeOH afforded the advanced phenol intermediates63,64 on which the varied aryl substituent was added. A Suzuki coupling with phenylboronic acid via the corresponding triflate intermediate, a Mitsunobu alkylation65 of the phenol with benzyl alcohol and Ph3P–diethyl azodicarboxylate (DEAD), and a modified Ullmann reaction66 of the phenol with phenylboronic acid yielded the corresponding 6-phenyl, 6-benzyloxy, and 6-phenoxy-1,2,3,4-tetrahydronapthalenes and indanes, respectively. Reduction of the methyl ester to the primary alcohol using LiAlH4 followed by oxidation with Dess–Martin periodinane67 gave the corresponding aldehyde. Vedejs oxazole metalation68 and condensation with the various C2 side chain aldehydes was followed by tert-butyldimethylsilyl (TBS) ether protection of the resulting alcohols. Selective C5-oxazole lithiation69 of these intermediates followed by treatment with Bu3SnCl afforded the corresponding C5 tributylstannanes. Stille coupling70 of the stannane intermediates with pyridine halides produced the C5-substituted oxazoles, which could be readily converted to the corresponding ketones by TBS ether deprotection (Bu4NF) and oxidation of the liberated alcohol with Dess–Martin periodinane. These candidate inhibitors were separated into their two enantiomers by resolution on a semipreparative Chiracel OD or AD column. The candidate inhibitors containing a methyl ester were then converted to their corresponding carboxylic acid using (CH3)3SnOH.71 This reagent and the conditions employed resulted in minimal racemization of the chiral center whereas the conventional use of LiOH (1 equiv, THF/H2O 3:2, 25 °C) resulted in more extensive racemization.
The synthesis of candidate inhibitors that bear a non-aromatic oxazole C5-substituent is summarized in Scheme 2. Following oxazole C5-lithiation, treatment with Mander’s reagent (NCCO2Me) provided the corresponding C5-substituted oxazoles bearing a methoxycarbonyl group in good conversions. In each case, deprotection of the TBS ether followed by Dess–Martin periodinane oxidation of the liberated alcohol yielded the corresponding α-ketooxazole. The methyl esters were also converted to the corresponding carboxamides by treatment with NH3–CH3OH and the carboxamides were dehydrated with trifluoroacetic anhydride (TFAA) and pyridine to provide the C5 nitriles that were converted to the α-ketooxazoles as well. These derivatives were separated into their two enantiomers by resolution on a semipreparative Chiracel OD or AD column.
Scheme 2.

Enzyme Assay
Enzyme assays were performed at 20–23 °C with purified recombinant rat FAAH expressed in Escherichia coli72 or with solubilized COS-7 membrane extracts from cells transiently transfected with human FAAH cDNA2 (where specifically indicated) in a buffer of 125 mM Tris/1 mM EDTA/0.2% glycerol/0.02% Triton X-100/0.4 mM Hepes, pH 9.0. The initial rates of hydrolysis (≤10–20% reaction) were monitored using enzyme concentrations (typically 1 nM) at least three times below the measured Ki by following the breakdown of 14C-oleamide, and Ki values (standard deviations are provided in the Supporting Information tables) were established as described (Dixon plot).29 Lineweaver–Burk analysis of 12 and 14 established that they behave as reversible, competitive inhibitors analogous to 253 and related inhibitors (see experimental).
Results and Discussion
In early work, some of the most potent inhibitors that emerged contained conformational constraints in the flexible C2 acyl side chain of 2 removing many of its rotatable bonds and introducing additional π-unsaturation. This portion of the inhibitors binds in a hydrophobic channel of the FAAH active site reserved for the unsaturated lipid chain of the fatty acid amide substrates. Additional restriction in this C2 acyl side chain may contribute to an enhancement in binding affinity while simultaneously improving the drug-like characteristics of the candidate inhibitors. Three improved C2 side chains previously identified57 (Figure 3) were now incorporated into the candidate inhibitors that contain a 1,2,3,4-tetrahydronaphthalene or indane core and were combined with representative or optimized C5 oxazole substituents that were found to impact inhibitor potency, selectivity, and physical properties.
Figure 3.

Prior constraints in the C2 acyl side chain and impact of the terminal aryl substituent.57
Tetrahydronaphthalene Series
The series examined include the 6-phenyl, 6-phenoxy, and 6-benzyloxy-1,2,3,4-tetrahydronaphthalene C2 acyl side chains combined with a set of representative oxazole C5-substituents. For each derivative, the racemic mixture as well as the pure enantiomers were prepared and examined, but only the results of the examination of the individual enantiomers are reported in Figure 4. In each instance, it was the slower eluting, second enantiomer obtained from chromatographic resolution (Chiralcel OD or AD) that was found to be more potent and, as detailed later, it was established to be the (S)-enantiomer. Thus, the inhibitors displayed a consistently more potent activity for the assigned (S)-enantiomer that was of a magnitude and range (10–400 fold, avg = 70-fold) that suggests the observed activity for the less active (R)-enantiomer is not distinguishable from that potentially derived from contaminant (S)-enantiomer in the assayed samples derived directly from the chromatographic resolutions. At this level of enantiomeric purity achieved by the chromatographic resolutions, we can confidently state that the reported activity of the active (S)-enantiomers is accurate and unaffected by any trace amounts of contaminate (R)-enantiomer (≥98% ee), whereas that of the less interesting (less active) or inactive (R)-enantiomer is not easy to distinguish from the potential presence of contaminant (S)-enantiomer. Moreover and from these and later studies (see Scheme 3), we can establish that these samples do not significantly epimerize under the conditions of the FAAH assay (pH = 9). For each oxazole C5-substituent, the potency of the (S)-enantiomers of the C2 acyl side chain aryl substituents consistently followed the order phenoxy > benzyloxy > phenyl indicating that the added conformational constraints in the C2 acyl linking chain has subtly reordered this aryl substituent preference (compare Figure 3). Here the distinctions between a phenoxy and benzyloxy substituent are small (typically 1.4–4 fold), whereas the differences with the less active and more rigid phenyl substituent are larger and more easily distinguished. Interestingly and unlike observations made with 2 itself, the impact of the oxazole C5-substituent on the activity in each series is much more modest, although it is most significant in the less active biaryl series. However, the trends for the oxazole C5-substituent, but not their magnitude, are maintained in these series and most significant is the enhanced potency observed with inhibitors that lack the C5 substituent. This suggests that beneficial enhancements in binding affinity in this series are gained by the C2 acyl side conformational restriction and the added hydrophobic interactions of the tetrahydronaphthalene.
Figure 4.

FAAH inhibitors with 1,2,3,4-tetrahydronaphthalene C2 acyl side chain, Ki (nM).
Scheme 3.
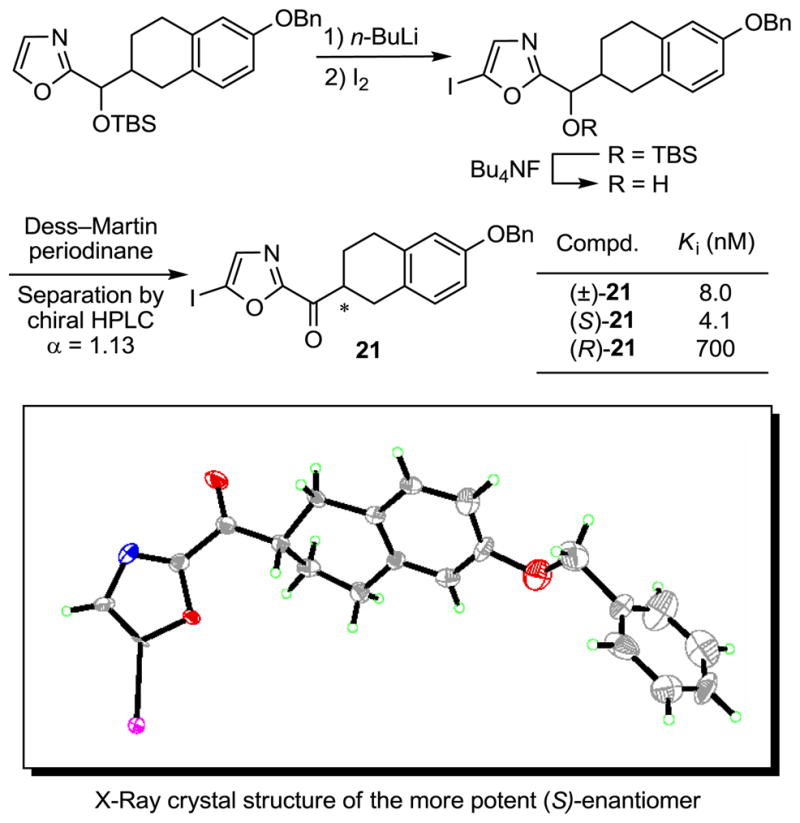
In order to unambiguously establish the absolute stereochemical assignment for the active enantiomer, an inhibitor in 6-benzyloxy-1,2,3,4-tetrahydronaphthalene series was prepared with an iodo substituent at the oxazole C5 position following the general procedure described earlier, Scheme 3. The racemic mixture and the pure enantiomers were tested for FAAH inhibition and one enantiomer was found to be ca. 200-fold more active. Its structure and absolute stereochemistry were established with an X-ray crystal structure determination73 indicating that the most potent enantiomer is the (S)-enantiomer, Scheme 3. With 21, the activity of the less potent (R)-enantiomer cannot be distinguished from potential contaminate (S)-enantiomer (0.5%) in the assay sample, and this comparison indicates that the active (S)-enantiomers may be ≥200-fold more active than the corresponding (R)-enantiomers.
Indane Series
An analogous, but smaller series of related inhibitors containing the 5-phenyl, 5-phenoxy, and 5-benzyloxyindane acyl chains were examined, Figure 5.
Figure 5.

FAAH inhibitors with indane C2 acyl side chain, Ki (nM).
This series exhibited an analogous enantiomeric selectivity with the tentatively assigned (S)-enantiomers being on average 60-fold (10–320 fold, avg = 60-fold) more potent than the corresponding (R)-enantiomers, and they approached or matched the potency of the corresponding inhibitors bearing the 1,2,3,4-tetrahydronaphthalene C2 acyl chain core. Although the number of comparisons is more limited in this series, the expected significant improvement in potency with the introduction of the oxazole C5 pyridyl substitutent (avg. 9-fold enhancement) is observed, and there appears to be less of a distinction between choices of acyl side chain aryl substituent in this series. Interestingly, and again with the caveat that the number of comparisons is limited, this latter observation results from small improvements in the potency of the inhibitors bearing the acyl chain C5 phenyl and benzyloxy substituents, whereas those bearing the C5 phenoxy substituent appear to be reduced relative to the corresponding inhibitors bearing a 1,2,3,4-tetrahydronaphthalene C2 acyl chain core. Since all are potent inhibitors of FAAH, this suggests that there are minor reorientation differences in the binding of the C2 acyl side chains in the two series (tetrahydronaphthalene vs indane) that may subtly impact the inhibitor affinity.
The Electrophilic Carbonyl
In several instances, the diastereomeric mixture of racemic alcohols used as the penultimate precursors to the α-ketoheterocycles 15, 19, 30, and 32 were also assessed for FAAH inhibition and all were found to be inactive (Ki > 10 μM) confirming the importance of the electrophilic carbonyl.
Additional Derivatives
A set of additional derivatives in the 6-phenoxy-1,2,3,4-tetrahydronaphthalene series were examined that incorporate a 1,3,4-oxadiazole as the central activating heterocycle. In earlier studies, comprehensive systematic changes in the central activating heterocycle of 2 were examined and found to significantly influence the inhibitor activity54,59 with the 1,3,4-oxadiazole derivatives providing extraordinarily potent inhibitors. Accordingly, the preparation of a representative small series of 1,3,4-oxadiazoles containing the 6-phenoxy-1,2,3,4-tetrahydronaphthalene side chain was conducted and is illustrated in Scheme 4. Reaction of the C2 side chain aldehyde with KCN afforded the corresponding cyanohydrin and was followed by conversion of the nitrile to the methyl ester. TBS protection of the alcohol followed by saponification of the methyl ester yielded the carboxylic acid that was condensed with a series of hydrazides in a reaction promoted by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI) to provide the diacyl hydrazide intermediates. These intermediates were cyclized to the corresponding 1,3,4-oxadiazoles upon treatment with p-toluenesulfonyl chloride (TsCl) and Et3N. The desired α-keto-1,3,4-oxadiazoles were obtained after TBS ether deprotection (Bu4NF or TASF) and oxidation of the liberated alcohol with Dess–Martin periodinane. These derivatives wereseparated into their two enantiomers by resolution on a semipreparative Chiracel OD column.
Scheme 4.

Finally and for comparison purposes, a series of derivatives were prepared and examined that lack an aryl substituent on the acyl side chain 1,2,3,4-tetrahydronaphthalene or indane core. Their synthesis entailed Vedejs C2-lithiation of oxazole followed by condensation of the corresponding aldehyde and TBS protection of the resulting alcohol. Selective oxazole C5-lithiation (n-BuLi) followed by treatment with Bu3SnCl afforded the corresponding tributylstannane intermediates. Subsequent Stille coupling with 2-bromopyridine produced the C5 substituted oxazoles, which were converted to the corresponding ketones by TBS ether deprotection (Bu4NF) and oxidation of the liberated alcohols using Dess–Martin periodinane, Scheme 5. The 1,2,3,4-tetrahydronaphthalene derivatives were separated into their two enantiomers by resolution on a semipreparative Chiracel OD column. The indane derivatives are meso compounds and no resolution is required.
Scheme 5.

The results of the examination of these derivatives are summarized in Figure 6. The incorporation of the activating 1,3,4-oxadiazole heterocycle in the 6-phenoxy-1,2,3,4-tetrahydronaphthalene series further enhanced the activity of the candidate inhibitors providing extraordinarily potent inhibitors. This was most evident with 34, lacking a 1,3,4-oxadiazole C5-substituent, that exhibited a Ki of 500 pM (0.5 nM) for the more potent (S)-enantiomer. Analogous to prior observations,54,59 this represents a 4–5 fold improvement in potency relative to the corresponding oxazole 9. In this series, the measured differences between the (S)- and (R)-enantiomers proved modest, indicating that a well-precedented74 enhanced racemization of the 1,3,4-oxadiazole most likely occurs under the conditions (pH 9) and time course of the assay because of the stronger electron-withdrawing properties of the activating heterocycle.
Figure 6.

Additional FAAH inhibitors, Ki (nM).
For the candidate inhibitors without an acyl side chain aryl substituent, a substantial loss in potency (from nM to μM range) is observed highlighting the importance of its anchoring interaction within the enzyme active site, Figure 6. Notably, 38 (2-fold, Ki = 430 nM) and 40 (5-fold, Ki = 1.1 μM) approach the activity of the FAAH inhibitor lacking the conformational constraints (Ki = 200 nM, Figure 3) indicating that their introduction may subtly, but not seriously, affect their active site affinity. More interestingly, the enantiomer distinctions with 37 were modest (5.1 vs 12 μM, ca. 2-fold). As revealed in the X-ray structure of 12 bound to FAAH, it is the spatial relationship of the dominant anchoring C6-phenoxy substituent of 12 with the chiral center that imposes the enantiomeric selectivity observed in the tetrahydronaphthalene or indane series. Additionally, the introduction of the oxazole C5 pyridyl substituent increased the potency 7–12 fold compared to their unsubstituted counterparts in this series (38 vs 37 and 40 vs 39), analogous to prior observations,53 suggesting the hydrogen-bond capability of the weakly basic pyridyl substitutent is responsible for their enhanced affinities.
Inhibition of Recombinant Human FAAH
Rat and human FAAH are very homologous (82% sequence identity),2 exhibit near identical substrate selectivity and inhibitor sensitivity in our studies with the α-ketoheterocycles disclosed to date, and embody an identical amidase signature sequence, suggesting the observations made with rat FAAH (rFAAH) would be analogous to those made with human FAAH (hFAAH). Consequently, the active (S)-enantiomers of two representative inhibitors, 12 and 14, were examined with hFAAH and the results are summarized in Figure 7. As observed with 2, no significant distinction was observed for 12, whereas 14 exhibited a modest 4-fold reduction in activity against the human enzyme.
Figure 7.
Inhibition of human versus rat FAAH (hFAAH vs rFAAH), Ki (nM).
X-ray Structure of the Active Enantiomer of Inhibitor 12 Bound to FAAH
The X-ray structure of the active enantiomer of the α-ketooxazole 12 bound to h/rFAAH75 was solved at 1.9 Å resolution and the data processing and refinement statistics are summarized in Table S1 (Supporting Information). Compound 12 was found covalently attached to the catalytic Ser241 residue through its electrophilic carbonyl bound as a deprotonated hemiketal mimicking the enzymatic tetrahedral intermediate, a hallmark of the α-ketoheterocycle class of FAAH inhibitors.76,77 The electron density of the bound inhibitor and its excellent resolution provide an unambiguous depiction of the chiral center of 12 confirming that it is the (S)-enantiomer, Figure 8. A structural overlay of the X-ray crystal structure of 2, whose interactions with FAAH have been described in detail,76 and 12 is provided in Figure 9. The terminal phenyl ring of the acyl side chain of 12 is positioned in the same plane and only slightly shifted (0.6 Å) from the analogous group of 2, Figure 9. This phenyl group serves as a key anchoring interaction with FAAH, which adopts an active site conformation that leads to a broadened and open membrane access channel with truncation of the acyl chain-binding pocket.76 Beautifully, the α, β, and γ carbons of the intervening flexible hydrophobic linker in the acyl side chain of 2, which adopt a gauche conformation, nearly superimpose with C2–C4 of the tetrahydronaphthalene core of 12, Figure 9.
Figure 8.

Top: View of 12 in the binding pocket of FAAH and its interactions. Bottom: An enlarged view of the chiral center within the tetrahydronaphthalene is shown. Electron density at 1.5σ contour is shown with white meshes.
Figure 9.

Two views of the superposition of the FAAH–12 (in yellow/orange) complex with FAAH–2 complex76 (in green). The rearrangement of Phe192 and Met495 residues are shown.
The backbone of the enzyme bound to 12 did not exhibit any major conformational changes relative to previously published structures, although two residues lining the active site undergo rearrangement. The most significant change is the relocation of the side chain of the Phe192 residue that rotates and shifts away from the compound acyl chain due to the size of the tetrahydronaphthalene core of 12, Figure 9. This reorganization of the lining of the hydrophobic binding pocket, also observed in other structures,77 confirms that this residue is highly flexible and is likely a major adapter residue when binding different substrates and inhibitors. As a consequence of this rearrangement, the inhibitor oxazole C5-pyridyl substituent is pushed towards Ile238 and Leu278. In spite of this, the pyridyl nitrogen remains hydrogen-bonded to an ordered cytosolic port water molecule that in turn is hydrogen-bonded to Thr236, a feature that is conserved in all related structures analyzed to date.76,77 Interestingly, the space generated by the shift of the pyridine is compensated by the appearance of a new water molecule in the cytosolic port that had not appeared in earlier structures. It sits above the aromatic ring of the pyridine at a distance of 3.6 Å, and is coordinated, perhaps polarized, by the backbone amide of Cys269, previously identified as a key residue forming part of an anion binding site, Figure 8.77 The activating oxazole and its attached pyridine substituent are bound in the cytosolic port, adopting bound orientations and a biaxial twist (18°) analogous to those found with 2 (15°) and related inhibitors. However, the unusual Ser217 OH hydrogen bond to the π-system of activating oxazole observed with 2 is now replaced with a hydrogen bond directly from Ser217 OH to the oxazole nitrogen in a fashion observed for histidine in serine proteases,78 Figure 9. This change is not derived from a significant relocation of the Ser217 residue, but rather is a result of the reorientation of activating heterocycle (oxazole) and the displacement of its attached C5-pyridyl substituent. These offsetting changes, which still provide inhibitors more potent than 2, may account for the reason that the introduction of the pyridine substituent did not improve affinity in this series (12 vs 9 and 18 vs 15) although it still enhances their selectivity for FAAH.31 An additional change unique to this structure is the rearrangement of the distal rotamer of Met495, which now points towards the inhibitor and sits over the acyl chain linker aromatic ring at a distance of 4.4 Å, Figure 9.
In order to establish the origin of the inhibitor enantiomer selectivity extrapolated from the crystal structure, molecular modeling79 studies were performed and used to calculate the relative energies involved in the binding of the two enantiomers. Covalent docking (ICM, Molsoft Inc.) of 12 with Ser241 and Monte Carlo simulations for sampling bound conformations were used to compare the two enantiomers of 12. As empirically predicted from the crystal structure analysis and established in the enzymatic assays, the (R)-enantiomer binding is destabilized relative to the (S)-enantiomer suffering from a large penalty in the van der Waals term (steric clash). Analysis of the contributions of single amino acids to the total binding energy established that this coincides predominately with a van der Waals repulsion with Ser193. When bound with the same orientation as (S)-12 and in order to maintain phenoxy binding at the terminus of the truncated acyl chain binding pocket, (R)-12 suffers a destabilizing steric clash with Ser193 proximal to the chiral center (Figure 8: bottom). If (R)-12 is flipped 180° to avoid this steric interaction and arrange the tetrahydronaphthalene core in a disposition similar to (S)-12 (chiral C2-H down as found in Figure 8: bottom), the terminal phenoxy group is no longer positioned to bind in this key region of the acyl chain binding pocket. Thus, it is not surprising that while the two enantiomers of 37 and 38 that lack the phenoxy group bind FAAH with comparable, albeit weak affinities, the two enantiomers of 12 and related inhibitors exhibit much more pronounced differences.
Preliminary In Vivo Characterization
In initial efforts to establish in vivo inhibition of FAAH and the potential pharmacological effects, a select set of the conformationally restricted inhibitors (12, 14, 27, and 29) were examined alongside 2 for their ability to increase the endogenous levels of a series of lipid amide signaling molecules in the brain (CNS effect) and liver (peripheral effect). This includes monitoring the effects of the inhibitors on the endogenous levels of the FAAH substrates anandamide (N-arachidonoyl ethanolamine), N-oleoyl ethanolamine (OEA), and N-palmitoyl ethanolamine (PEA), as well as the key lipids 2-arachidonoylglycerol (2-AG) and arachidonic acid that are not endogenous substrates for FAAH. Notably, it is the increase in endogenous levels of anandamide and its subsequent action at CB1 and CB2 receptors that are thought to be responsible for the antinociceptive and anti-inflammatory effects of FAAH inhibitors although both PEA and 2-AG are also known to exhibit anti-inflammatory and CB receptor-mediated antinociceptive effects, respectively. Pharmacological effects were initially established 1 h after intraperitoneal (i.p.) administration of 30 mg/kg inhibitor in a single mouse for the initial screen and the results are summarized in Figure 10. The inhibitors 12 and 27 increased the endogenous levels of key lipid amides thought to be responsible for antinociceptive effects (anandamide, PEA) without impacting the endogenous levels of 2-AG, consistent with selective FAAH inhibition in vivo. They also had a minimal impact on reducing the endogenous levels of arachidonic acid, the hydrolysis product of anandamide and a key proinflammatory fatty acid.80 The effects of 12 (and presumably 27) were observed both in the brain (CNS) and liver (peripheral) matching or exceeding the effects of 2, whereas the impact of the more polar inhibitor 14 (and presumably 29) was principally seen peripherally (liver) with more muted effects in the CNS (brain).
Figure 10.

Initial screen for the effects of FAAH inhibitors on brain and liver lipid levels following in vivo inhibitor treatment; n = 1 per group.
Given the results of this initial screen, both 12 and 14 were examined side-by-side not only with i.p. (30 mg/kg), but also oral (p.o., 50 mg/kg) dosing with 3 mice per group to provide the results presented in Figure 11. Significantly, the oral dosing matched and even improved on the results observed with i.p. administration.
Figure 11.

Brain lipid amide levels following oral versus intraperitoneal dosing.
Given the oral activity established in the PD model with 12, a follow up dose- and time-dependent study of its effects on the endogenous brain levels of anandamide, PEA and OEA was conducted alongside 2 and the irreversible FAAH inhibitor 41 (PF-3845).42c In the first of these studies, each comparison standard (2 and 41) was administered orally at 50 mg/kg, 12 was administered orally at 10, 25, and 50 mg/kg, and the resulting impact on the endogenous brain levels of fatty acid amides was measured at a single time point (1 h). Compound 12 increased the levels of anandamide (12–13 fold at 25–50 mg/kg) in a dose dependent manner more efficaciously and more potently than 2 (11-fold at 50 mg/kg), but not as effectively as the irreversible FAAH inhibitor 41, Figure 12a. In contrast, the increases in the brain levels of PEA and OEA observed with 12 at 1 h were substantial (5.5–7.8 fold) and essentially the same at all three administered doses. These increases exceeded the effects observed with 2 at 50 mg/kg and matched the effects of the irreversible inhibitor 41 administered at 50 mg/kg, Figure 12b. These data are consistent with reports showing that partial blockade of FAAH can cause elevations in PEA and OEA, but that >90% FAAH blockade is required to elevate anandamide levels.42c In the second of the studies, 12 was administered to mice (50 mg/kg, p.o.) and the animals were sacrificed at various time points up to 9 h post-administration. Brains from these mice were analyzed for levels of anandamide and the additional FAAH substrates PEA and OEA. Administration of 12 caused dramatic accumulations of all three N-acylethanolamines in brain, with peak levels of anandamide achieved between 1.5–3 h (Figure 12c and d). Remarkably, the elevations in these lipids were maintained over the 9 h time course, similar to the time course reported for the irreversible FAAH inhibitor 41 and significantly longer than that reported for the irreversible carbamate inhibitor URB597.42c These time course data suggest that the reversible inhibitor 12 remains in the brain after its initial oral dosing at sufficiently high concentrations to completely inhibit FAAH (>90%) for a prolonged period.
Figure 12.

Dose (panels a and b, analysis performed 1 h post-treatment) and time (panels c and d at 50 mg/kg 12) dependent impact on brain lipid amide levels following oral dosing of 12. AEA = anandamide, PEA = N-palmitoyl ethanolamine, OEA = N-oleoyl ethanolamine.
An additional feature of the α-ketoheterocycle FAAH inhibitors that we have not disclosed previously is that they rapidly establish an equilibirum mixture of active ketone and reduced alcohol in vivo. Although this is likely a general feature of the entire class of α-ketoheterocycles that have been examined as enzyme inhibitors,81 we are not aware of its discussion elsewhere. Consequently, triaging screens for compound development using rat or human liver microsomes (rlm and hlm) may often misleadingly suggest rapid metabolic reduction that is not representative of the true metabolic fate of such candidate inhibitors (e.g., rlm and hlm t1/2 for 2 and 18 are 2–4 min and 12–15 min, respectively). Rather, we have found that they are subject to competitive reduction/reoxidation metabolism that sets up a steady-state equilibrium between the two states (ketone/alcohol) with the true in vivo fate of the candidate inhibitors being determined by other features of the molecule. For 2, this steady state equilibrium is established within 15 min and was found to be 1/3 (ketone/alcohol) independent of the means of administration.82 For 12, we also measured the compound levels in the brain following its oral administration (50 mg/kg) and could detect both the parent ketone and the reduced alcohol in inhibitor-treated, but not vehicle-treated, mice. The relative ratio of ketone/alcohol was 3–3.5/1 at early stages following compound administration (0.5–1 h) and slowly equilibrated to 1–1.5/1 at the longer times following administration (1.5–9 h) where both the ratio and brain levels persisted. Although preliminary, the results seem to indicate that the added conformational constraints in the C2 acyl chain and the increased steric hindrance surrounding the electrophilic ketone both slow the rate of equilibrationand improve the ketone/alcohol rati oin vivo.
Concurrent with these studies, the two prototypical inhibitors 12 (not shown) and 14 were examined for antinociceptive activity in a preclinical model of acute thermal pain (tail flick at 52 °C) in mice alongside 2 and morphine (i.v. administration). The results of the dose-response studies for 14, the more soluble of the two inhibitors, are summarized in Figure 13 alongside those of morphine. Compound 14 produced the same maximal effect (efficacy) observed with morphine requiring only a 2-fold higher dose, reached its maximal effect at 30–60 min, and exhibited a duration of effect matching that of morphine (150 min).
Figure 13.

Antinociception in the tail flick assay (52 °C) following i.v. administration of 14, 2 and morphine.
In an important extension of the studies and following the observation of the long acting in vivo effects of 12 on endogenous anandamide levels following oral administration, mice were subjected to chronic constriction injury (CCI) and tested 10 days later for signs of neuropathic pain. Inhibitor 12 administered orally (50 mg/kg) significantly attenuated mechanical allodynia [F(1,51) = 17.1; p < 0.001; Figure 14A] and cold allodynia [F(1–51) = 26.4; p < 0.0001; Figure 14B], in paws ipsilateral to CCI surgery. In the control paws of the same mice, 12 had no effect on mechanical (p = 0.16) or cold allodynia (p = 0.48), indicating a lack of sedative effects. Significantly, the effects of 12 following its oral dosing were sustained, lasting >6 h in the mechanical allodynia and >9 h in the cold allodynia, consistent with its long acting effects in raising the endogenous concentration of anandamide (see Fig. 12c).
Figure 14.

FAAH inhibition by 12 significantly attenuated neuropathic pain for up to 9 hours. (A) Male C57BL/6 mice were subjected to chronic constriction injury (CCI) of the sciatic nerve and tested 10 days later for mechanical allodynia, as measured with von Frey filaments, and (B) acetone-induced cold allodynia. Inhibitor 12 (50 mg/kg, p.o.) significantly attenuated CCI-induced mechanical allodynia, as well as cold allodynia, in paws ipsilateral to CCI surgery, but had no effect in paws contralateral to CCI surgery. Circles, vehicle treatment; triangles, 12 treatment; Open shapes, control paws; filled shapes, CCI paws. Data expressed as mean ± SEM. (n = 9–10). * p < 0.05, ** p < 0.01, vs vehicle.
Finally and significantly, inhibitor 12 was also examined for cannabimimetic side effects following oral administration at a dose that provided analgesic effects (50 mg/kg). Like 2 and earlier FAAH inhibitors in this class,61 12 did not produce catalepsy, hypothermia, or hypomotility (data not shown) indicating that it does not produce THC-like effects characteristic of a classical CB receptor agonist.
Conclusions
A series of α-ketooxazoles containing conformational constraints in the C2 acyl side chain of 2 were prepared and examined as inhibitors of FAAH. Members of this new series exhibited comparable or improved enzyme inhibition potency relative to 2, indicating that the additional conformational restriction in the C2 acyl side chain is achievable and beneficial. A cocrystal X-ray structure of the prototypical -ketoheterocycle 12 bound to a humanized variant of rat FAAH confirmed that the (S)-enantiomer is the bound active inhibitor, shed light on the structural origin of the enantiomeric selectivity, and confirmed that the active site catalytic Ser241 is covalently bound to the electrophilic carbonyl, mimicking the enzymatic tetrahedral intermediate. Preliminary in vivo characterization of the prototypical inhibitors 12 and 14 in mice was reported demonstrating that they raise endogenous anandamide levels with either intraperitoneal or oral administration, and that the oral adminstration of 12 caused sustained accumulation of three major N-acylethanolamines (anandamide, OEA, and PEA) in the brain with peak levels of anandamide achieved between 1.5–3 h with elevations that were maintained over the 9 h time course of the examination. This duration of action with oral administration of 12 proved similar to that reported for the irreversible urea FAAH inhibitor 41 and significantly longer than that reported for the irreversible carbamate inhibitor URB597, suggesting that the reversible inhibitor 12 remains in the brain after its initial oral dosing at sufficiently high concentrations to completely inhibit FAAH (>90%) for a prolonged period. Preliminary in vivo studies with the two representative members of the series (12 and 14) indicate that they exhibit robust analgesic activity in mouse models of thermal hyperalgesia and neuropathic pain including the demonstration that oral administration of 12 (50 mg/kg) significantly attenuated both mechanical (>6 h) and cold (>9 h) allodynia for sustained periods consistent with its long acting effects in raising the endogenous concentration of anandamide.
Experimental Section
General Experimental
The purity of each inhibitor (>95%) was determined on an Agilent 1100 LC/MS instrument on a ZORBAX® SB-C18, 3.5 mm, 4.6×50, a flow rate of 0.75 mL/min, detection at 220 and 254 nm, with a 10–98% acetonitrile/water/0.1% formic acid gradient and a 50–98% acetonitrile/water/0.1% formic acid gradient (see Table S2 in Supporting Information).
(6-Phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)(5-(pyridin-2-yl)oxazol-2-yl)methanone (12)
A solution of oxazole (0.226 mL, 3.44 mmol) in anhydrous THF (20 mL) was treated with BH3·THF (1 M, 3.74 mL, 3.74 mmol) and the solution was stirred at room temperature for 1 h before being cooled to −78 °C and treated with 2.16 M n-BuLi (2 mL, 4.47 mmol) dropwise. The reaction mixture was stirred at −78 °C for 40 min before a solution of 6-phenoxy-1,2,3,4-tetrahydronaphthalene-2-carboxaldehyde (870 mg, 3.44 mmol) in THF (20 mL) was added. The reaction mixture was stirred at −78 °C for 2 h before being warmed to room temperature. A 5% HOAc–EtOH solution (50 mL) was added and this mixture was stirred at room temperature for 12 h. The solvent was removed under reduced pressure and the residue was dissolved in EtOAc, washed with H2O, saturated aqueous NaHCO3 and saturated aqueous NaCl before the organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 40% EtOAc–hexanes) afforded oxazol-2-yl(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methanol (740 mg, 67%) as colorless oil: 1H NMR (CDCl3, 600 MHz) δ 7.65 (s, 1H), 7.32–7.31 (m, 2H), 7.10–6.98 (m, 5H), 6.78–6.75 (m, 2H), 4.78–4.74 (m, 1H), 2.88–2.78 (m, 4H), 2.61–2.59 (m, 0.5H), 2.34 (m, 0.5H), 2.14–2.12 (m, 0.5H), 1.80–1.77 (m, 0.5H), 1.62–1.51 (m, 1H); 13C NMR (CDCl3, 150 MHz) δ 165.3, 157.5, 154.8, 138.9, 137.9, 137.7, 130.5, 130.3, 130.2, 130.1, 129.5 (2C), 126.5, 122.8, 122.7, 118.86 (2C), 118.82, 118.49, 118.42, 116.78, 116.73, 71.2, 71.0, 39.8, 39.7, 30.8, 30.2, 29.6, 28.9, 28.8, 25.1, 24.4.
A solution of oxazol-2-yl(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methanol (400 mg, 1.24 mmol), TBSCl (450 mg, 2.98 mmol) and imidazole (421 mg, 6.2 mmol) in DMF (20 mL) was stirred at room temperature for 16 h before it was diluted with EtOAc, washed with H2O, and saturated aqueous NaCl. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 10% EtOAc–hexanes) yielded 2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)oxazole (459 mg, 85%) as a thick colorless oil: 1H NMR (CDCl3, 500 MHz) δ 7.65 (s, 1H), 7.31 (t, 2H, J = 8.5 Hz), 7.12 (d, 1H, J = 8.0 Hz), 7.01–6.97 (m, 4H), 6.81–6.75 (m, 2H), 4.80 (d, 0.5H, J= 7.0 Hz), 4.74 (d, 0.5H, J= 7.0 Hz), 2.99–2.73 (m, 3H), 2.55–2.51 (m, 1H), 2.39–2.25 (m, 1H), 1.74–1.71 (m, 1H), 1.53–1.49 (m, 1H), 0.96 (s, 4.5H), 0.93 (s, 4.5H), −0.03 (s, 1.5H), −0.04 (s, 1.5H), −0.05 (s, 1.5H), −0.06 (s, 1.5H); 13C NMR (CDCl3, 125 MHz) δ 164.4, 164.3, 157.6, 157.5, 154.7, 154.6, 138.47, 138.41, 138.0, 137.7, 130.7, 130.4, 130.3, 130.1 (2C), 129.4, 126.7, 122.6, 122.5, 118.8, 118.3, 118.2, 116.7, 116.6, 72.2, 72.1, 40.3, 30.67, 30.62, 28.8, 28.7, 25.6 (3C), 25.1, 24.8, 18.0, −5.3, −5.41, −5.44, −5.6.
A solution of 2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-oxazole (459 mg, 1.05 mmol) in anhydrous THF (15 mL) was cooled to −78 °C before it was treated with 2.16 M n-BuLi (0.6 mL, 1.15 mmol) dropwise. The reaction mixture was stirred at −78 °C for 2 h, and treated with Bu3SnCl (0.6 mL, 2.1 mmol) and stirred for 5 min. The solution was warmed to room temperature, diluted with EtOAc, and washed with saturated aqueous NaCl. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 10% EtOAc–hexanes) yielded 2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-5-(tributylstannyl)oxazole (500 mg, 78%) as a thick colorless oil: 1H NMR (CDCl3, 500 MHz) δ 7.30 (t, 2H, J = 7.5 Hz), 7.12 (d, 1H, J = 6.0 Hz), 7.06 (t, 2H, J = 7.0 Hz), 6.97 (m, 2H), 6.77 (dd, 2H, J = 2.5, 8.5 Hz), 4.80 (d, 0.5H, J = 7.0 Hz), 4.75 (d, 0.5H, J = 7.0 Hz), 2.96–2.92 (m, 2H), 2.82–2.70 (m, 2H), 2.52–2.22 (m, 2H), 1.58–1.46 (m, 8H), 1.36–1.30 (m, 6H), 1.15–1.11 (m, 5H), 0.94–0.90 (s, 18H), 0.08 (s, 1.5H), 0.06 (s, 1.5H), −0.11 (s, 1.5H), −0.12 (s, 1.5H); 13C NMR (CDCl3, 125 MHz) δ 168.5, 168.4, 157.8, 154.9, 154.8, 154.7, 154.6, 138.2, 138.0, 137.1, 131.2, 130.8, 130.4, 130.2, 129.5 (2C), 122.67, 122.64, 118.9, 118.4, 118.3, 116.8, 116.6, 72.5, 72.3, 40.69, 40.64, 30.85, 30.81, 29.3, 29.2 (3C), 29.1, 29.0, 28.98, 28.90, 28.8, 28.5, 27.6, 27.4, 27.3, 27.2 (3C), 27.1, 27.0, 26.8, 25.7, 25.2, 25.0, 18.1, 13.69, 13.60 (3C), 11.6, 10.7, 10.2 (3C), 9.98, −5.3, −5.4, −5.61, −5.62.
2-((tert-Butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-5-(tributylstannyl)oxazole (5.0 g, 6.89 mmol), Pd(PPh3)4 (800 mg, 0.689 mmol) and 2-bromopyridine (0.9 mL, 8.96 mmol) were dissolved in anhydrous 1,4-dioxane (30 mL) and the mixture was warmed at reflux for 16 h under Ar. The mixture was diluted with EtOAc, washed with saturated aqueous NaCl, and dried over Na2SO4. Flash chromatography (SiO2, 20% EtOAc–hexanes) yielded 2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-5-(pyridin-2-yl)oxazole (1.49 g, 42%; typically 42–61%) as a colorless oil: 1H NMR (CDCl3, 500 MHz) δ 8.64 (d, 1H, J = 4.5 Hz), 7.78–7.76 (m, 1H), 7.71–7.67 (m, 2H), 7.30 (t, 2H, J = 7.5 Hz), 7.24–7.22 (m, 1.5H), 7.07–7.04 (m, 1.5H), 6.98–6.95 (m, 2H), 6.78–7.72 (m, 2H), 4.81 (d, 0.5H, J= 7.0 Hz), 4.75 (d, 0.5H, J= 7.0 Hz), 2.96–2.73 (m, 2H), 2.58–2.55 (m, 1H), 2.39–2.34 (m, 1H), 2.26–2.20 (m, 1H), 1.81–1.77 (m, 1H), 1.58–1.53 (m, 1H), 0.90 (s, 9H), 0.11 (s, 1.5H), 0.09 (s, 1.5H), −0.05 (s, 1.5H), −0.04 (s, 1.5H); 13C NMR (CDCl3, 125 MHz) δ 157.7, 157.6, 154.8, 154.7, 149.6, 138.1, 137.9, 137.18, 137.14, 132.1, 130.8, 130.5, 130.4 (2C), 130.3, 129.5, 128.5 (2C), 125.5, 125.4, 122.8, 122.78, 122.73, 119.1, 118.9, 118.4, 118.3, 116.8, 116.7, 72.5, 72.4, 40.5, 30.9, 30.5, 29.0, 28.9, 25.7 (3C), 25.3, 24.9, 18.2, −5.0, −5.23, −5.26.
2-((tert-Butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-5-(pyridin-2-yl)oxazole (1.49 g, 2.90 mmol) was dissolved in THF (30 mL), treated with Bu4NF (1 M in THF, 4 mL, 3.48 mmol) and the solution was stirred at room temperature for 2 h under Ar. The reaction mixture was diluted with EtOAc, washed with saturated aqueous NaCl, and dried over Na2SO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 50–100% EtOAc–hexanes) yielded (6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)(5-(pyridin-2-yl)oxazol-2-yl)methanol (740 mg, 64%) as a yellow oil: 1H NMR (CDCl3, 600 MHz) δ 8.63 (d, 1H, J= 4.2 Hz), 7.78 (t, 1H, J= 7.8 Hz), 7.71–7.65 (m, 2H), 7.30 (t, 2H, J = 7.2 Hz), 7.27–7.25 (m, 2H), 7.07–6.96 (m, 3H), 6.77–6.73 (m, 2H), 4.87 (d, 0.5H, J = 7.0 Hz), 4.82 (d, 0.5H, J = 7.0 Hz), 2.86–2.68 (m, 4H), 2.45–2.42 (m, 1H), 2.17–2.15 (m, 1H), 1.92–1.89 (m, 1H), 1.66–1.61 (m, 1H); 13C NMR (CDCl3, 150 MHz) δ 157.6, 154.8, 149.5, 146.7, 137.9, 137.7, 137.3, 130.5, 130.4, 130.3, 130.2, 129.6 (2C), 125.37, 125.34, 123.1, 122.8, 119.4, 118.9, 118.8, 118.4 (2C), 116.85, 116.81, 71.5, 71.3, 39.9, 39.8, 30.9, 30.0, 29.6, 28.98, 28.94, 25.3, 24.3.
(6-Phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)(5-(pyridin-2-yl)oxazol-2-yl)methanol (740 mg, 1.85 mmol) was dissolved in CH2Cl2 (40 mL) and Dess–Martin periodinane (1.0 g, 2.22 mmol) was added. The mixture was stirred at room temperature for 2 h and the reaction mixture was evaporated in vacuo. Flash chromatography (SiO2, 20% EtOAc–hexanes) yielded (6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)(5-(pyridin-2-yl)oxazol-2-yl)methanone (12, 650 mg, 88%) as a yellow oil: 1H NMR (CDCl3, 600 MHz) δ 8.68 (d, 1H, J = 4.2 Hz), 7.93 (s, 1H), 7.90–7.83 (m, 2H), 7.34–7.31 (m, 3H), 7.19–7.14 (m, 4H), 6.88–6.78 (m, 2H), 3.92–3.90 (m, 1H), 3.10–2.90 (m, 4H), 2.32–2.30 (m, 1H), 1.95–1.93 (m, 1H); 13C NMR (CDCl3, 150 MHz) δ 190.5, 157.5, 156.8, 155.1, 153.3, 150.0, 146.1, 137.2, 137.0, 130.2, 129.7, 129.6 (2C), 127.0, 124.2, 122.9, 120.4, 118.9, 118.5 (2C), 116.9, 43.5, 30.6, 28.8, 25.7; HRMS-ESI-TOF m/z 397.1551 ([M + H]+, C25H20N2O3 requires 397.1547). The enantiomers were separated using a semipreparative chiral phase HPLC column (Daicel ChiraCel OD, 10 μm, 2 × 25 cm, 10% EtOH hexanes, 7 mL/min, α= 1.35).
(S)-12: [α]23D −2.0 (c 0.1, THF).
(R)-12: [α]23D +1.8 (c 0.1, THF).
Methyl 6-(2-(6-Phenoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl)oxazol-5-yl)picolinate (13)
2-((tert-Butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)-5-(tributylstannyl)oxazole (5.0 g, 6.89 mmol), Pd(PPh3)4 (800 mg, 0.68 mmol), and methyl 6-bromopicolinate (2.0 g, 8.96 mmol) were dissolved in anhydrous 1,4-dioxane (30 mL) and the mixture was warmed at reflux for 16 h under Ar. The reaction mixture was diluted with EtOAc, washed with saturated aqueous NaCl, dried over Na2SO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 30% EtOAc hexanes) yielded methyl 6-(2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)oxazol-5-yl)picolinate (2.88 g, 73%) as a colorless oil: 1H NMR (CDCl3, 500 MHz) δ 8.01 (dd, 1H, J = 4.5, 7.0 Hz), 7.99–7.97 (m, 1H), 7.89–7.85 (m, 1H), 7.80–7.78 (m, 1H), 7.65–7.59 (m, 1H), 7.25–7.22 (m, 2H), 7.01–6.97 (m, 1H), 6.92–6.90 (m, 2H), 6.73–6.66 (m, 2H), 4.80 (d, 0.5H, J = 7.0 Hz), 4.77 (d, 0.5H, J = 7.0 Hz), 3.96 (s, 1.5H), 3.93 (s, 1.5H), 2.91–2.87 (m, 1H), 2.78–2.76 (m, 3H), 2.73–2.71 (m, 1H), 2.38–2.33 (m, 0.5H), 2.23–2.20 (m, 0.5H), 1.62–1.52 (m, 1H), 0.90 (s, 9H), 0.11 (s, 1.5H), 0.09 (s, 1.5H), −0.05 (s, 1.5H), −0.04 (s, 1.5H); 13C NMR (CDCl3, 125 MHz) δ 165.0, 164.9, 164.8, 164.0, 157.47, 157.40, 154.6, 154.5, 149.9, 149.8, 148.4, 148.0, 147.38, 147.35, 141.8, 138.9, 137.8, 137.6, 131.8, 131.7, 131.5, 130.5, 130.2, 130.1, 130.0, 129.3 (2C), 128.3, 128.23, 126.20, 126.1, 123.8, 123.7, 122.56, 122.52, 121.8 (2C), 118.7, 118.2, 118.1, 72.3, 72.1, 52.8, 52.6, 40.2, 30.7, 30.3, 28.7, 28.6, 27.6, 26.5, 25.5 (3C), 25.1, 24.6, 17.9, 17.3, 13.3, −5.2, −5.40, −5.44.
Methyl 6-(2-((tert-butyldimethylsilyloxy)(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)oxazol-5-yl)picolinate (2.88 g, 5.04 mmol) was dissolved in THF (50 mL), treated with Bu4NF (1 M in THF, 6 mL, 6.05 mmol) and the solution was stirred at room temperature for 2 h under Ar. The reaction mixture was diluted with EtOAc, washed with saturated aqueous NaCl, dried over Na2SO4 and the solvent was removed under reduced pressure. Flash chromatography (SiO2, 50–100% EtOAc hexanes) yielded methyl 6-(2-(hydroxy(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)oxazol-5-yl)picolinate (2.0 g, 86%) as a yellow oil: 1H NMR (CDCl3, 400 MHz) δ 8.20 (dd, 1H, J = 1.2, 7.6 Hz), 8.05 (t, 1H, J = 8.0 Hz), 7.98–7.96 (m, 2H), 7.48 (t, 2H, J = 7.2 Hz), 7.25–7.12 (m, 4H), 6.95–6.90 (m, 2H), 5.06 (d, 0.5H, J = 6.8 Hz), 5.01 (d, 0.5H, J = 6.8 Hz), 4.18 (s, 3H), 3.08–2.95 (m, 3H), 2.84–2.81 (m, 1H), 2.65–2.61 (m, 1H), 2.38–2.03 (m, 1H), 1.81–1.45 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 165.8, 165.7, 165.1, 157.4, 154.77, 154.74, 150.1, 148.0, 147.1, 137.8, 137.6, 130.4, 130.3, 130.2, 130.1, 129.49 (2C), 129.47, 125.9, 123.9, 122.6, 122.2, 118.79, 117.74, 118.33, 118.30, 116.7, 116.6, 71.2, 71.0, 64.2, 52.8, 39.69, 39.65, 30.9, 30.1, 28.8, 25.2, 24.4, 18.9, 17.4, 13.4.
Methyl 6-(2-(hydroxy(6-phenoxy-1,2,3,4-tetrahydronaphthalen-2-yl)methyl)oxazol-5-yl)picolinate (2.0 g, 4.38 mmol) was dissolved in CH2Cl2 (60 mL) and Dess Martin periodinane (2.7 g, 6.25 mmol) was added. The mixture was stirred at room temperature for 2 h before the reaction mixture was evaporated in vacuo. Flash chromatography (SiO2, 30% EtOAc hexanes) yielded methyl 6-(2-(6-phenoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl)oxazol-5-yl)picolinate (13, 1.67 g, 70%) as a white solid: 1H NMR (CDCl3, 500 MHz) δ 8.09 (dd, 1H, J = 1.0, 8.0 Hz), 8.03 (s, 1H), 8.01 (dd, 1H, J = 1.5, 8.0 Hz), 7.95 (t, 1H, J= 7.5 Hz), 7.29 (t, 2H, J= 7.5 Hz), 7.06 (t, 2H, J= 7.5 Hz), 6.98–6.96 (m, 2H), 6.79–6.77 (m, 2H), 4.01 (s, 3H), 3.91–3.86 (m, 1H), 3.08 (d, 2H, J= 8.0 Hz), 2.93–2.87 (m, 2H), 2.31–2.27 (m, 1H), 1.94–1.89 (m, 1H); 13C NMR (CDCl3, 125 MHz) δ 190.3, 164.9, 157.4, 156.8, 154.9, 152.3, 148.4, 146.3, 138.1, 137.1, 130.0, 129.6 (2C), 129.5, 127.8, 125.0, 123.1, 122.7, 118.8, 118.4 (2C), 116.8, 52.9, 43.4, 30.4, 28.6, 25.6; HRMS-ESI-TOF m/z 455.1617 ([M + H]+, C27H22N2O5 requires 455.1601). The enantiomers were separated using a semipreparative chiral phase HPLC column (Daicel ChiraCel OD, 10 μm, 2 × 25 cm, 40% EtOH–hexanes, 7 mL/min, α= 1.19).
(S)-13: [α]23D −0.7 (c 0.8, THF).
(R)-13: [α]23D +0.5 (c 0.8, THF).
6-(2-(6-Phenoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl)oxazol-5-yl)picolinic Acid (14)
Each pure enantiomer (S)-13 and (R)-13 (0.010 mmol) were dissolved in 1,2-dichloroethane and after addition of trimethyltin hydroxide (3 equiv), the mixture was warmed at 70 °C for 16 h. The mixture was concentrated in vacuo and diluted with EtOAc and the organic layer was washed with aqueous 0.01 N KHSO4, saturated aqueous NaCl, and dried over Na2SO4. Evaporation in vacuo yielded the crude acid that was purified by flash chromatography (SiO2, 5% HOAc–EtOAc) to yield 6-(2-(6-phenoxy-1,2,3,4-tetrahydronaphthalene-2-carbonyl)oxazol-5-yl)picolinic acid (14, 70%) as a yellow solid: 1H NMR (CDCl3 + 0.1% TFA, 600 MHz) δ 8.34 (d, 1H, J = 6.0 Hz), 8.22–8.19 (m, 2H), 7.98 (s, 1H), 7.36 (t, 2H, J = 8.0 Hz), 7.13–7.10 (m, 2H), 7.03 (d, 2H, J = 7.8 Hz), 6.85–6.78 (m, 2H), 3.85–3.84 (m, 1H), 3.13–3.09 (m, 2H), 2.96–2.90 (m, 2H), 2.34–2.31 (m, 1H), 1.97–1.94 (m, 1H); 13C NMR (CDCl3 + 0.1% TFA, 150 MHz) δ 191.0, 157.2, 156.8, 155.3, 151.2, 145.0, 140.5, 136.8, 130.2, 129.7 (2C), 128.9, 127.9, 125.8, 125.2, 123.2, 118.9 (2C), 118.6, 117.1, 43.9, 30.2, 28.5, 25.7; HRMS-ESI-TOF m/z441.1451 ([M + H] +, C26H20N2O5requires 441.1445).
(S)-14: [α]23D −5.5 (c 0.7, THF).
(R)-14: [α]23D +5.4 (c 0.6, THF).
FAAH Inhibition
14C-labeled oleamide was prepared from 14C-labeled oleic acid as described.13 The truncated rat FAAH (rFAAH) was expressed in E. coli and purified as described.71 The purified recombinant rFAAH was used in the inhibition assays unless otherwise indicated. The full-length human FAAH (hFAAH) was expressed in COS-7 cells as described,2 and the lysate of hFAAH-transfected COS-7 cells was used in the inhibition assays where explicitly indicated. The inhibition assays were performed as described.13 In brief, the enzyme reaction was initiated by mixing 1 nM of rFAAH (800, 500, or 200 pM rFAAH for inhibitors with Ki ≤ 1–2 nM) with 20 μM of 14C-labeled oleamide in 500 μL of reaction buffer (125 mM TrisCl, 1 mM EDTA, 0.2% glycerol, 0.02% Triton X-100, 0.4 mM Hepes, pH 9.0) at room temperature in the presence of three different concentrations of inhibitor. The enzyme reaction was terminated by transferring 20 μL of the reaction mixture to 500 μL of 0.1 N HCl at three different time points. The 14C-labeled oleamide (substrate) and oleic acid (product) were extracted with EtOAc and analyzed by TLC as detailed.13 The Ki of the inhibitor was calculated using a Dixon plot as described (standard deviations are provided in the Supporting Information tables). Lineweaver–Burk analysis was performed as described53 confirming competitive, reversible inhibition for 12 and 14, Figure 15.
Figure 15.

Lineweaver Burk analysis of 12 and 14 illustrating reversible, competitive inhibition of FAAH.
In vivo Pharmacodynamic Studies with Inhibitors
Inhibitors were prepared as a saline-emulphor emulsion for i.p. administration by vortexing, sonicating, and gently heating neat compound directly into an 18:1:1 v/v/v solution of saline:ethanol:emulphor, or as a homogeneous PEG solution for p.o. administration by vortexing, sonicating, and gently heating neat compound directly into PEG300 (Fluka). Male C57Bl/6J mice (<6 months old, 20–28 g) were administered inhibitors in saline-emulphor emulsion or an 18:1:1 v/v/v saline:emulphor:ethanol vehicle i.p. at a volume of 10 μL/g weight, or alternatively inhibitors in PEG300 or a PEG300 vehicle p.o. at a volume of 4 μL/g weight. After the indicated amount of time, mice were anesthetized with isofluorane and killed by decapitation. Total brains (~400 mg) and a portion of the liver (~100 mg) were removed and flash frozen in liquid N2. Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Measurement of Brain Lipids
Tissue was weighed and subsequently Dounce homogenized in 2:1:1 v/v/v CHCl3:MeOH:Tris pH 8.0 (8 mL) containing standards for lipids (50 pmol d4-PEA, 2 pmol d4-anandamide, 0.5 nmol d5-2-AG, and 10 nmol pentadecanoic acid). The mixture was vortexed and then centrifuged (1,400 × g, 10 min). The organic layer was removed, dried under a stream of N2, resolubilized in 2:1 v/v CHCl3:MeOH (120 μL), and 10 μL of this resolubilized lipid was injected onto an Agilent G6410B QQQ instrument. LC separation was achieved with a Gemini reverse-phase C18 column (5 μm, 4.6 mm × 50 mm, Phenomonex) together with a pre-column (C18, 3.5 μm, 2 mm × 20 mm). Mobile phase A was composed of a 95:5 v/v H2O:MeOH, and mobile phase B was composed of a 65:35:5 v/v/v i-PrOH:MeOH:H2O. 0.1% Formic acid or 0.1% ammonium hydroxide was included to assist in ion formation in positive and negative ionization mode, respecitvely. The flow rate for each run started at 0.1 mL/min with 0% B. At 5 min, the solvent was immediately changed to 60% B with a flow rate of 0.4 mL/min and increased linearly to 100% B over 10 min. This was followed by an isocratic gradient of 100% B for 5 min at 0.5 mL/min before equilibrating for 3 min at 0% B at 0.5 mL/min (23 min total per sample). MS analysis was performed with an electrospray ionization (ESI) source. The following MS parameters were used to measure the indicated metabolites in positive mode (precursor ion, product ion, collision energy in V): anandamide (348, 62, 11), OEA (326, 62, 11), PEA (300, 62, 11), d4-anandamide (352, 66, 11), d4-PEA (304, 62, 11), 2-AG (379, 287, 8), d5-2-AG (384, 287, 8). For negative polarity, the analysis was performed in MS2 scan mode from 100–1000 m/z. The capillary was set to 4 kV, the fragmentor was set to 100 V, and the delta EMV was set to 0. Lipids were quantified by measuring the area under the peak in comparison to the standards.
Mouse Tail Flick Assay
Male CD-1 (25–35 g, Charles River) mice were housed in groups of five in Plexiglas chambers with food and water available ad libitum. All animals were maintained on a 12 h light/dark cycle (lights on at 7:00 AM) in a temperature- and humidity-controlled animal colony. All tail-flick experiments were performed under an approved University of New England animal protocol in accordance with institutional guidelines and in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. Efficacy of the test compound was assessed using the 52 °C warm water tail-flick test. The latency to the first sign of a rapid tail-flick was taken as the behavioral endpoint. Each mouse was first tested for baseline latency by immersing its tail in the water and recording the time to response. Mice not responding within 5 s were excluded from further testing. Mice were then administered the test compound and tested for antinociception at various time points afterwards. Antinociception was calculated by the following formula: % Antinociception = 100 × (test latency-control latency)/(10-control latency). A maximum score was assigned (100%) to animals not responding within 10 sec to avoid tissue damage.
Chronic Constriction Injury (CCI)
Surgery was performed as described previously.83 Briefly, the right hind leg of male C57BL/6 mice was shaved and swabbed with betadine solution and ethanol. Posterior to the femur, an incision was made and the sciatic nerve was visualized and isolated, following muscle separation. The nerve was ligated twice with 5-0 (1.0 metric) black silk braided suture (Surgical Specialties Corporation, Reading, PA). The surrounding muscle and skin were then sutured with 6-0 nylon. Mice were recovered in a heated cage and observed for approximately 2 h before being returned to the vivarium. Anesthesia was maintained by constant inhalation of 1.5% isoflurane. In addition, mice were administered acetaminophen (2.4 mg/mL in drinking water) from 24 h before surgery through 48 h post surgery.
Allodynia Assays
Allodynia was initially tested 10 days after surgery. Male C57BL/6 mice were habituated to the test apparatus for 2 h on the 2 days prior to testing. On the test day, the mice were brought into the test room, weighed, and allowed to acclimate for at least 1 h before the start of the experiment. Mice were administered inhibitor 12 (50 mg/kg) in PEG300 (4 μL/g body weight) or vehicle (p.o.), then placed in ventilated polycarbonate cylinders on a mesh table. Mechanical (von Frey) and cold (acetone-induced) allodynia were tested at 1, 3, 6, and 9 h post drug administration. Testing was carried out by a separate observer who was blinded to treatment conditions. Mechanical allodynia was assessed with von Frey filaments (North Coast Medical, Morgan Hill, CA), using the “up-down” method.61d,83 Each hind paw was stimulated 5 times per filament (0.16–6.0 g), starting with the 0.6 g filament and increasing weight. Paw clutching or lifting, in response to three or more stimulations, was coded as a positive response. Once a positive response was detected, sequentially lighter-weight filaments were used to assess paw withdrawal threshold. Approximately 30 min after completing the von Frey test, cold allodynia was tested by propelling 10 μL of acetone (Fisher Bioscience) via air burst, from a 200 μL pipette (Rainin Instruments, Oakland, CA) onto the plantar surface of each hind paw. Total time lifting or clutching each paw was recorded, with a maximum time of 20 s.84
Data Analyses
Behavioral data were analyzed using a two-way mixed factorial analysis of variance (ANOVA) for each paw, with drug treatment as the between subjects measure, and time as the within subjects measure. Follow-up comparisons were made using the Bonferroni test. All animals were included in the analyses. Differences between groups were considered statistically significant at p < 0.05.
FAAH Production, Crystallization, and Crystal Structure Determination
The N-terminal transmembrane-deleted humanized version of FAAH (amino acids 32–579) was expressed in E. coli and purified as previously described,75 using 0.08% n-undecyl-β-D-maltoside in the ion exchange and size exclusion chromatography steps of the purification. Samples of pure protein were concentrated up to 35 mg/mL and supplemented with 13% xylitol and 2% benzyldimethyl(2-dodecyloxyethyl)ammonium chloride (Aldrich). Large crystals of h/rFAAH were obtained using a reservoir buffer (ratio 1:1) containing 100 mM MES pH 5.5, 100 mM KCl, 30% PEG400, and 8% polypropylene glycol-P400 by sitting drop vapor diffusion at 14 °C in 96-well plates (Innovaplate SD-2, Innovadyne Technologies, Inc.). The crystals were frozen directly in liquid nitrogen and complete datasets were collected from a single crystal at the Stanford Synchrotron Radiation Laboratory (SSRL, Menlo Park, CA) on beamline 11-1 at a temperature of 100K. The cocrystal structure of FAAH bound to 12 was solved at 1.90 Å resolution (Table S1). Data processing was performed using the XDS software package and the structure solved by molecular replacement (Phaser, CCP4 package85) using the coordinates from a previous h/rFAAH structure (PDB code 2WJ1) as a search model and refined using programs Phenix suite, coot, Refmac5, and BUSTER.86 Chemical parameters for the inhibitors were calculated by the Dundee PRODRG Web server.87 For the last steps of refinement, TLS (Translation/Libration/Screw) parameterization has been applied by dividing each monomer in 8 partitions (16 partitions in the crystallographic asymmetric unit).
Molecular Modeling and Binding Energy Evaluation
The program ICM-Pro (Molsoft, L.L.C.), employing a Monte Carlo minimization algorithm for energy stochastic optimization of ligands, has been previoulsy adopted for covalent docking simulation and binding energy calculation of FAAH inhibitors. Here, the coordinates of h/rFAAH crystal structure with bound 12 (PDB 3OJ8) were used for the simulation of both the (R)- and (S)-enantiomers (X and Y) and binding energies were calculated. The energy functions included the following ICM terms: van der Waals (‘vw’) and 1–4 van der Waals, hydrogen bonding (‘hb’), electrostatics (‘el’), entropic free energy, and constant surface tension (‘sf’). Estimation of the electrostatic energy was accurately calculated with analytical molecular surface as dielectric boundary.88 After adding hydrogens, applying global energy-minimization, and assigning partial charges, the inhibitor was removed from the model and each enantiomer was created in silico and manually conjugated to the γ-oxygen of the catalytic Ser241. The docking of (S)-12 was, as expected, fully superimposable to the experimentally (X-ray) determined structure.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (DA015648, DLB; DA009789 and DA017259, BFC; DA017259, RCS; NS052727-01A2, EJB; DA007027, DA009789, and DA017259, AHL) and thank Raj K. Chadha for the X-ray crystal structure of 21.
Abbreviations
- 2-AG
2-arachidonylglycerol
- CB
cannabinoid
- CCI
chronic constriction injury
- DEAD
diethyl azodicarboxylate
- EDCI
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
- FAAH
fatty acid amide hydrolase
- hERG
human ether-a-go-go related gene
- MTI
mild thermal injury
- hlm
human liver microsomes
- OEA
N-oleoyl ethanolamine
- PEA
N-palmitoyl ethanolamine
- rlm
rat liver microsomes
- SNL
spinal nerve ligation
- TBS
tert-butyldimethylsilyl
- TFAA
trifluoroacetic anhydride
- TGH
triacylglycerol hydrolase
- TsCl
p-toluenesulfonyl chloride
Footnotes
PDB deposition code: FAAH-12 (3OJ8)
Supporting Information Available. Full experimental details and characterization of the candidate inhibitors, FAAH assay measurement errors, purities of the FAAH inhibitors disclosed, table of data processing and refinement statstics for the FAAH–12 X-ray structure, and this material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular Characterization of an Enzyme that Degrades Neuromodulatory Fatty Acid Amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 2.Giang DK, Cravatt BF. Molecular Characterization of Human and Mouse Fatty Acid Amide Hydrolases. Proc Natl Acad Sci USA. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patricelli MP, Cravatt BF. Proteins Regulating the Biosynthesis and Inactivation of Neuromodulatory Fatty Acid Amides. Vit Hormones. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 4.Egertova M, Cravatt BF, Elphick MR. Comparative Analysis of Fatty Acid Amide Hydrolase and CB1 Cannabinoid Receptor Expression in the Mouse Brain: Evidence of a Widespread Role for Fatty Acid Amide Hydrolase in Regulation of Endocannabinoid Signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 5.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Fatty Acid Amide Hydrolase Substrate Specificity. Bioorg Med Chem Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 6.Ezzili C, Otrubova K, Boger DL. Fatty Acid Amide Signaling Molecules. Bioorg Med Chem Lett. 2010;20:5959–5968. doi: 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and Structure of a Brain Constituent that Binds to the Cannabinoid Receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 8.Martin BR, Mechoulam R, Razdan RK. Discovery and Characterization of Endogenous Cannabinoids. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- 9.Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Martin BR. Cannabimimetic Fatty Acid Derivatives: The Anandamide Family and Other “Endocannabinoids”. Curr Med Chem. 1999;6:721–744. [PubMed] [Google Scholar]
- 10.Schmid HHO, Schmid PC, Natarajan V. N-Acylated Glycerophospholipids and Their Derivatives. Prog Lipid Res. 1990;29:1–43. doi: 10.1016/0163-7827(90)90004-5. [DOI] [PubMed] [Google Scholar]
- 11.Boger DL, Henriksen SJ, Cravatt BF. Oleamide: An Endogenous Sleep-Inducing Lipid and Prototypical Member of a New Class of Lipid Signaling Molecules. Curr Pharm Des. 1998;4:303–314. [PubMed] [Google Scholar]
- 12.Cravatt BF, Lerner RA, Boger DL. Structure Determination of an Endogenous Sleep-Inducing Lipid, cis-9-Octadecenamide (Oleamide): A Synthetic Approach to the Chemical Analysis of Trace Quantitites of a Natural Product. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 13.(a) Cravatt BF, Prospero-Garcia O, Suizdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Chemical Characterization of a Family of Brain Lipids that Induce Sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]; (b) Lerner RA, Siuzdak G, Prospero Garcia O, Henriksen SJ, Boger DL, Cravatt BF. Cerebrodiene: A Brain Lipid Isolated from Sleep-Deprived Cats. Proc Natl Acad Sci USA. 1994;91:9505–9508. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Patricelli MP, Cravatt BF. Fatty Acid Amide Hydrolase Competitively Degrades Bioactive Amides and Esters Through a Nonconventional Catalytic Mechanism. Biochemistry. 1999;38:14125–14130. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]; (b) Patricelli MP, Cravatt BF. Clarifying the Catalytic Roles of Conserved Residues in the Amidase Signature Family. J Biol Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]; (c) Patricelli MP, Lovato MA, Cravatt BF. Chemical and Mutagenic Investigations of Fatty Acid Amide Hydrolase: Evidence for a Family of Serine Hydrolases with Distinct Catalytic Properties. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 15.McKinney MK, Cravatt BF. Evidence for Distinct Roles in Catalysis for Residues of the Serine-Serine-Lysine Catalytic Triad of Fatty Acid Amide Hydrolase. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- 16.McKinney MK, Cravatt BF. Structure and Function of Fatty Acid Amide Hydrolase. Ann Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 17.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural Adaptations in a Membrane Enzyme that Terminates Endocannabinoid Signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 18.Fowler CJ, Jonsson K-D, Tiger G. Fatty Acid Amide Hydrolase: Biochemistry, Pharmacology, and Therapeutic Possibilities for an Enzyme Hydrolyzing Anandamide, 2-Arachidonoylglycerol, Palmitoylethanolamide, and Oleamide. Biochem Pharmacol. 2001;62:517–526. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 19.(a) Cravatt BF, Lichtman AH. Fatty Acid Amide Hydrolase: An Emerging Therapeutic Target in the Endocannabinoid System. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]; (b) Ahn K, McKinney MK, Cravatt BF. Enzymatic Pathways that Regulate Endocannabinoid Signaling in the Nervous System. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahn K, Johnson DS, Cravatt BF. Fatty Acid Amide Hydrolase as a Potential Therapeutic Target for the Treatment of Pain and CNS Disorders. Exp Opin Drug Discov. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert DM, Fowler CJ. The Endocannabinoid System: Drug Targets, Lead Compounds, and Potential Therapeutic Applications. J Med Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 21.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to Anandamide and Enhanced Endogenous Cannabinoid Signaling in Mice Lacking Fatty Acid Amide Hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice Lacking Fatty Acid Amide Hydrolase Exhibit a Cannabinoid Receptor-Mediated Phenotypic Hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 23.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional Disassociation of the Central and Peripheral Fatty Acid Amide Signaling Systems. Proc Natl Acad Sci USA. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karsak M, Gaffal E, Date R, Wang-Eckhardt L, Rehnelt J, Petrosino S, Starowicz K, Steuder R, Schlicker E, Cravatt BF, Mechoulam R, Buettner R, Werner S, Di Marzo V, Tueting T, Zimmer A. Attenuation of Allergic Contact Dermatitis Through the Endocannabinoid System. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- 25.(a) Huitrón-Reséndiz S, Gombart L, Cravatt BF, Henriksen SJ. Effect of Oleamide on Sleep and Its Relationship to Blood Pressure, Body Temperature, and Locomotor Activity in Rats. Exp Neurol. 2001;172:235–243. doi: 10.1006/exnr.2001.7792. [DOI] [PubMed] [Google Scholar]; (b) Huitrón-Reséndiz S, Sanchez-Alavez M, Wills DN, Cravatt BF, Henriksen SJ. Characterization of the Sleep-Wake Patterns in Mice Lacking Fatty Acid Amide Hydrolase. Sleep. 2004;27:857–865. doi: 10.1093/sleep/27.5.857. [DOI] [PubMed] [Google Scholar]
- 26.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased Seizure Susceptibility and Proconvulsant Activity of Anandamide in Mice Lacking Fatty Acid Amide Hydrolase. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patricelli MP, Patterson JP, Boger DL, Cravatt BF. An Endogenous Sleep-Inducing Compound is a Novel Competitive Inhibitor of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 1998;8:613–618. doi: 10.1016/s0960-894x(98)00073-0. [DOI] [PubMed] [Google Scholar]
- 28.Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG. Inhibitors of Arachidonoyl Ethanolamide Hydrolysis. J Biol Chem. 1994;269:22937–22940. [PubMed] [Google Scholar]
- 29.Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong C-H, Lerner RA. Inhibition of Oleamide Hydrolase Catalyzed Hydrolysis of the Endogenous Sleep-Inducing Lipid cis-9-Octadecenamide. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- 30.Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Trifluoromethyl Ketone Inhibitors of Fatty Acid Amide Hydrolase: A Probe of Structural and Conformational Features Contributing to Inhibition. Bioorg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 31.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering Potent and Selective Reversible Inhibitors of Enzymes in Complex Proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 32.De Petrocellis L, Melck D, Ueda N, Maurelli S, Kurahashi Y, Yamamoto S, Marino G, Di Marzo V. Novel Inhibitors of Brain, Neuronal, and Basophilic Anandamide Amidohydrolase. Biochem Biophys Res Commun. 1997;231:82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- 33.Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl Arachidonyl Fluorophosphonate: A Potent Irreversible Inhibitor of Anandamide Amidase. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 34.Deutsch DG, Lin S, Hill WAG, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A. Fatty Acid Sulfonyl Fluorides Inhibit Anandamide Metabolism and Bind to the Cannabinoid Receptor. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- 35.Edgemond WS, Greenberg MJ, McGinley PJ, Muthians S, Campbell WB, Hillard CJ. Synthesis and Characterization of Diazomethylarachidonyl Ketone: An Irreversible Inhibitor of N-Arachidonylethanolamine Amidohydrolase. J Pharmacol Exp Ther. 1998;286:184–190. [PubMed] [Google Scholar]
- 36.Fernando SR, Pertwee RG. Evidence that Methyl Arachidonyl Fluorophosphonate is an Irreversible Cannabinoid Receptor Antagonist. Br J Pharmacol. 1997;121:1716–1720. doi: 10.1038/sj.bjp.0701303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du W, Hardouin C, Cheng H, Hwang I, Boger DL. Heterocyclic Sulfoxide and Sulfone Inhibitors of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 2005;15:103–106. doi: 10.1016/j.bmcl.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 38.(a) Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of Anxiety Through Blockade of Anandamide Hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]; (b) Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like Activity and Modulation of Brain Monaminergic Transmission by Blockage of Anandamide Hydrolase. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH Inhibitor URB597 in Neuropathic and Inflammatory Chronic Pain Models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic Acid 3′- or 4′-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantitative Structure Activity Relationships, and Molecular Modeling Studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]; (b) Biancalani C, Giovanni MP, Pieretti S, Cesari N, Graziano A, Vergeli C, Cilibrizzi A, Di Gianuario A, Colucci M, Mangano G, Garrone B, Polezani L, Dal Piaz V. Further Studies on Acylpiperazinyl Alkyl Pyridazinones: Discovery of an Exceptionally Potent, Orally Active, Antinociceptive Agent in Thermally Induced Pain. J Med Chem. 2009;52:7397–7409. doi: 10.1021/jm900458r. [DOI] [PubMed] [Google Scholar]
- 41.(a) Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, Synthesis, and Structure–Activity Relationships of Alkylcarbamic Acid Aryl Esters, a New Class of Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]; (b) Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. Synthesis and Structure–Activity Relationships of FAAH Inhibitors: Cyclohexylcarbamic Acid Biphenyl Esters with Chemical Modulation at the Proximal Phenyl Ring. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]; (c) Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. A Second Generation of Carbamate-based Fatty Acid Amide Hydrolase Inhibitors with Improved Activity in vivo. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Normanbhoy T, Alexander JP, Cravatt BF. A Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]; (b) Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, Liimatta M, Beidler D, Smtih S, Nomanbhoy TK, Cravatt BF. Benzothiophene Piperazine and Piperidine Urea Inhibitors of Fatty Acid Amide Hydrolase (FAAH) Bioorg Med Chem Lett. 2009;19:2865–2869. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and Characterization of a Highly Selective FAAH Inhibitor that Reduces Inflammatory Pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abouab–Dellah A, Burnier P, Hoornaert C, Jeunesse J, Puech F. 2004/099176 WO. Derivatives of Piperidinyl- and Piperazinyl-alkyl Carbamates, Preparation Methods and Application in Therapeutics (Sanofi) Abouab–Dellah A, Almario GA, Froissant J, Hoornaert C. 2005/077898 WO. Aryloxyalkylcarbamate Derivatives, Including Piperidine Carbamates, Their Preparation and Use as Fatty Acid Amide Hydrolase (FAAH) Inhibitors for Treating FAAH-related Pathologies (Sanofi) Abouab–Dellah A, Almario GA, Hoornaert C, Li AT. 2005/070910 WO. 1-Piperazine and 1-Homopiperazine Derivatives, Their Preparation and Use as Fatty Acid Amide Hydrolase (FAAH) Inhibitors for Treating FAAH-related Pathologies (Sanofi). Abouab Dellah A, Almario GA, Hoornaert C, Li AT. 2007/027141 WO. Alkyl(homo)piperazine-carboxylate Derivatives, Their Preparation and Use as Fatty Acid Amide Hydrolase (FAAH) Inhibitors for Treating FAAH-related Pathologies (Sanofi).
- 44.Sit S-Y, Xie K, Deng H. 2003/06589 WO. Preparation of (Hetero)aryl Carbamates and Oximes as Fatty Acid Amide Hydrolase Inhibitors (Bristol–Myers Squibb). Sit S-Y, Xie K. 2002/087569 WO. Preparation of Bis Arylimidazolyl Fatty Acid Amide Hydrolase Inhibitors for Treatment of Pain (Bristol–Myers Squibb). Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. Novel Inhibitors of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009.Sit SY, Conway CM, Xie K, Bertekap R, Bourin C, Burris KD. Oxime Carbamates: Discovery of a Series of Novel FAAH Inhibitors. Bioorg Med Chem Lett. 2010;20:1272–1277. doi: 10.1016/j.bmcl.2009.11.080.
- 45.Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao WJJ. Piperazinyl and Piperidinyl Ureas as Modulators of Fatty Acid Amide Hydrolase. 2006/01731842006 US Patent. Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao WJJ. Preparation of Thiadiazolylpiperazine Carboxamides as Modulators of Fatty Acid Amide Hydrolase (FAAH) 2007/0047412007 US Patent. Keith JM, Apocada R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham C-S, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Thiadiazolopiperazinyl Ureas as Inhibitors of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 2008;18:4838–4843. doi: 10.1016/j.bmcl.2008.07.081.Karbarz MJ, Luo L, Chang L, Tham C-S, Palmer JA, Wilson SJ, Wennerholm ML, Brown SM, Scott BP, Apocada RL, Keith JM, Wu J, Breitenbucher JG, Chaplan SR, Webb M. Biochemical and Biological Properties of 4-(3-Phenyl[1,2,4]thiadiazol-5-yl)piperazine-1-carboxylic Acid Phenylamide. A Mechanism-based Inhibitor of Fatty Acid Amide Hydrolase. Anesth Analg. 2009;108:316–329. doi: 10.1213/ane.0b013e31818c7cbd.
- 46.Matsumoto T, Kori M, Miyazaki J, Kiyota Y. Preparation of Piperidinecarboxamides and Piperazinecarboxamides as Fatty Acid Amide Hydrolase (FAAH) Inhibitors. 2006054652. Patent WO. 2006 Takeda.Matsumoto T, Kori M, Kouno M. Preparation of Piperazine-1-carboxamide Derivatives as Brain/Neuronal Cell-protecting Agents, and Therapeutic Agents for Sleep Disorder. 2007020888. Patent WO. 2007 Takeda.
- 47.Ishii T, Sugane T, Maeda J, Narazaki F, Kakefuda A, Sato K, Takahashi T, Kanayama T, Saitoh C, Suzuki J, Kanai C. Preparation of Pyridyl Non-Aromatic Nitrogenated Heterocyclic-1-carboxylate Ester Derivatives as FAAH Inhibitors. 2006/088075. Patent WO. 2006 Astellas.
- 48.(a) Moore SA, Nomikos GG, Dickason-Chesterfield AK, Sohober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, Iyengar S, Felder CC. Identification of a High-Affinity Binding Site Involved in the Transport of Endocannabinoids (LY2183240) Proc Natl Acad Sci USA. 2005;102:17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alexander JP, Cravatt BF. The Putative Endocannabinoid Transport Blocker LY2183240 is a Potent Inhibitor of FAAH and Several Other Brain Serine Hydrolases. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- 49.Alexander JP, Cravatt BF. Mechanism of Carbamate Inactivation of FAAH: Implications for the Design of Covalent Inhibitors and In vivo Functional Probes for Enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.(a) Minkkila A, Myllymaki MJ, Saario SM, Castillo-Melendez JA, Koskinen AMP, Fowler CJ, Leppanen J, Nevalainene T. The Synthesis and Biological Evaluation of para-Substituted Phenolic N-Alkyl Carbamates as Endocannabinoid Hydrolyzing Enzyme Inhibitors. Eur J Med Chem. 2009;44:2294–3008. doi: 10.1016/j.ejmech.2009.01.007. [DOI] [PubMed] [Google Scholar]; (b) Kasnanen H, Myllymaki MJ, Minkkila A, Kataja AO, Saario SM, Nevalainene T, Koskinen AMP, Poso A. 3-Heterocycle-phenyl N-Alkylcarbamates as FAAH Inhibitors: Design, Synthesis and 3D-QSAR Studies. ChemMedChem. 2010;5:213–231. doi: 10.1002/cmdc.200900390. [DOI] [PubMed] [Google Scholar]
- 51.Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Exceptionally Potent Inhibitors of Fatty Acid Amide Hydrolase: The Enzyme Responsible for Degradation of Endogenous Oleamide and Anandamide. Proc Natl Acad Sci USA. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boger DL, Miyauchi H, Hedrick MP. α-Keto Heterocycle Inhibitors of Fatty Acid Amide Hydrolase: Carbonyl Group Modification and α-Substitution. Bioorg Med Chem Lett. 2001;11:1517–1520. doi: 10.1016/s0960-894x(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 53.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaráes CRW, Jorgensen WL, Cravatt BF. Discovery of a Potent, Selective, and Efficacious Class of Reversible α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase Effective as Analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Discovery of an Exceptionally Potent and Selective Class of Fatty Acid Amide Hydrolase Inhibitors Enlisting Proteome-Wide Selectivity Screening: Concurrent Optimization of Enzyme Inhibitor Potency and Selectivity. Bioorg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 55.Romero FA, Hwang I, Boger DL. Delineation of a Fundamental α-Ketoheterocycle Substituent Effect for Use in the Design of Enzyme Inhibitors. J Am Chem Soc. 2006;68:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher BJ, Cravatt BF, Boger DL. Potent and Selective α-Ketoheterocycle-based Inhibitors of the Anandamide and Oleamide Catabolizing Enzyme, Fatty Acid Amide Hydrolase. J Med Chem. 2007;50:1058–1068. doi: 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. Structure–Activity Relationships of α-Ketooxazole Inhibitors of Fatty Acid Amide Hydrolase. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimball FS, Romero FA, Ezzili C, Garfunkle J, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. Optimization of α-Ketooxazole Inhibitors of Fatty Acid Amide Hydrolase. J Med Chem. 2008;51:937–947. doi: 10.1021/jm701210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garfunkle J, Ezzili C, Rayl TJ, Hochsttater DG, Hwang I, Boger DL. Optimization of the Central Heterocycle α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase. J Med Chem. 2008;51:4393–4403. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeMartino JK, Garfunkle J, Hochstatter DG, Cravatt BF, Boger DL. Exploration of a Fundamental Substituent Effect of α-Ketoheterocycle Enzyme Inhibitors: Potent and Selective Inhibitors of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 2008;18:5842–5846. doi: 10.1016/j.bmcl.2008.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.(a) Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible Inhibitors of Fatty Acid Amide Hydrolase that Promote Analgesia: Evidence for an Unprecedented Combination of Potency and Selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]; (b) Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of Fatty Acid Amide Hydrolase Produces Analgesia by Multiple Mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH. Endocannabinoid Modulation of Scratching Response in an Acute Allergenic Model: New Prospective Neural Therapeutic Target for Pruritus. J Pharmacol Exp Ther. 2009;329:314–323. doi: 10.1124/jpet.108.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kinsey SG, Long JZ, O’Neal ST, Abdulla RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of Endocannabinoid-degrading Enzymes Attenuates Neuropathic Pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.For additional studies, see: Onnis V, Congiu C, Bjorklund E, Hempel F, Soderstrom E, Fowler CJ. Synthesis and Evaluation of Paracetamol Esters As Novel Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2010;53:2286–2298. doi: 10.1021/jm901891p.Vincent F, Nguyen MT, Emerling DE, Kelly MG, Duncton MA. Mining Biologically-Active Molecules for Inhibitors of Fatty Acid Amide Hydrolase (FAAH): Identification of Phenmediphan and Amperozide as FAAH Inhibitors. Bioorg Med Chem Lett. 2009;19:6793–6796. doi: 10.1016/j.bmcl.2009.09.086.Wang JL, Bowen SJ, Schweitzer BA, Madsen HM, McDonald J, Pelc MJ, Tenbrink RE, Beidler D, Thorarensen A. Structure-based Design of Novel Irreversible FAAH Inhibitors. Bioorg Med Chem Lett. 2009;19:5970–5974. doi: 10.1016/j.bmcl.2009.07.101.Feledziak M, Michaix C, Urbach A, Labar G, Muccioli GG, Lambert DM, Marchand-Brynaert J. Beta-lactams Derived From A Carbapenem Chiron Are Selective Inhibitors of Human Fatty Acid Amide Hydroalse versus Monoacylglycerol Lipase. J Med Chem. 2009;52:7054–7068. doi: 10.1021/jm9008532.Hart T, Macias AT, Benwell K, Brooks T, D’Alessandro J, Dokurno P, Francis G, Gibbons B, Haymes T, Kennet G, Lightowler S, Mansell H, Matassova N, Misra A, Padfield A, Parsons R, Pratt R, Robertson A, Walls S, Wong M, Roughley S. Fatty Acid Amide Hydrolase Inhibitors. Surprising Selectivity of Chiral Azetidine Ureas. Bioorg Med Chem Lett. 2009;19:4241–4244. doi: 10.1016/j.bmcl.2009.05.097.Wang X, Sarris K, Kage K, Zhang D, Brown SP, Kolasa T, Surowy C, El Kouhen OF, Muchmore SW, Briono JD, Stewart AO. Synthesis and Evaluation of Benzothiazole-based Analogues as Novel, Potent, and Selective Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2009;52:170–180. doi: 10.1021/jm801042a.Minkkila A, Saario SM, Kasnanen H, Leppanene J, Poso A, Nevalainen T. Discovery of Boronic Acids as Novel and Potent Inhibitors of Fatty Acid Amide Hydrolase. J Med Chem. 2008;51:7057–7060. doi: 10.1021/jm801051t.Urbach A, Muccioli GG, Stern E, Lambert DM, Marchand-Brynaert J. 3-Alkenyl-2-azetidinones as Fatty Acid Amide Hydrolase Inhibitors. Bioorg Med Chem Lett. 2008;18:4163–4167. doi: 10.1016/j.bmcl.2008.05.081.Muccioli GG, Fazio N, Scriba GKE, Poppitz W, Cannata F, Poupaert JH, Wouters J, Lambert DM. Substituted 2-Thioxoimidazolidin-4-ones and Imidazolidine-2,4-diones as Fatty Acid Amide Hydrolase Inhibitors Templates. J Med Chem. 2006;49:417–425. doi: 10.1021/jm050977k.Saario SM, Poso A, Juvonen RO, Jarvinen T, Salo-Ahen OMH. Fatty Acid Amide Hydrolase Inhibitors from Virtual Screening of the Endocannabinoid System. J Med Chem. 2006;49:4650–4656. doi: 10.1021/jm060394q.Myllymaki MJ, Saario SM, Kataja AO, Castillo-Melendez JA, Navalainen T, Juvonen RO, Jarvinen T, Koskinen AMP. Design, Synthesis, and In vitro Evaluation of Carbamate Derivatives of 2-Benzoxazolyl- and 2-Benzothiazolyl-(3-hydroxyphenyl)-methanones as Novel Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2007;50:4236–4242. doi: 10.1021/jm070501w.Gattinoni S, De Simone C, Dallavalle S, Fezza F, Nannei R, Amadio D, Minetti P, Quattrociocchi G, Caprioli A, Borsini F, Cabri W, Penco S, Merlini L, Maccarrone M. Enol Carbamates as Inhibitors of Fatty Acid Amide Hydrolase (FAAH) Endowed with High Selectivity for FAAH Over the Other Targets of the Endocannabinoid System. ChemMedChem. 2010;5:357–360. doi: 10.1002/cmdc.200900472.
- 63.Pedregal-Tercero C, Siegel MG, Stucky RD, Takeuchi K. Preparation of 6-Substituted Nicotamides as Opioid Receptor Antagonists for Treating Obesity and Related Diseases. 2004080968. Patent WO. 2004 Eli Lilly.
- 64.Dey S, Mal D. Total Synthesis of BE-23254, a Chlorinated Angucycline Antibiotic. Tetrahedron Lett. 2005;46:5483–5486. [Google Scholar]
- 65.Mitsunobu O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformations of Natural Products. Synthesis. 1981:1–28. [Google Scholar]
- 66.Ley SV, Thomas AW. Modern Synthetic Methods for Copper-Mediated C(aryl)–O, C(aryl)–N, and C(aryl)–S Bond Formation. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 67.Dess DB, Martin JC. A Useful 12-I-5 Triacetoxyperiodinane (the Dess–Martin Periodinane) for the Selective Oxidation of Primary or Secondary Alcohols and a Variety of Related 12-I-5 Species. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 68.Vedejs E, Monahan SD. Metalation of Oxazole–Borane Complexes: A Practical Solution to the Problem of Electrocyclic Ring Opening of 2-Lithiooxazoles. J Org Chem. 1996;61:5192–5193. [Google Scholar]
- 69.Hari Y, Obika S, Sakaki M, Morio K, Yamagata Y, Imanishi T. Effective Synthesis of C-Nucleosides with 2′,4′-BNA Modification. Tetrahedron. 2002;58:3051–3063. [Google Scholar]
- 70.Farina V, Krishnamurthy V, Scott WJ. The Stille Reaction. Org React. 1997;50:1–652. [Google Scholar]
- 71.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. A Mild and Selective Method for the Hydrolysis of Esters with Trimethyltin Hydroxide. Angew Chem Int Ed. 2005;44:1378–1382. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 72.Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF. Comparative Characterization of a Wild Type and Transmembrane Domain-deleted Fatty Acid Amide Hydrolase: Identification of the Transmembrane Domain as a Site for Oligomerization. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
- 73.The structure and absolute stereochemistry of 21 were established with a single-crystal X-ray structure determination conducted on a colorless needle grown from MeOH with the active enantiomer (CCDC 790167).
- 74.Ohmoto K, Yamamoto T, Okama M, Horiuchi T, Imanishi H, Odagnki Y, Kawabata K, Sekioka T, Itirota Y, Matsuoka S, Nakai H, Toda M, Cheronis JC, Spruce LW, Gyorkos A, Wieczorek M. Development of Orally Active Nonpeptide Inhibitors of Human Neutrophil Elastase. J Med Chem. 2001;44:1268–1285. doi: 10.1021/jm000410y. [DOI] [PubMed] [Google Scholar]
- 75.Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided Inhibitor Design for Human FAAH by Interspecies Active Site Conversion. Proc Natl Acad Sci USA. 2008;105:12820–12824. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Cocrystal Structures. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mileni M, Garfunkle J, Kimball FS, Cravatt BF, Stevens RC, Boger DL. X-ray Crystallographic Analysis of α-Ketoheterocycle Inhibitors Bound to a Humanized Variant of Fatty Acid Amide Hydrolase. J Med Chem. 2010;53:230–240. doi: 10.1021/jm9012196.See also: Mileni M, Garfunkle J, Ezzili C, Cravatt BF, Stevens RC, Boger DL. Fluoride-mediated Capture of a Noncovalent Bound State of a Reversible Covalent Enzyme Inhibitor: X-ray Crystallographic Analysis of an Exceptionally Potent -Ketoheterocycle Inhibitor of Fatty Acid Amide Hydrolase. J Am Chem Soc. 2011;133 doi: 10.1021/ja110877y. in press.
- 78.(a) Edwards PD, Meyer EFJ, Vijayalakshmi J, Tuthill PA, Andisik DA, Gomes B, Strimpler A. Design, Synthesis, and Kinetic Evaluation of a Unique Class of Elastase Inhibitors, the Peptidyl α-Ketobenzoxazoles, and the X-ray Crystal Structure of the Covalent Complex Between Porcine Pancreatic Elastase and Ac-Ala-Pro-Val-2-benzoxazole. J Am Chem Soc. 1992;114:1854–1863. [Google Scholar]; (b) Edwards PD, Bernstein PR. Synthetic Inhibitors of Elastase. Med Res Rev. 1994;14:127–194. doi: 10.1002/med.2610140202. [DOI] [PubMed] [Google Scholar]; (c) Edwards PD, Zottola MA, Davis M, Williams J, Tuthill PA. Peptidyl α-Ketoheterocycle Inhibitors of Human Neutrophil Elastase. 3. In vitro and In vivo Potency of a Series of Peptidyl α-Ketobenzoxazoles. J Med Chem. 1995;38:3972–3982. doi: 10.1021/jm00020a011. [DOI] [PubMed] [Google Scholar]; (d) Edwards PD, Andisik DW, Bryant CA, Ewing B, Gomes B, Lewis JJ, Rakiewicz D, Steelman G, Strimpler A, Trainor DA, Tuthill PA, Mauger RC, Veale CA, Wildonger RA, Williams JC, Wolanin DJ, Zottola M. Discovery and Biological Activity of Orally Active Peptidyl Trifluoromethyl Ketone Inhibitors of Human Neutrophil Elastase. J Med Chem. 1997;40:1876–1885. doi: 10.1021/jm960819g. [DOI] [PubMed] [Google Scholar]
- 79.For prior modeling studies, see: Guimaraes CRW, Boger DL, Jorgensen WL. Elucidation of Fatty Acid Amide Hydrolase Inhibition by Potent α-Ketoheterocycle Derivatives from Monte Carlo Simulations. J Am Chem Soc. 2005;127:17377–17384. doi: 10.1021/ja055438j.Tubert-Brohman I, Acevedo O, Jorgensen WL. Elucidation of Hydrolysis Mechanisms for Fatty Acid Amide Hydrolase and its Lys142Ala Varaint via QM/MM Simulations. J Am Chem Soc. 2006;128:16904–16913. doi: 10.1021/ja065863s.
- 80.Long JZ, Nomura DK, Cravatt BF. Characterization of Monoacylglycerol Lipase Inhibition Reveals Differences in Central and Peripheral Enodocannabinoid Metabolism. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maryanoff BE, Costanzo MJ. Inhibitors of Proteases and Amide Hydrolases that Employ an α-Ketoheterocycle as a Key Enabling Functionality. Bioorg Med Chem. 2008;16:1562–1595. doi: 10.1016/j.bmc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 82.Cameron M. The Scripps Research Institute. Florida: Unpublished studies. [Google Scholar]
- 83.Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase Inhibitors Produce Anti-Allodynic Effects in Mice through Distinct Cannabinoid Receptor Mechanisms. J Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Decosterd I, Woolf CJ. Spared Nerve Injury: an Animal Model of Persistent Peripheral Neuropathic Pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 85.Collaborative Computational Project Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 86.(a) Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of Severely Incomplete Structures with Maximum Likelihood in BUSTER-TNT. Acta Crystallogr. 2004;D60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]; (b) Kabsch WJ. Automatic Processing of Rotation Diffraction Data from Crystals of Initially Unknown Symmetry and Cell Constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 87.Schuettelkopf AW, van Aalten DMF. PRODRG: a Tool for High-throughput Crystallography of Protein-ligand Complexes. Acta Crystallogr. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 88.Totrov M, Abagyan R. Rapid Boundary Element Solvation Electrostatics Calculations in Folding Simulations: Successful Folding of a 23-Residue Peptide. Biopolymers. 2001;60:124–133. doi: 10.1002/1097-0282(2001)60:2<124::AID-BIP1008>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.