

Abstract
Rheumatoid arthritis is associated with the development of autoantibodies to citrullinated self-proteins. Citrullinated synovial proteins, which are generated via the actions of the protein arginine deiminases (PADs), are known to develop in the murine collagen-induced arthritis (CIA) model of inflammatory arthritis. Given these findings, we evaluated whether N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide (Cl-amidine), a recently described pan-PAD inhibitor, could affect the development of arthritis and autoimmunity by treating mice in the CIA model with Cl-amidine on days 0–35. Cl-amidine treatment reduced total synovial and serum citrullination, decreased clinical disease activity by ∼50%, and significantly decreased IgG2a anti-mouse type II collagen Abs. Additionally, histopathology scores and total complement C3 deposition were significantly lower in Cl-amidine–treated mice compared with vehicle controls. Synovial microarray analyses demonstrated decreased IgG reactivity to several native and citrullinated epitopes compared with vehicle controls. Cl-amidine treatment had no ameliorative effect on collagen Ab-induced arthritis, suggesting its primary protective mechanism was not mediated through effector pathways. Reduced levels of citrullinated synovial proteins observed in mice treated with Cl-amidine are consistent with the notion that Cl-amidine derives its efficacy from its ability to inhibit the deiminating activity of PADs. In total, these results suggested that PADs are necessary participants in the autoimmune and subsequent inflammatory processes in CIA. Cl-amidine may represent a novel class of disease-modifying agents that modulate aberrant citrullination, and perhaps other immune processes, necessary for the development of inflammatory arthritis.
Rheumatoid arthritis (RA) is an autoimmune disorder that is estimated to affect nearly 1% of the population. RA is characterized by chronic inflammation of the synovium, resulting in pannus formation and joint destruction. The presence of autoantibodies designated rheumatoid factors, which recognize Ig Fc domains, have been well-characterized in RA. However, a new class of autoantibodies, designated Abs to citrullinated protein Ags (ACPA), have proven to be more specific than rheumatoid factors for the presence of RA (1–3). Indeed, several studies showed that ACPA are present in the sera of individuals who ultimately develop seropositive RA for an average of 4–5 y prior to the onset of clinically apparent disease (4–6), suggesting that the development of these autoantibodies may be an early event in the onset and progression of RA.
ACPA preferentially recognize citrullinated proteins. These epitopes are generated via the posttranslational modification of peptidyl-arginine residues to peptidyl-citrulline. This hydrolytic reaction, which is variably termed deimination or citrullination, is catalyzed by protein arginine deiminases (PADs), a small family of five calcium-dependent enzymes (PAD1, 2, 3, 4, and 6; there is no PAD5) (7–9). Consistent with a role for aberrant PAD activity in RA is the fact that genetic studies have identified single nucleotide polymorphisms within the PAD4 gene that are associated with an increased risk for developing RA, although this linkage has only been definitively established in Asian populations (10–12).
In general, the functional roles of PAD enzymes and of citrullinated peptides and proteins in normal human physiology are poorly understood. However, recent studies suggested that PADs play key roles in apoptosis, differentiation, and murine oogenesis (13–15). PAD4 is also known to play a role in regulating eukaryotic gene transcription; for example, citrullination of histones H3 and H4 was shown to downregulate the expression of genes under the control of the estrogen receptor, thyroid hormone receptor (16–19) and p53 (20, 21). Citrullination is also essential for the formation of neutrophil extracellular traps (22), and site-specific citrullination was reported to alter chemokine function (23–25). Interestingly, one report also demonstrated that when incubated with unmodified hen egg lysozyme, dendritic cells and peritoneal macrophages presented citrullinated peptides and stimulated citrulline-specific T cell responses (26).
There are five PAD isozymes expressed in humans and mice; enzymatic activity has been detected for all enzymes, with the exception of PAD6. PADs show varied tissue-expression patterns in humans and in mice: PAD1 is highly expressed in the skin and uterus, PAD2 is widely expressed, PAD3 is found in the skin and hair follicles, and PAD6 is expressed in human leukocytes and in mouse oocytes and embryos (9, 27). PAD4 is primarily expressed in WBCs (macrophages, neutrophils, and eosinophils); this localization is consistent with a role for PAD4 in modulating the immune response. With regard to RA pathogenesis, the subcellular localization of PADs is noteworthy because at sites of inflammation, intracellular and extracellular proteins show elevated levels of citrullination. In addition, PAD2 and PAD4 are present in RA synovial fluid (28). These observations suggest that the normal intracellular localization of PADs is somehow perturbed in the RA synovium (29).
The pathophysiologic significance of protein citrullination in RA is unclear, because it is unknown whether citrullination reflects ongoing inflammation or plays a causal role in the pathogenesis of disease. We examined whether a recently described pan-PAD inhibitor, N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide (Cl-amidine), could ameliorate the signs and symptoms of murine collagen-induced arthritis (CIA) by blocking the development of citrullinated epitopes and ACPA generation and/or affecting other PAD-dependent cellular processes. Cl-amidine is a mechanism-based PAD inactivator that irreversibly inhibits all of the known active PAD isozymes (i.e., PADs 1–4) with low micromolar potency; the ability of Cl-amidine to inhibit PAD6 has not been assessed because in vitro activity has not been detected for this isozyme. The structure of Cl-amidine is similar to benzoyl arginine amide, a small molecule PAD substrate, except that it incorporates a reactive haloacetamidine warhead in place of the substrate guanidinium. This warhead reacts with the active site cysteine, which is critical for catalysis, to form a thioether adduct; the existence of this adduct has been verified by crystallography (30, 31). In addition to Cl-amidine's ability to inhibit PADs in vitro, it has been evaluated in a cell-based assay of PAD4 activity. This is a mammalian two-hybrid assay that monitors the enhanced interaction between the glucocorticoid receptor-interacting protein 1 binding domain of p300 and glucocorticoid receptor-interacting protein 1 on intracellular PAD4. This assay demonstrated that Cl-amidine is cell permeable, can transit membranes, and inhibit PAD4 activity in the nucleus (31).
The CIA model of inflammatory arthritis was used for these studies because these mice are known to develop serum Ab reactivity to citrullinated epitopes, as determined by synovial proteome-array analysis (32, 33). Additionally, citrullinated peptide tolerance and passive anti-citrulline–specific mAb-transfer methods were shown to modulate arthritis severity in the CIA model (32). In this study, we showed that PAD inhibition resulted in decreased generation of synovial citrulline; a decreased Ab response to citrullinated proteins and other autoantigens; and a substantially reduced arthritis severity, as measured by clinical and histological assessments. Determination that the levels of citrullinated synovial proteins are reduced with Cl-amidine treatment is consistent with the notion that Cl-amidine derives its efficacy from its ability to inhibit the deiminating activity of PADs. In total, these results demonstrated a key role for PAD activity in CIA and suggested that PADs may be a therapeutic target for the treatment of inflammatory arthritis.
Materials and Methods
Collagen-induced arthritis
For induction of CIA, 6–8-wk-old DBA/1J mice (The Jackson Laboratory) were injected intradermally with 100 μl IFA containing 200 μg bovine type II collagen (CII) (Elastin Products) and 200 μg inactivated Mycobacterium tuberculosis (H37Ra; Difco) on days 0 and 21 (34). All animal experiments were approved by the University of Colorado School of Medicine Institutional Animal Care and Use Committee.
Treatment groups
Each experimental group consisted of eight mice that were treated daily throughout the entire experiment (i.e., day 0 of the first injection through sacrifice on day 35) with one of five interventions: no injection, PBS (vehicle control), 1 mg/kg Cl-amidine, 10 mg/kg Cl-amidine, or 50 mg/kg Cl-amidine. All injections were given i.p. in a standard volume of 200 μl containing the appropriate concentration of Cl-amidine. Doses were calculated for the average weight of the group (20 g). After the day-21 booster injection, clinical disease activity was independently evaluated by two trained laboratory personnel blinded to treatment status. A 3-point scale was used for each paw: 0 = normal joint; 1 = slight inflammation and redness; 2 = severe erythema and swelling affecting the entire paw; and 3 = deformed paw or joint with ankylosis, joint rigidity, and loss of function. The maximum score of 12 for each animal was based on the total score for all four paws. At day 35, all animals were sacrificed by anesthesia with 2,2,2-tribromoethanol and cervical dislocation. One mouse in the 1-mg/kg treatment group died on day 8 of the experiment of causes unrelated to treatment.
Histological examination
On day 35, both forepaws and the right hind limb (including the paw, ankle, and knee) were surgically removed and immediately fixed in 10% buffered formalin (Biochemical Sciences). Tissue samples were embedded in paraffin, sectioned, and stained with H&E for histopathology and with toluidine blue for specific evaluation of cartilage changes (35). Joint sections were scored by an individual blinded to treatment status for levels of synovial inflammation, pannus, cartilage damage, and bone damage; each was scored on a scale of 0–5. The compiled data are expressed as the mean ± SEM based upon a set of five joints per animal. Quantitative immunohistological analysis for mouse C3 deposition in the synovium and cartilage was scored on a scale of 0–3 (36).
Measurement of total citrulline content
Mice were administered one i.p. injection of CII and then were treated for 21 d with PBS or three different doses of Cl-amidine. On day 21, the entire knee joint was surgically removed from control and Cl-amidine–treated DBA/1J CIA mice (n = 5 per group) and flash-frozen in liquid nitrogen. Synovial tissue was enriched from the joints by further dissection. Tissue homogenates were prepared according to the method of Moscarello and colleagues (37, 38). Briefly, individual tissue samples were resuspended in 50 mM HEPES (pH 7.6), 1.0 mM EDTA, 0.5 mM DTT, and 0.43 mM PMSF at a final concentration of 20 mg tissue/0.1 ml buffer. Cell extracts were prepared with a Dounce homogenizer and subsequently clarified by centrifugation. Total citrulline content was analyzed by adding 60 μl synovial lysate or 20 μl serum lysate to a reaction buffer containing 50 mM NaCl, 10 mM CaCl2, 2 mM DTT, and 100 mM Tris (pH 7.6). A color-development solution that detects citrullinated proteins (200 μl) was added (COLDER) (39). The sample was vortexed and incubated at 95°C for 30 min. The absorbance at 540 nm was measured and compared to a standard curve of known citrulline concentrations. Measurements were made in duplicate, and the data were normalized using the protein concentrations for each sample.
Flow cytometric analysis of immune cell populations
To determine how Cl-amidine treatment affects immune cells in vivo, flow cytometry analysis was performed on splenocytes obtained from control and treatment group mice. Mice were immunized with bovine CII on day 0 and were treated daily with PBS (vehicle control) or 50 mg/kg Cl-amidine i.p. At day 21, mice were sacrificed, and spleens were surgically removed and placed in media on ice. Splenocytes were isolated through a cell strainer and stained for the cell surface markers listed in Table I and Table II. All Abs used for flow cytometry were obtained from BD Pharmingen.
Table I. Frequency of immune cells in Cl-amidine–treated and control mice with CIA.
| Treatment | CD4 (%) | CD8 (%) | CD19+B220+ (%) | DX5 (%) | CD11b (%) | CD11c (%) |
|---|---|---|---|---|---|---|
| PBS (n = 5) | 10.1 ± 0.9 | 4.0 ± 0.4 | 59.8 ± 1.6 | 2.7 ± 0.3 | 1.6 ± 0.2 | 1.6 ± 0.2 |
| CL50 (n = 4) | 10.4 ± 0.8 | 3.8 ± 0.3 | 56.3 ± 1.7 | 2.8 ± 0.1 | 1.6 ± 0.2 | 1.9 ± 0.1 |
Splenocytes from CIA mice treated with Cl-amidine or PBS for 21 d were stained with the listed cell surface markers, and the frequency of each population was determined. Data are mean ± SEM.
CL50, 50 mg/kg/d Cl-amidine.
Table II. Absolute number (×106) of immune cells in Cl-amidine–treated and control mice with CIA.
| Treatment | Spl | CD4 | CD8 | CD19+B220+ | DX5 | CD11b | CD11c |
|---|---|---|---|---|---|---|---|
| PBS (n = 5) | 74 ± 14 | 6.8 ± 0.78 | 2.7 ± 0.34 | 44 ± 7.9 | 2.0 ± 0.42 | 1.2 ± 0.29 | 1.2 ± 0.29 |
| CL50 (n = 4) | 96 ± 24 | 9.0 ± 1.7 | 3.3 ± 0.67 | 52 ± 15 | 2.6 ± 0.63 | 1.5 ± 0.43 | 1.8 ± 0.46 |
Splenocytes from CIA mice treated with Cl-amidine or PBS for 21 d were stained with the listed cell surface markers, and the absolute number was determined for each population. Data are mean ± SEM.
CL50, 50 mg/kg/d Cl-amidine; Spl, splenocyte.
Induction of collagen Ab-induced arthritis
Six to eight-week-old male DBA/1J mice (The Jackson Laboratory) were injected i.p. with PBS or 1, 10, or 50 mg/kg Cl-amidine daily for 7 d, after which collagen Ab-induced arthritis (CAIA) was induced by injecting the mice i.p. with Arthrogen (Chondrex; 4 mg/mouse). Three days later, mice were injected i.p. with 50 μg LPS and scored daily for disease severity on days 3–10, as described above. Daily treatment with PBS or Cl-amidine was continued on days 0–10.
Anti-collagen Ab determination
Serum was collected from all mice by retro-orbital aspiration on days 0 and 21 before each injection of CII, as well as by cardiac puncture at the end of the study on day 35. IgG2a and IgG1 Abs to murine and bovine CII were measured in triplicate by specific ELISA by coating 96-well plates with 5 μg/ml murine or bovine CII (Chondrex). A standard pool of anti-CII Abs was obtained by combining sera from several mice with severe CIA; the levels of IgG1 and IgG2a Abs in this pool were considered 100 U/ml. A standard curve was obtained using serum dilutions, and the Michaelis–Menton equation was used to convert OD values into units (34).
Synovial proteome microarray
Synovial proteome arrays containing 191 proteins and peptides representing candidate autoantigens in RA were used to perform multiplex characterization of autoantibody responses in sera derived from Cl-amidine–treated mice and control mice (40, 41). The synovial proteome arrays were previously validated with a panel of mAbs and reference sera specific for many of the spotted proteins (40). Significance analysis of microarrays (SAM; version 3.08) was used to identify autoantibody reactivities that exhibited significant differences between Cl-amidine– and PBS-treated CIA mice. The mice and their Ag array reactivities were arranged using hierarchical cluster analysis (Cluster 3.0 software) and displayed as a heatmap (Java TreeView software version 1.1.3 created by Alok) (42, 43). An accession number (GSE23731) for complete microarray data has been assigned and approved in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23731).
Autoantigen ELISA
The levels of murine IgG reactive with putative autoantigens [HCgp39 (130–145), HCgp39 (160–175), DEAE pool 1 and biglycan (227–246)] from the peptide array were measured in duplicate by coating 96-well plates with 10 μg/ml Ag in PBS overnight at 4°C. After washing with PBS, 0.05% Tween 20, plates were blocked in PBS, 1% BSA for 4 h at 4°C. Following blocking, 50 μl/well a 1:150 dilution of day 0 or 35 serum in PBS was added to plates and incubated overnight at 4°C. Plates were washed in PBS, 0.05% Tween 20, and Ab binding was detected using HRP-conjugated anti-mouse IgG visualized with tetramethylbenzidine substrate.
Statistical analysis
ANOVA, with tests for multiple comparisons, was used to examine the data for CIA and CAIA clinical disease activity scores, histology, anti-CII Ab levels, and synovial and serum citrulline content. Flow cytometry, grouped synovial citrulline, and autoantigen ELISA data were analyzed by the Student t test. The Pearson correlation coefficient was calculated for selected comparisons between potentially related datasets. The Shapiro–Wilks “W” test was used to test all histological and disease activity data for normal distribution before applying subsequent parametric tests. Data are expressed as the mean ± SEM, with p < 0.05 considered significant.
Results
Cl-amidine treatment reduces clinical disease activity in CIA
We hypothesized that if citrullination is an important mechanism in the pathogenesis of inflammatory arthritis, inhibition of PAD activity would lead to decreased disease severity because of reduced evolution of ACPA epitopes and autoantibody production. To test this hypothesis, we used Cl-amidine, a recently described first-generation cell-permeable inhibitor of all known active PAD isozymes, in the CIA mouse model of inflammatory arthritis. Mice were immunized on days 0 and 21 with bovine CII, and clinical disease activity was monitored on days 21–35. Although all of the mice developed disease, mice receiving daily i.p. injections of Cl-amidine in PBS, beginning on day 0, exhibited reduced clinical disease activity ∼50% on days 24–36 (p < 0.05 for all three doses of Cl-amidine at day 35) (Fig. 1A). These results indicated that Cl-amidine treatment reduces clinical disease activity in CIA.
FIGURE 1.
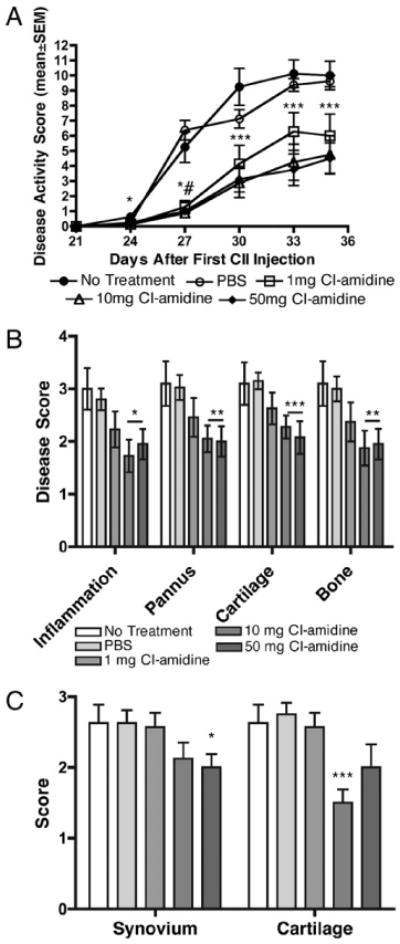
Cl-amidine treatment reduces clinical disease activity and joint destruction in CIA. Arthritis was induced by two injections of bovine CII in CFA at days 0 and 21. Mice were treated as described in Materials and Methods and assessed three times weekly for clinical disease activity. A, All treatment groups showed significantly reduced clinical disease activity scores compared with the PBS vehicle control beginning on day 27 and continuing through day 35. *p < 0.05 at day 24 and p < 0.03 at day 27 for 10-mg group, #p < 0.01 for 50-mg group, ***p < 0.03 for 10- and 50-mg groups, and p < 0.05 for 1-mg group. (n = 8 for all groups, except n = 7 for 1-mg group.) B, Histologic sections were scored for changes in synovial inflammation, pannus, cartilage damage, and bone damage, all on a scale of 0–5. C, Sections were scored for C3 deposition in the synovium and cartilage. Data are expressed as the mean ± SEM based upon a set of five joints per animal. Treatment with Cl-amidine resulted in decreased histopathology scores for all parameters in the 10- and 50-mg/kg/d treatment groups compared with the PBS vehicle group. (n = 8 for all groups, except n = 7 for 1-mg group.) *p ≤ 0.03, **p < 0.02, ***p < 0.01.
Treatment with Cl-amidine reduces joint damage in CIA
Histological analyses of joints were performed at the end point of the experiment (day 35) to examine the effects of treatment with Cl-amidine on joint destruction. Histopathology scores for synovial inflammation, pannus, cartilage damage, and bone damage were significantly lower in the 10 and 50 mg/kg/d treatment groups compared with the PBS vehicle group (p < 0.03) (Fig. 1B). The clinical disease activity and histology scores showed a significant correlation with each other (R2 = 0.8; p < 0.001). Cl-amidine treatment at 50 and 10 mg/kg also reduced C3 deposition in the synovium and the cartilage, respectively (Fig. 1C). No differences in neutrophil or macrophage infiltration of CIA joints was observed in mice treated with Cl-amidine (data not shown), suggesting that recruitment of these cells was not blocked by Cl-amidine. These results suggested that Cl-amidine treatment prevents joint destruction and reduces C3 deposition in CIA.
Cl-amidine treatment reduces serum and synovial citrulline content in CIA
To help confirm that Cl-amidine derives its effects from its ability to inhibit PAD activity, we measured changes in synovial citrulline content (i.e., the product of PAD reaction) as a proxy of PAD inhibition. As expected, synovial citrulline content trended downward in response to increasing Cl-amidine dose (Fig. 2A, left panel). We then compared the synovial citrulline content in all Cl-amidine–treated mice (all doses combined) with all control mice (mice receiving no treatment or PBS). Mice treated daily for 21 d with Cl-amidine, beginning with one CII immunization, exhibited significantly lower synovial citrulline content compared with PBS-treated or untreated control mice with CIA (p < 0.05), and they showed similar synovial citrulline content compared with unimmunized mice (Fig. 2A, right panel). These results indicated that treatment with daily injections of Cl-amidine for 21 d decreased citrulline content in the joints.
FIGURE 2.
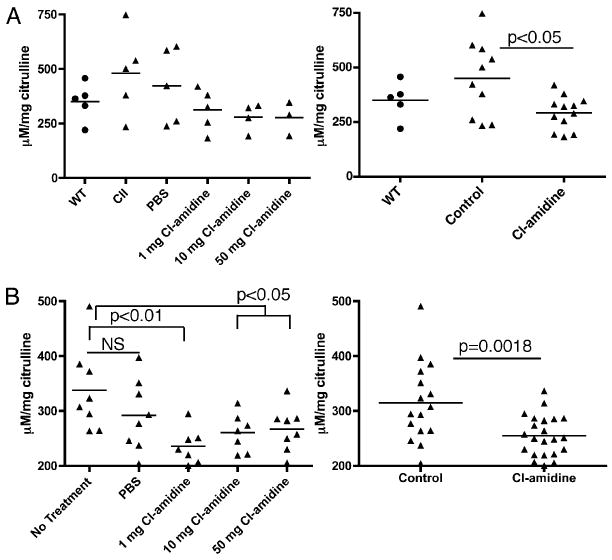
Cl-amidine treatment reduces serum and synovial citrulline content in vivo. A, Synovial citrulline content. Arthritis was induced in control and Cl-amidine–treated mice by a single injection of bovine CII in CFA at day 0. Mice were treated daily, beginning on day 0, for 21 d with Cl-amidine (1, 10, or 50 mg/kg) in PBS or PBS alone or were left untreated. Results are displayed by dose (left panel) and grouped (right panel) as unimmunized (WT), control (PBS and untreated), and Cl-amidine (1, 10, or 50 mg/kg)-treated mice. B, Serum citrulline content was measured in mice at day 35. Results are displayed by dose (left panel) and grouped (right panel) as control (untreated and PBS) and Cl-amidine (1, 10, or 50 mg/kg)-treated mice.
Additionally, we measured the serum citrulline content in the mice with CIA (Fig. 1) at day 35 to verify the activity of Cl-amidine. As expected, serum citrulline content trended downward in response to increasing Cl-amidine dose (Fig. 2B, left panel). We then compared serum citrulline content in all Cl-amidine treated mice (all doses combined) with all control mice (mice receiving no treatment or PBS). Mice treated daily for 35 d with Cl-amidine, beginning with their first CII immunization, exhibited significantly lower serum citrulline content compared with PBS-treated control mice with CIA (Fig. 2B, right panel) (p = 0.0018). These data indicated that Cl-amidine treatment reduces PAD-mediated citrullination.
Cl-amidine treatment does not alter immune cell populations
To examine whether Cl-amidine treatment has effects on the number and distribution of immune cell populations, flow cytometry was performed on splenocytes from mice with CIA treated with Cl-amidine daily for 21 d, beginning with CII immunization (Table I and Table II). No differences were seen in the frequency or absolute number of T, B, or NK cells or monocytes compared with cells from control mice treated with PBS. Therefore, we concluded that the beneficial effects of Cl-amidine on CIA are not accompanied by a depletion of major subsets of immune cells.
Cl-amidine treatment does not ameliorate the effector phase of experimental inflammatory arthritis
We studied the CAIA mouse model of inflammatory arthritis to examine the possibility that the beneficial effects of Cl-amidine in CIA are mediated at the Ab-mediated effector phase. Unlike CIA, the CAIA mouse model bypasses the initial Ag-recognition phases of the immune response through i.p. administration of a mixture of mAb to CII. This is followed by an i.p. injection of LPS on day 3, leading to the rapid and consistent development of inflammatory arthritis.
DBA/1J mice were administered 1, 10, or 50 mg/kg Cl-amidine in PBS or PBS daily i.p. for 7 d prior to receiving Arthrogen (4 mg/ mouse; i.p.) to induce disease. Mice continued receiving daily treatment with PBS or Cl-amidine until sacrifice 10 d after disease induction. Cl-amidine pretreatment in mice with CAIA had no effect on disease activity (Fig. 3A) or histologically evaluated joint damage (Fig. 3B). Clinical disease activity and histology scores showed a significant correlation with each other (R2 = 0.68; p = 0.0063). These results in CAIA contrast with the decrease in disease activity and joint destruction in CIA resulting from treatment with Cl-amidine (Fig. 1). We concluded that Cl-amidine treatment does not ameliorate inflammation and joint tissue destruction in CIA by altering the effector phase of disease.
FIGURE 3.
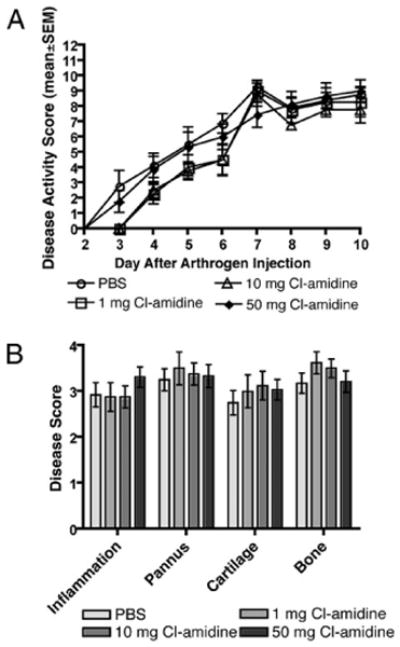
Cl-amidine treatment does not affect passively transferred arthritis. Mice were given daily i.p. injections of PBS or 1, 10, or 50 mg/kg Cl-amidine for 7 d prior to receiving 4 mg Arthrogen i.p. Three days later, mice were injected i.p. with 50 μg LPS. Mice continued to receive PBS or Cl-amidine daily on days 0–10 and were scored for disease severity daily. A, Disease severity scores. B, Histologic sections were scored for changes in synovial inflammation, pannus, cartilage damage, and bone damage, all on a scale of 0–5. (n = 8, PBS; n = 4, Cl-amidine 1 or 10 mg; n = 9, Cl-amidine 50 mg.)
Treatment with Cl-amidine decreases IgG2a and IgG1 anti-mouse CII Abs
Xeno (bovine) and self (mouse) anti-CII Ab evolve over time in CIA, reflecting an expanding loss in tolerance to self-epitopes. To gain a better understanding of how Cl-amidine treatment affects self-tolerance, sera from each treatment group were incubated on 96-well plates coated with bovine or mouse CII. The sera were analyzed by ELISA specific for mouse IgG1 and IgG2a Ab to bovine and murine CII.
Cl-amidine treatment did not alter the anti-bovine CII IgG1 or IgG2a Ab levels at any dose (Fig. 4A). However, there were significantly lower (p ≤ 0.05) IgG1 and IgG2a anti-mouse CII Ab levels in the sera from mice treated with 50 and 10 mg/kg/d Cl-amidine, respectively, compared with treatment with PBS alone at day 35 (Fig. 4B). Notably, Cl-amidine treatment at 10 mg/kg reduced the titer of IgG2a autoantibodies, which are known to be pathogenic in CIA (Fig. 4C) (44, 45). These results demonstrated that Cl-amidine treatment decreases the humoral response to native, but not foreign, CII.
FIGURE 4.
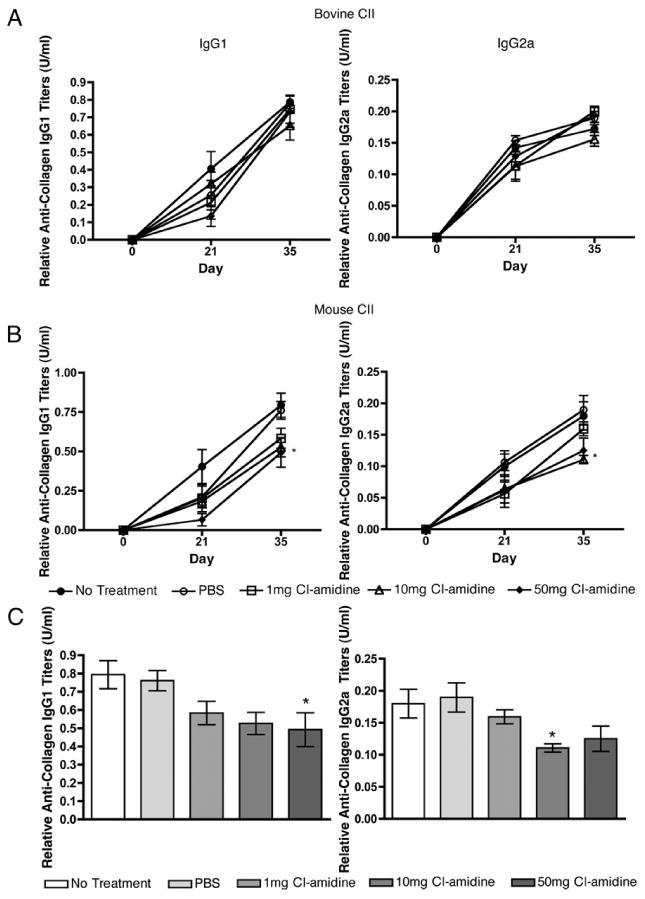
Cl-amidine treatment reduces IgG autoantibody response to mouse but not bovine CII. Sera from individual mice were incubated on a 96-well plate coated with bovine (A) or mouse (B) CII. The presence of anti-mouse IgG1 or IgG2a Ab was detected by ELISA. The 50-mg/kg/d group showed significantly lower IgG1 anti-mouse CII Ab titers, whereas the 10-mg/kg/d group had significantly lower IgG2a anti-mouse CII Ab titers compared with the PBS vehicle control group at day 35. C, IgG1 and IgG2a Ab titers at day 35. *p ≤ 0.05. (n = 8 for all group, except n = 7 for 1-mg group.)
Treatment with Cl-amidine decreases Ab responses to native and citrullinated synovial epitopes
To examine the effects of Cl-amidine treatment on Ab responses, a synovial Ag-array analysis was performed using day-35 sera derived from mice with CIA treated with 50 mg/kg/d Cl-amidine or PBS. These synovial arrays contain 191 peptides and proteins representing candidate Ags in RA, including overlapping peptides spanning the full length of the α- and β-chains of fibrinogen in citrullinated and unmodified forms, as well as peptides from other candidate citrullinated autoantigens, notably vimentin (46). SAM identified eight autoantibody reactivities that were statistically decreased in CIA mice treated with Cl-amidine compared with PBS treatment (Fig. 5A). More than 150 native and citrullinated epitopes on the arrays did not exhibit significant differences in reactivity. Sera from mice treated with 50 mg/kg/d Cl-amidine demonstrated decreased IgG reactivity to native epitopes, including human cartilage gp39, biglycan, and hnRNP-A, as well as decreased autoantibody reactivity to citrullinated filaggrin peptides cfc2 and cfc4, compared with sera from controls treated with PBS. There were no differences between treatment with Cl-amidine or PBS alone in the levels of autoantibodies to 38 other citrullinated proteins in the array. Reactivity to a subset of Ags in Fig. 5A was evaluated by ELISA (Fig. 5B). Mice treated with 50 mg/kg/d Cl-amidine showed a trend of lower reactivity to all Ags tested [HCgp39 (130–145), HCgp39 (160–175), and DEAE pool 1] and a statistically significant decrease to biglycan (227–246) compared with controls. These data demonstrated that Cl-amidine treatment decreases the development of autoantibodies in CIA and suggested that Cl-amidine treatment only modestly impairs epitope spreading.
FIGURE 5.
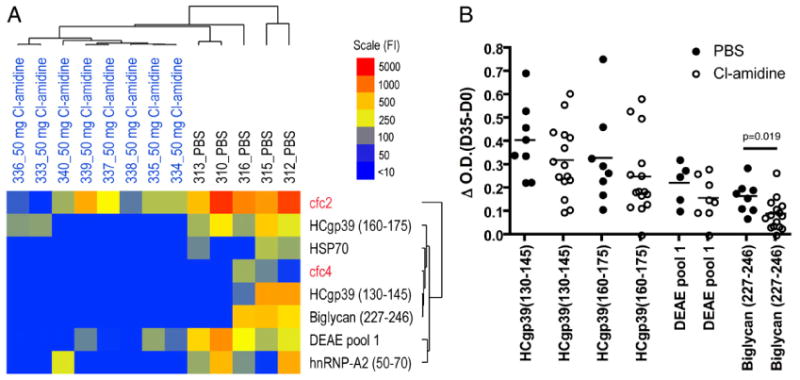
Cl-amidine treatment reduces autoantibody responses in CIA. Sera were collected from Cl-amidine–treated and control mice, and autoantibodies in these samples were profiled using synovial Ag arrays and an anti-IgG/M goat anti-mouse secondary Ab (A) or specific ELISA (B). A, SAM was used to analyze the Ag-array datasets and identified eight Ags with a significant difference in autoantibody reactivity (false discovery rate [q] < 5%). Hierarchical clustering was performed to elucidate the relationships between the autoantibody profiles. The results are displayed as a heatmap (blue, negative; yellow, intermediate; red, strong positive). The Cl-amidine–treated mice clustered together (on the left side of the heatmap) and exhibited lower autoantibody titers against all eight of the Ags identified by SAM, including autoantibody reactivity to native epitopes derived from cartilage and gp39 (native Ags in black type), as well as citrulline-modified filaggrin peptides (red type). B, Serum IgG reactivity to the indicated putative autoantigens from the array was measured by specific ELISA using serum from PBS- and Cl-amidine (10 or 50 mg/kg/d)-treated mice. Day-0 serum reactivity was treated as baseline and subtracted from day-35 serum reactivity. Cl-amidine–treated mice showed a statistically significant decrease in reactivity to Biglycan (227–246) (p = 0.019).
Discussion
Our data indicated that mice treated with Cl-amidine, a novel small molecule PAD inhibitor, exhibit a decrease in severity of clinical disease activity, as well as in joint inflammation and destruction in CIA. In contrast, Cl-amidine treatment had no effect on CAIA, suggesting that it does not alter the Ab-mediated effector phase of disease. Furthermore, Cl-amidine treatment led to a decrease in citrulline content in the joint and serum, as well as a decrease in Ab levels to a subset of potential human citrullinated synovial Ags in RA. We propose that treatment with Cl-amidine ameliorates disease, at least in part, by reducing development of a subset of ACPA, possibly through limiting ACPA epitope availability. In addition, treatment with Cl-amidine led to a decrease in Ab responses to murine CII and to a variety of synovial proteins that are candidate Ags in RA. Notably, our data indicated that the ACPA humoral response is not uniquely affected by Cl-amidine treatment. These results suggested that Cl-amidine treatment may exert broader effects on autoantibody responses than those resulting from decreased citrullinated epitope generation.
We found that Cl-amidine treatment returned CIA synovial citrulline content to unimmunized levels. This suggested that Cl-amidine treatment, at the doses used in this study, does not inhibit all citrullination. It is not likely that insufficient Cl-amidine was bioavailable, because the 10- and 50-mg/kg/d doses showed similar beneficial clinical effects. However, because Cl-amidine preferentially binds activated PAD enzymes (31), it is possible that it is only able to inhibit “active” PADs (i.e., PADs that are present in a sufficiently calcium-rich environment [e.g., extracellularly]). Because all PADs are normally intracellular, and increased levels of citrullination are observed on extracellular proteins during disease (28, 29), it is possible that despite its cell permeability, Cl-amidine mainly inhibits extracellular or aberrant PAD activity.
It is of note that Cl-amidine treatment only partially affected CIA development. If Cl-amidine treatment reduces the availability of citrullinated epitopes then why isn't disease prevented? One possible explanation is that citrulline-reactive T and B cells could be activated in a bystander manner as part of a nonself-specific response, as was described previously (47). Autoreactive citrulline-specific clones activated in the inflammatory environment following immunization with CFA-containing Ag would be primed to react with intracellularly citrullinated molecules (e.g., histones) that are released during cell turnover. These clones could then initiate a break in self-tolerance if Cl-amidine is unable to inhibit intracellular citrullination. The observed decrease, but not elimination, of disease activity in this model would represent the balance between Cl-amidine dose and citrullinated epitope availability.
An alternate explanation for the inability of Cl-amidine treatment to prevent CIA onset completely is that anticitrulline reactivity is not responsible for the initial break in tolerance in CIA, but it amplifies disease severity after disease onset. This is consistent with our previously published data, whereby infusion of ACPA did not initiate disease in mice, but it augmented the severity of CAIA induced by a submaximal dose of anti-CII Abs (32). This was also demonstrated in another citrulline-related autoimmune disease model (48), experimental autoimmune encephalomyelitis (EAE). In EAE, it was shown that citrullinated Ag-reactive T cells were unable to induce disease, but they were able to augment disease in the presence of disease-initiating noncitrulline autoreactive clones. Likewise, it is possible that Cl-amidine prevents activation of citrulline-reactive but not disease-initiating cells, resulting in decreased disease activity but not prevention.
In our study, Cl-amidine treatment impaired the IgG1 and IgG2a response to mouse CII without affecting normal humoral responses to bovine CII (Fig. 3). The mechanism by which Cl-amidine affects the humoral autoimmune response to uncitrullinated epitopes remains to be determined. However, our results may reflect the effects of Cl-amidine on the expression of co-stimulatory or other molecules. Although Cl-amidine had no apparent effect on immune cell frequency or numbers (Table I and Table II), we cannot rule out the possibility that it alters a regulatory cell population (e.g., regulatory T or B cells) to specifically affect the humoral autoimmune response. The balance of regulatory and pathogenic cells is clearly altered in autoimmune arthritis in mice and in RA (49–51). This balance is controlled, in part, by soluble cytokines and chemokines. For example, CXCL12 was reported to alter the balance between regulatory T cells and Th17 cells in EAE (52). Interestingly, citrullination of inflammatory chemokines, including CXCL12 (23–25), was shown to alter immune responses in vitro. Thus, if Cl-amidine is affecting regulatory, effector, or other lymphocytes, it could do so directly or indirectly. Additionally, Cl-amidine may have an effect on any number of targets involved in maintaining tolerance, like IDO, which results in enhanced autoreactive cell responses (53).
Although our data cannot exclude the possibility that Cl-amidine affects any number of autoimmune regulators, it seems that it impairs the humoral autoimmune response, at least in part, by decreasing autoepitope availability. For example, synovial proteome-array analysis of sera from Cl-amidine–treated CIA mice revealed decreased reactivity to several native peptides and a decreased response to a subset of citrullinated peptides compared with controls. These arrays have been particularly useful in studies of CIA, because they demonstrated citrulline-specific reactivity with peptides and epitope spreading during the evolution of disease (32). Previously, we detected ACPA in CIA using anti-cyclic citrullinated peptide (CCP)2 ELISA. However, further investigations by us and other investigators revealed that DBA/1 mice immunized with CII do not produce specific anti-CCP Abs (54, 55). These results are in agreement with our subsequent findings using newly available arginine-containing control CCP2 peptide plates. These findings indicated that anti-CCP2 ELISA kit plate reactivity is not citrulline specific in DBA/1 mice (data not shown), and it is not a useful tool in this strain. Thus, to evaluate ACPA reactivity, we used array-based technologies, as noted above. Our array data also agree with previous studies that suggested that citrulline-related epitope spreading occurs in RA as a result of neoantigen formation as a consequence of inflammatory citrullination (33). This limiting of the spread of autoreactivity may be another explanation for how Cl-amidine treatment reduces disease severity in CIA.
Elevated levels of PAD2 and 4 are found in arthritic synovial tissue and fluid (28, 56). The main source of these PADs in the joint is thought to be inflammatory cells, primarily macrophages and neutrophils. Therefore, it is notable that Cl-amidine did not ameliorate CAIA, a disease model dependent on the function of these cells.
Although it is possible that Cl-amidine treatment affects autoimmunity by inhibiting PAD activities unrelated to epitope generation or through effects on an unanticipated or unknown target(s), addressing these questions was beyond the scope of the current study. However, our data argue that PAD-mediated citrullinated-epitope generation plays an important role in the pathogenesis of inflammatory arthritis, suggesting PADs as a novel therapeutic target for the treatment of RA, and identify Cl-amidine as a candidate therapy for this disease.
Acknowledgments
This work was supported by an American College of Rheumatology Research and Foundation Within Our Reach Grant, a Predoctoral Training Grant in Molecular Biology (National Institutes of Health-National Institute of General Medical Sciences T32 GM008730), in part by National Institutes of Health Grant GM079357, and a Physician Scientist Development Award from the American College of Rheumatology Research and Education Foundation. This work was also supported by National Institutes of Health/National Center for Research Resources Colorado Clinical and Translational Sciences Institute Grant TL1 RR025778.
The contents are the authors' sole responsibility and do not necessarily represent official views of the National Institutes of Health.
Abbreviations used in this article
- ACPA
Abs to citrullinated protein Ags
- CAIA
collagen Ab-induced arthritis
- CCP
cyclic citrullinated peptide
- CIA
collagen-induced arthritis
- CII
type II collagen
- Cl-amidine
N-a-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide
- EAE
experimental autoimmune encephalomyelitis
- PAD
protein arginine deaminase
- RA
rheumatoid arthritis
- SAM
significance analysis of microarrays
Footnotes
The sequences presented in this article have been submitted to Gene Expression Omnibus under accession number GSE23731.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Goldbach-Mansky R, Lee J, McCoy A, Hoxworth J, Yarboro C, Smolen JS, Steiner G, Rosen A, Zhang C, Ménard HA, et al. Rheumatoid arthritis associated autoantibodies in patients with synovitis of recent onset. Arthritis Res. 2000;2:236–243. doi: 10.1186/ar93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bizzaro N, Mazzanti G, Tonutti E, Villalta D, Tozzoli R. Diagnostic accuracy of the anti-citrulline antibody assay for rheumatoid arthritis. Clin Chem. 2001;47:1089–1093. [PubMed] [Google Scholar]
- 3.Avouac J, Gossec L, Dougados M. Diagnostic and predictive value of anti-cyclic citrullinated protein antibodies in rheumatoid arthritis: a systematic literature review. Ann Rheum Dis. 2006;65:845–851. doi: 10.1136/ard.2006.051391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majka DS, Holers VM. Can we accurately predict the development of rheumatoid arthritis in the preclinical phase? Arthritis Rheum. 2003;48:2701–2705. doi: 10.1002/art.11224. [DOI] [PubMed] [Google Scholar]
- 5.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 6.Rantapää-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 7.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000;2:249–251. doi: 10.1186/ar95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavanas S, Méchin MC, Takahara H, Kawada A, Nachat R, Serre G, Simon M. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene. 2004;330:19–27. doi: 10.1016/j.gene.2003.12.038. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 11.Kang CP, Lee HS, Ju H, Cho H, Kang C, Bae SC. A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheum. 2006;54:90–96. doi: 10.1002/art.21536. [DOI] [PubMed] [Google Scholar]
- 12.Takata Y, Inoue H, Sato A, Tsugawa K, Miyatake K, Hamada D, Shinomiya F, Nakano S, Yasui N, Tanahashi T, Itakura M. Replication of reported genetic associations of PADI4, FCRL3, SLC22A4 and RUNX1 genes with rheumatoid arthritis: results of an independent Japanese population and evidence from meta-analysis of East Asian studies. J Hum Genet. 2008;53:163–173. doi: 10.1007/s10038-007-0232-4. [DOI] [PubMed] [Google Scholar]
- 13.Liu GY, Liao YF, Chang WH, Liu CC, Hsieh MC, Hsu PC, Tsay GJ, Hung HC. Overexpression of peptidylarginine deiminase IV features in apoptosis of haematopoietic cells. Apoptosis. 2006;11:183–196. doi: 10.1007/s10495-006-3715-4. [DOI] [PubMed] [Google Scholar]
- 14.Nakashima K, Hagiwara T, Ishigami A, Nagata S, Asaga H, Kuramoto M, Senshu T, Yamada M. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxy-vitamin D(3) J Biol Chem. 1999;274:27786–27792. doi: 10.1074/jbc.274.39.27786. [DOI] [PubMed] [Google Scholar]
- 15.Esposito G, Vitale AM, Leijten FP, Strik AM, Koonen-Reemst AM, Yurttas P, Robben TJ, Coonrod S, Gossen JA. Peptidylarginine deiminase (PAD) 6 is essential for oocyte cytoskeletal sheet formation and female fertility. Mol Cell Endocrinol. 2007;273:25–31. doi: 10.1016/j.mce.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 17.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 18.Hagiwara T, Hidaka Y, Yamada M. Deimination of histone H2A and H4 at arginine 3 in HL-60 granulocytes. Biochemistry. 2005;44:5827–5834. doi: 10.1021/bi047505c. [DOI] [PubMed] [Google Scholar]
- 19.Nakashima K, Hagiwara T, Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem. 2002;277:49562–49568. doi: 10.1074/jbc.M208795200. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, Gilmour DS, Wang Y. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol. 2008;28:4745–4758. doi: 10.1128/MCB.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao H, Li P, Venters BJ, Zheng S, Thompson PR, Pugh BF, Wang Y. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J Biol Chem. 2008;283:20060–20068. doi: 10.1074/jbc.M802940200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Struyf S, Noppen S, Loos T, Mortier A, Gouwy M, Verbeke H, Huskens D, Luangsay S, Parmentier M, Geboes K, et al. Citrullination of CXCL12 differentially reduces CXCR4 and CXCR7 binding with loss of inflammatory and anti-HIV-1 activity via CXCR4. J Immunol. 2009;182:666–674. doi: 10.4049/jimmunol.182.1.666. [DOI] [PubMed] [Google Scholar]
- 24.Proost P, Loos T, Mortier A, Schutyser E, Gouwy M, Noppen S, Dillen C, Ronsse I, Conings R, Struyf S, et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med. 2008;205:2085–2097. doi: 10.1084/jem.20080305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loos T, Mortier A, Gouwy M, Ronsse I, Put W, Lenaerts JP, Van Damme J, Proost P. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: a naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood. 2008;112:2648–2656. doi: 10.1182/blood-2008-04-149039. [DOI] [PubMed] [Google Scholar]
- 26.Ireland J, Herzog J, Unanue ER. Cutting edge: unique T cells that recognize citrullinated peptides are a feature of protein immunization. J Immunol. 2006;177:1421–1425. doi: 10.4049/jimmunol.177.3.1421. [DOI] [PubMed] [Google Scholar]
- 27.Wright PW, Bolling LC, Calvert ME, Sarmento OF, Berkeley EV, Shea MC, Hao Z, Jayes FC, Bush LA, Shetty J, et al. ePAD, an oocyte and early embryo-abundant peptidylarginine deiminase-like protein that localizes to egg cytoplasmic sheets. Dev Biol. 2003;256:73–88. doi: 10.1016/s0012-1606(02)00126-4. [DOI] [PubMed] [Google Scholar]
- 28.Kinloch A, Lundberg K, Wait R, Wegner N, Lim NH, Zendman AJ, Saxne T, Malmström V, Venables PJ. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 2008;58:2287–2295. doi: 10.1002/art.23618. [DOI] [PubMed] [Google Scholar]
- 29.De Rycke L, Nicholas AP, Cantaert T, Kruithof E, Echols JD, Vandekerckhove B, Veys EM, De Keyser F, Baeten D. Synovial intracellular citrullinated proteins colocalizing with peptidyl arginine deiminase as pathophysiologically relevant antigenic determinants of rheumatoid arthritis-specific humoral autoimmunity. Arthritis Rheum. 2005;52:2323–2330. doi: 10.1002/art.21220. [DOI] [PubMed] [Google Scholar]
- 30.Knuckley B, Bhatia M, Thompson PR. Protein arginine deiminase 4: evidence for a reverse protonation mechanism. Biochemistry. 2007;46:6578–6587. doi: 10.1021/bi700095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–973. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kidd BA, Ho PP, Sharpe O, Zhao X, Tomooka BH, Kanter JL, Steinman L, Robinson WH. Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Res Ther. 2008;10:R119. doi: 10.1186/ar2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banda NK, Kraus D, Vondracek A, Huynh LH, Bendele A, Holers VM, Arend WP. Mechanisms of effects of complement inhibition in murine collagen-induced arthritis. Arthritis Rheum. 2002;46:3065–3075. doi: 10.1002/art.10591. [DOI] [PubMed] [Google Scholar]
- 35.Bendele AM, Chlipala ES, Scherrer J, Frazier J, Sennello G, Rich WJ, Edwards CK., III Combination benefit of treatment with the cytokine inhibitors interleukin-1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum. 2000;43:2648–2659. doi: 10.1002/1529-0131(200012)43:12<2648::AID-ANR4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 36.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–1912. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 37.Moscarello MA, Pritzker L, Mastronardi FG, Wood DD. Peptidylarginine deiminase: a candidate factor in demyelinating disease. J Neurochem. 2002;81:335–343. doi: 10.1046/j.1471-4159.2002.00834.x. [DOI] [PubMed] [Google Scholar]
- 38.Pritzker LB, Nguyen TA, Moscarello MA. The developmental expression and activity of peptidylarginine deiminase in the mouse. Neurosci Lett. 1999;266:161–164. doi: 10.1016/s0304-3940(99)00276-1. [DOI] [PubMed] [Google Scholar]
- 39.Knipp M, Vasák M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem. 2000;286:257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 40.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HE, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 41.Hueber W, Kidd BA, Tomooka BH, Lee BJ, Bruce B, Fries JF, Sønderstrup G, Monach P, Drijfhout JW, van Venrooij WJ, et al. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–2655. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]
- 42.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J Exp Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watson WC, Townes AS. Genetic susceptibility to murine collagen II autoimmune arthritis. Proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J Exp Med. 1985;162:1878–1891. doi: 10.1084/jem.162.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hill JA, Bell DA, Brintnell W, Yue D, Wehrli B, Jevnikar AM, Lee DM, Hueber W, Robinson WH, Cairns E. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J Exp Med. 2008;205:967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nickdel MB, Conigliaro P, Valesini G, Hutchison S, Benson R, Bundick RV, Leishman AJ, McInnes IB, Brewer JM, Garside P. Dissecting the contribution of innate and antigen-specific pathways to the breach of self-tolerance observed in a murine model of arthritis. Ann Rheum Dis. 2009;68:1059–1066. doi: 10.1136/ard.2008.089300. [DOI] [PubMed] [Google Scholar]
- 48.Carrillo-Vico A, Leech MD, Anderton SM. Contribution of myelin autoantigen citrullination to T cell autoaggression in the central nervous system. J Immunol. 2010;184:2839–2846. doi: 10.4049/jimmunol.0903639. [DOI] [PubMed] [Google Scholar]
- 49.Benito-Miguel M, García-Carmona Y, Balsa A, Pérez de Ayala C, Cobo-Ibáñez T, Martín-Mola E, Miranda-Carús ME. A dual action of rheumatoid arthritis synovial fibroblast IL-15 expression on the equilibrium between CD4+CD25+ regulatory T cells and CD4+CD25- responder T cells. J Immunol. 2009;183:8268–8279. doi: 10.4049/jimmunol.0900007. [DOI] [PubMed] [Google Scholar]
- 50.Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford) 2009;48:602–606. doi: 10.1093/rheumatology/kep028. [DOI] [PubMed] [Google Scholar]
- 51.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med. 2008;205:2643–2655. doi: 10.1084/jem.20080730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott GN, DuHadaway J, Pigott E, Ridge N, Prendergast GC, Muller AJ, Mandik-Nayak L. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J Immunol. 2009;182:7509–7517. doi: 10.4049/jimmunol.0804328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vossenaar ER, Nijenhuis S, Helsen MM, van der Heijden A, Senshu T, van den Berg WB, van Venrooij WJ, Joosten LA. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 2003;48:2489–2500. doi: 10.1002/art.11229. [DOI] [PubMed] [Google Scholar]
- 55.Cantaert T, Teitsma CA, Tak PP, Baeten DL. Detection of genuine anti-citrullinated protein antibodies in mice reveals their presence in BALB/c but not DBA/1 and SJL mice hyperimmunised with citrullinated collagen. Ann Rheum Dis. 2010;69:A5. [Google Scholar]
- 56.Foulquier C, Sebbag M, Clavel C, Chapuy-Regaud S, Al Badine R, Méchin MC, Vincent C, Nachat R, Yamada M, Takahara H, et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007;56:3541–3553. doi: 10.1002/art.22983. [DOI] [PubMed] [Google Scholar]