

Although more than 100 years of study have demonstrated that the aldol reaction is one of the most fundamental and effective methods for the construction of complex molecules, in particular natural products, its full potential has not been realized.[1] Polyketides, a family of natural products, have provided chemists with a rich source of molecular architectures and biologically significant compounds.[2] Polyketides often contain the 1,3-polyol motif, and unsurprisingly the aldol reaction has been the preferred method to access these structures.[1,2] Unfortunately, however, the most selective aldol products afford ketones or esters[3] and not aldehydes. Thus, the reported biomimetic routes require additional protection and redox steps for each iteration, thus making the preparation of long-chain polyketides excessively lengthy (poor redox economy).[4] Therefore, interest in one-pot polyaldol cascade reactions has increased.[5] Although, several elegant stereoselective approaches have been reported, all the methods inevitably stop after the second aldol reaction because of the cyclization of the hydroxyaldehydes or -ketones. This cyclization could be blocked if the pendant hydroxy groups were rendered non-nucleophilic by an in situ generated blocking variant, which, importantly, would afford aldehydes 2 as products. We recently reported the first high-yielding triple aldol reaction (Scheme 1A).[6] This cascade results in high 1,3-stereoinduction, generated from the extreme bulk of the tris(trimethylsilyl)silyl (supersilyl) group, which also retards undesired polymerization.[7]
Scheme 1.
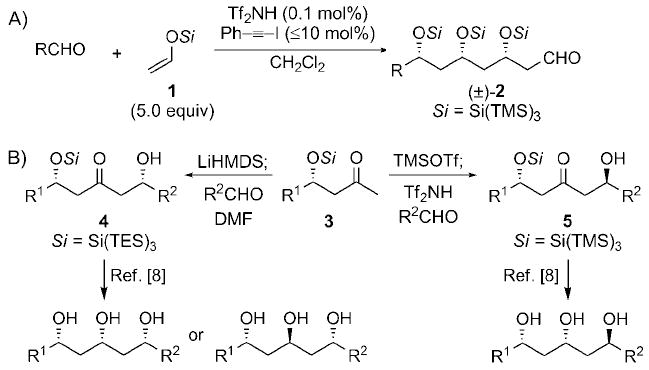
Supersilyl-directed aldol reactions. HMDS = 1,1,1,3,3,3-hexamethyldisilazane, TES = triethylsilyl, Tf = trifluoromethanesulfonyl, TMS = trimethylsilyl.
The use of the bulky β-silyloxy methyl ketones 3 in 1,5-stereoselective aldol reactions with aldehydes produces 4 or 5 with high diastereoselectivity (Scheme 1B).[8] Importantly, all three 1,3,5-triol stereoisomers can easily be prepared from 4 and 5.[8] The utilization of both of these strategies would allow for the facile synthesis of complex 1,3-polyols and spiroketals. Herein we report the rapid total syntheses of EBC-23 and polymethoxy-1-alkene 13 by these approaches.
As part of a screening program to identify new anticancer agents, spiroketal EBC-23 (Scheme 2A) was isolated from the fruit of Cinnamomum laubatii.[9a] This natural product was active in vitro against several human cancer cell lines, and more importantly, inhibited the growth of a human prostate cancer xenograft in mice with no observable side effects.[9c] The structure and absolute stereochemistry of EBC-23 was determined by the Williams research group (in their 15-step total synthesis, 11 steps for the longest linear sequence).[9b]
Scheme 2.
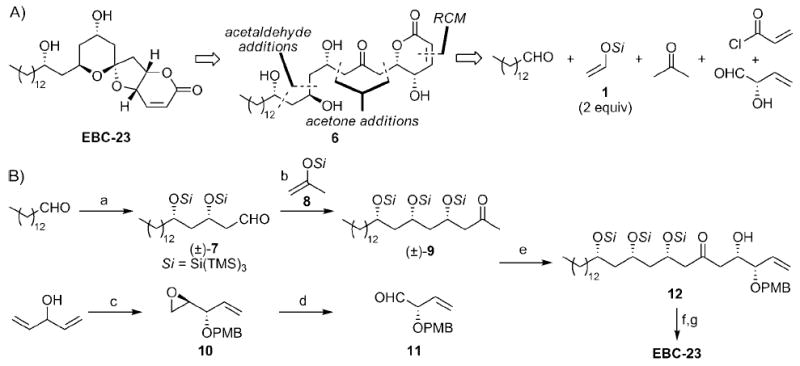
Asymmetric retro- and forward syntheses of EBC-23: a) 1 (2.3 equiv), Tf2NAlMe2 (0.20 mol%), CH2Cl2, −40°C, 85%, 15:1 d.r.; b) 8 (1.8 equiv), Tf2NH (0.30 mol%), 1-iodo-2-phenylacetylene (10 mol%), CH2Cl2, −40°C, 97%, 77:16:6: <1 d.r.; c) see Ref. [10]; then PMBBr (1.4 equiv), NaH (1.2 equiv), nBu4Nl (2 mol%), THF, 0→23°C, 90%; d) KOH (4.0 equiv), H2O/DMSO (1:1), 75°C, 93%; then NalO4 (1.2 equiv), THF/H2O (2:3), 0→23°C, 90→95%; e) LiHMDS (1.2 equiv), toluene, −40→0°C; DMF, −78°C; 11, −78°C, 63% (95% based on recovered 9); f) LiHMDS (1.1 equiv), −78°C; acryloyl chloride (1.5 equiv), −78→23°C; then Zhan cat. 1B[15] (6 mol%), toluene, 95°C; then HF py (excess), py, THF, 0→23°C, 33% (from 12); g) DDQ (2.3 equiv), CH2Cl2, H2O, 23°C, 72%. DDQ = 2,3-dichloro-5,6-dicyano-para-benzoquinone, PMB = para-methoxybenzyl, py = pyridine, RCM = ring-closing metathesis.
Our retrosynthetic analysis of EBC-23 relies on supersilyl-directed aldol methods (Scheme 2A). Hydrolytic opening of the spiroketal reveals polyhydroxy ketone 6, which could be rapidly generated from tetradecanal, two equivalents of acetaldehyde silyl enol ether 1, acetone, an α-hydroxyaldehyde, and acryloyl chloride.
With this strategy in mind, ketone (±)-9 was prepared as shown in Scheme 2B. Tetradecanal[10] was treated with 1 and Tf2NAlMe2[11] to give aldehyde (±)-7 in 85% yield. This aldehyde was treated with silyl enol ether 8 in the presence of Tf2NH and 1-iodo-2-phenylacetylene[6] to furnish (±)-9 in 75% yield and a diastereomerically pure form after column chromatography. The preparation of alkoxyaldehyde 11 commenced with our asymmetric epoxidation of 3-hydroxy-1,4-pentadiene.[12] This epoxy alcohol was converted into 10 in 90% yield according to a literature procedure.[13] Aldehyde 11 was prepared from 10 by a hydrolysis/oxidative cleavage sequence in ≥ 84% (yield over two steps).
The endgame for the synthesis of EBC-23 commenced with the coupling of (±)-9 and 11 under standard conditions,[8] which produced 12 in only 6% yield, presumably because of the low solubility of (±)-9 in DMF (Table 1, entry 1). The addition of 10% (v/v) THF improved the yield slightly, but the solubility of (±)-9 and its putative lithium enolate was still insufficient (entry 2). When the reaction was performed with THF as the lone solvent, 12 was obtained in good yield but poor diastereoselectivity (entry 3). Therefore, the use of DMF as a cosolvent in the aldol reaction was explored (entries 4–8), since we postulate that two molecules coordinate to the lithium atom in the closed transition state.[8] The optimal result was obtained by using toluene as the major solvent (entry 7), which allowed for the preparation of 12 in high diastereoselectivity and 63% yield.[14] This alcohol was then subjected to a one-pot acylation/ring-closing metathesis[15]/HF py deprotection sequence to afford a mixture of anomers in 33% yield (Scheme 2). This mixture was treated with DDQ, which resulted in spiroketalization occurring spontaneously[16] to give EBC-23 in high enantiopurity in a total of ten steps, seven in the longest linear sequence.
Table 1.
Optimization of the coupling of (±)-9 and 11.
| Entry | Solvent(s) (v/v) | T [°C] | 12 [%][a] | d.r.[b] |
|---|---|---|---|---|
| 1 | DMF | −65 | 6 | 48:44:6:2 |
| 2 | DMF/THF (9:1)[a] | −65 | 10 | 48:44:6:2 |
| 3 | THF | −78 | 56 | 47:38:12:3 |
| 4 | Et2O/DMF (19:1)[a] | −78 | 43 | 47:40:10:3 |
| 5 | tBuOMe/DMF (19:1)[a] | −78 | 36 | 47:40:10:3 |
| 6 | CH2Cl2/DMF (19:1)[a] | −78 | 29 | 48:43:7:2 |
| 7 | toluene/DMF (19:1)[a] | −78 | 63 | 48:43:7:2 |
| 8 | CyMe/DMF (19:1)[a] | −78 | 50 | 48:44:6:2 |
Yield of the combined isolated diastereomers.
The diastereomeric ratios were determined by 1H NMR spectroscopic analysis of the crude product. Cy = cyclohexyl.
Polymethoxy-1-alkene 13 was isolated from tolytoxin-producing blue-green algae Tolypothrix conglutinate var.[17] This natural product and several closely related polymethoxy-1-alkenes have generated significant interest in the synthetic community.[17d,18] The structure of 13 was proved by the Mori research group in their 21-step total synthesis,[17d] and recently, it was prepared by Taylor and co-workers in 16 steps.[18g]
We anticipated that (±)-13 could be prepared rapidly, by using our methods, from acetone, 3-butenal, three equivalents of acetaldehyde silyl enol ether 1, and n-hexanal (Scheme 3A). The forward synthesis commenced with the preparation of β-siloxy methyl ketone (±)-15 in 75% yield from 3-butenal[19] and silyl enol ether 14 in the presence of Tf2NAlMe2 (Scheme 3B). Hexanal was treated with silyl enol ether 1 in the presence of Tf2NH and 1-iodo-2-phenylacetylene to afford (±)-16 in 79% yield with high syn selectivity. The next key step was the union of (±)-15 and (±)-16 by a 1,5-syn aldol reaction. Initial attempts to assemble (±)-17 under standard conditions gave the desired adduct in 21% yield, which left room for improvement. Presumably, the somewhat low yield was due to the extreme steric bulk of ketone (±)-15 and aldehyde (±)-16. Fortunately, the addition of lithium tetrafluoroborate[20] allowed for the generation of (±)-17 and other isomers in 64% yield.[21] Stereoselective reduction of the ketone with NaBH4 gave (±)-18 in 89% yield. Finally, removal of the silyl protecting groups by irradiation with UV light[8] and methylation gave natural product (±)-13 in a total of ten steps, seven in the longest linear sequence, from commercially available chemicals.
Scheme 3.
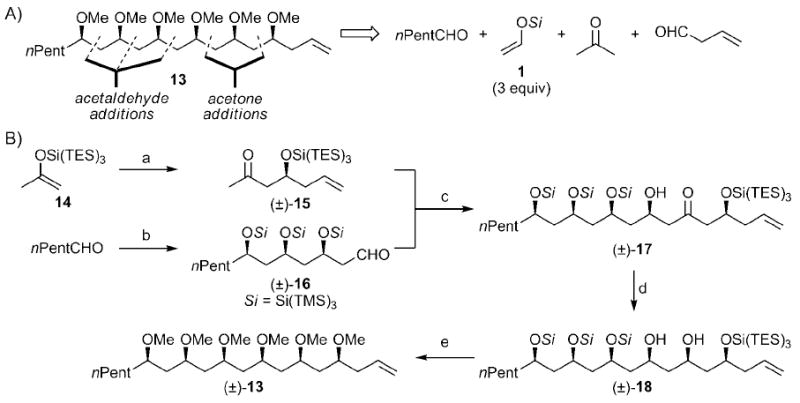
Retro- and forward syntheses of polymethoxy-1-alkene 13: a) 3-butenal (1.2 equiv), Me2AlNTf2 (0.5 mol%), CH2Cl2, 0°C, 75%; b) 1 (5.0 equiv), Tf2NH (0.05 mol%), 1-iodo-2-phenylacetylene (10 mol%), CH2Cl2, −40°C, 79%, 80:12:5:3 d.r.; c) LiHMDS (1.2 equiv), LiBF4 (5.0 equiv), DMF, −60°C, 64%; d) NaBH4 (10 equiv), MeOH, −20°C, 89%, > 10:1 d.r.; e) UV light, MeOH/CH2Cl2 (4:1), 23°C; then Mel (40 equiv), NaH (20 equiv), THF, 0→23°C, 61% (yield over two steps).
In summary, the concise stereoselective total synthesis of EBC-23 and (±)-polymethoxy-1-alkene 13 have been achieved by using supersilyl-directed aldol reactions. Some of the salient points of this report are: 1) the syntheses of EBC-23 and 13 are the shortest routes to date, made possible by supersilyl chemistry, 2) the syntheses are redox-economical, requiring few redox manipulations, and 3) the supersilyl methods should allow the ready synthesis of various stereoisomers.
Supplementary Material
Footnotes
This work was made possible by the generous support of the NIH (P50GM086 145-01) and a Uehara Foundation fellowship (Y.Y). We would additionally like to thank Antoni Jurkiewicz for his NMR expertise and Chang-Jin Qin for his assistance with mass spectrometry.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201007210.
References
- 1.For aldol reactions using ester, thioester, and ketone enolates as nucleophiles with aldehydes, see Mukaiyama T. In: Organic Reactions. Dauben WG, Boswell GA Jr, Danishefsky S, Gschwend HW, Heck RF, Hirshchmann RF, Kende AS, Paquette LA, Posner GH, Trost BM, Bittman R, Weinstein B, editors. Vol. 28. Wiley; New York: 1982. pp. 203–331.; Heathcock CH, Kim BM, Williams SF, Masamune S, Rathke MW, Weipert P, Paterson I. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Pergamon; Oxford: 1991. pp. 133–319.; Nelson SG. Tetrahedron: Asymmetry. 1998;9:357–389.; Mahrwald R. Chem Rev. 1999;99:1095–1120. doi: 10.1021/cr980415r..
- 2.Rohr J. Angew Chem. 2000;112:2967–2969.; Angew Chem Int Ed. 2000;39:2847–2849. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0.; Rychnovsky SD. Chem Rev. 1995;95:2021–2040.; Koskinen AMP, Karisalmi K. Chem Soc Rev. 2005;34:677–690. doi: 10.1039/b417466f..
- 3.Or derivatives of the ester oxidation state.
- 4.For the importance of redox economy, see Burns NZ, Baran PS, Hoffmann RW. Angew Chem. 2009;121:2896–2910. doi: 10.1002/anie.200806086.; Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086..
- 5.Gijsen HJM, Wong C-H. J Am Chem Soc. 1995;117:7585–7591.; Yun S-S, Suh I-H, Choi S-S, Lee S. Chem Lett. 1998:985–986.; Haeuseler A, Henn W, Schmittel M. Synthesis. 2003:2576–2589.; Northrup AB, MacMillan DWC. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710.; Casas J, Engqvist M, Ibrahem I, Kaynak B, Córdova A. Angew Chem. 2005;117:1367–1369. doi: 10.1002/anie.200461400.; Angew Chem Int Ed. 2005;44:1343–1345. doi: 10.1002/anie.200461400.; Wang X, Meng Q, Perl NR, Xu Y, Leighton JL. J Am Chem Soc. 2005;127:12806–12807. doi: 10.1021/ja053593s..
- 6.Albert BJ, Yamamoto H. Angew Chem. 2010;122:2807–2809.; Angew Chem Int Ed. 2010;49:2747–2749. doi: 10.1002/anie.200907076..
- 7.Boxer MB, Yamamoto H. J Am Chem Soc. 2006;128:48–49. doi: 10.1021/ja054725k. [DOI] [PubMed] [Google Scholar]
- 8.Yamaoka Y, Yamamoto H. J Am Chem Soc. 2010;132:5354–5356. doi: 10.1021/ja101076q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddell PW, Gordon VA. WO 2007070984A1 20070628 PCT Int Appl. 2007; Dong L, Gordon VA, Grange RL, Johns J, Parsons PG, Porzelle A, Reddell P, Schill H, Williams CM. J Am Chem Soc. 2008;130:15262–15263. doi: 10.1021/ja807133p.; Dong L, Schill H, Grange RL, Porzelle A, Johns JP, Parsons PG, Gordon VA, Reddell PW, Williams CM. Chem Eur J. 2009;15:11307–11318. doi: 10.1002/chem.200901525..
- 10.Obtained by the oxidation of 1-tetradecanol with IBX: Wiseman JM, McDonald FM, Liotta DC. Org Lett. 2005;7:3155–3157. doi: 10.1021/ol050829o..
- 11.Marx A, Yamamoto H. Angew Chem. 2000;112:182–184.; Angew Chem Int Ed. 2000;39:178–181..
- 12.Li Z, Zhang W, Yamamoto H. Angew Chem. 2008;120:7630–7632.; Angew Chem Int Ed. 2008;47:7520–7522. doi: 10.1002/anie.200802523..
- 13.Nakatsuka M, Ragan JA, Sammakia T, Smith DB, Uehling DE, Schreiber SL. J Am Chem Soc. 1990;112:5583–5601. [Google Scholar]
- 14.Noncoordinating solvents were found to be important. See the Supporting Information for full details.
- 15.The catalyst was purchased from Strem Chemicals. 2007/0043180A1 Patents US PCT, WO 2007/003135A1.
- 16.Williams and co-workers reported[9b] difficulties in acid-promoted spirocyclization, but success with a postulated ceriumtemplated preorganized spirocyclization. However, fortunately, greater than 50% spirocycliztion (additional cyclization during silica gel chromatography) occurred with DDQ under the acidic deprotection conditions.
- 17.a) Desikachary TV. Cyanophyta. Indian Council of Agricultural Research; New Delhi: 1959. p. 503. [Google Scholar]; b) Mynderse JS, Moore RE. Phytochemistry. 1979;18:1181–1183. [Google Scholar]; c) Carmeli S, Moore RE, Patterson GM, Mori Y, Suzuki M. J Org Chem. 1990;55:4431–4438. [Google Scholar]; d) Mori Y, Kohchi Y, Suzuki M, Carmeli S, Moore RE, Patterson GM. J Org Chem. 1991;56:631–637. [Google Scholar]
- 18.Previous syntheses of 13 and related compounds: Nakata T, Suenaga T, Oishi T. Tetrahedron Lett. 1989;30:6525–6528.; Nakata T, Suenaga T, Nakashima K, Oishi T. Tetrahedron Lett. 1989;30:6529–6532.; Priepke H, Weigand S, Brückner R. Liebigs Ann. 1997:1635–1644.; Priepke H, Brückner R. Liebigs Ann. 1997:1645–1655.; Weigand S, Brückner R. Liebigs Ann. 1997:1657–1666.; Allerheiligen S, Brückner R. Liebigs Ann. 1997:1667–1676.; Liu K, Arico JW, Taylor RE. J Org Chem. 2010;75:3953–3957. doi: 10.1021/jo100094d..
- 19.Obtained from glyoxal: Crimmins MT, Kirincich SJ, Wells AJ, Choy AL. Synth Commun. 1998;28:3675–3679..
- 20.LiBF4 was the best source of lithium screened. The use of LiCl, LiI, and LiNTf2 as additives gave 17 in 45, 23, and 41 % yields, respectively.
- 21.See the Supporting Information for full details of this reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.