

Abstract
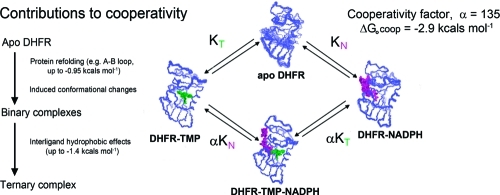
In order to examine the origins of the large positive cooperativity (ΔG0coop = −2.9 kcal mol−1) of trimethoprim (TMP) binding to a bacterial dihydrofolate reductase (DHFR) in the presence of NADPH, we have determined and compared NMR solution structures of L. casei apo DHFR and its binary and ternary complexes with TMP and NADPH and made complementary thermodynamic measurements. The DHFR structures are generally very similar except for the A−B loop region and part of helix B (residues 15−31) which could not be directly detected for L. casei apo DHFR because of line broadening from exchange between folded and unfolded forms. Thermodynamic and NMR measurements suggested that a significant contribution to the cooperativity comes from refolding of apo DHFR on binding the first ligand (up to −0.95 kcals mol−1 if 80% of A−B loop requires refolding). Comparisons of Cα−Cα distance differences and domain rotation angles between apo DHFR and its complexes indicated that generally similar conformational changes involving domain movements accompany formation of the binary complexes with either TMP or NADPH and that the binary structures are approaching that of the ternary complex as would be expected for positive cooperativity. These favorable ligand-induced structural changes upon binding the first ligand will also contribute significantly to the cooperative binding. A further substantial contribution to cooperative binding results from the proximity of the bound ligands in the ternary complex: this reduces the solvent accessible area of the ligand and provides a favorable entropic hydrophobic contribution (up to −1.4 kcal mol−1).
The enzyme dihydrofolate reductase (DHFR; 5,6,7,8-tetrahydrofolate:NADPH oxidoreductase, EC 1.5.1.3) catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) using NADPH as coenzyme.(1) Since THF and its metabolites are precursors of purine and pyrimidine bases, the normal functioning of this enzyme is essential for proliferating cells. This makes DHFR an excellent target for antifolate drugs such as methotrexate (anticancer), pyrimethamine (antimalarial), and trimethoprim (antibacterial). Such agents act by inhibiting the enzyme in parasitic or malignant cells.1,2 The cooperative binding of ligands to DHFR plays an important role not only in the enzyme catalytic cycle (negative cooperativity in THF/NADPH binding)(3) but also in enzyme inhibition (positive cooperativity in antifolate/NADPH binding).(4) The effects of positive cooperative binding in controlling enzyme inhibition are exemplified by trimethoprim (TMP) an effective antibacterial agent that binds to bacterial DHFR 135 times more tightly in the presence of NADPH (corresponding to a ΔG0coop = −RT ln 135 = −2.9 kcal mol−1).(5) Futhermore, TMP binds at least 3000 times more tightly to bacterial DHFR than it does to human DHFR,(4) and part of the decrease in TMP binding to the human enzyme can be attributed to the loss of the positive cooperative binding that is seen in the complex with the bacterial enzyme.(6) Several X-ray(7) and NMR8−12 structures of TMP complexes with bacterial and vertebrate DHFR have been studied previously.
In earlier studies of ligand binding cooperativity, Bystroff and Kraut have compared the crystal structure of E. coli apo DHFR with those of its complexes with folate, methotrexate, and NADP+ and have shown that there are no major differences in the overall structures except that part of the A−B loop region was disordered and could not be detected in the X-ray structure of the apo E.coli enzyme and its binary complexes.(13) By comparing pairs of structures, they detected ligand-induced changes in Cα−Cα distances (mostly less than 1 Å) and rotational angles (up to 6°) about a hinge axis between the adenosine binding domain (residues 38−88) and the major domain (residues 2−37 and 89−160)) involved in substrate binding and catalytic activity.
In this work we report the NMR solution structures of apo lc DHFR and its DHFR.NADPH binary complex and compare these with our previously determined structures of the DHFR.TMP binary(12) and DHFR.TMP.NADPH ternary(10) complexes. The availability of the structures and chemical shifts for all four components of this system now allows us to comment on the most likely contributions to the observed positive cooperativity. We have also examined 1H/15N chemical shifts for complexes which represent well-characterized examples of positive and negative cooperative binding5,14 to explore whether or not the ligand-induced shifts in regions remote from the ligand binding site provide any information about the putative shrinkage or expansion of the protein complexes showing positive and negative cooperativity.15,16
Materials and Methods
Sample Preparation
Lc DHFR was expressed in E. coli containing the lc DHFR gene, and the protein was isolated and purified as described earlier.17,18 The E. coli cells were grown in minimal media supplemented with l-tryptophan. The lc DHFR samples were uniformly enriched with 15N and 13C /15N by providing [15N]-ammonium sulfate (C. K. Gas Products Ltd.) as the sole nitrogen source and [13C6]-d-glucose (C. K. Gas Products Ltd.) as the sole carbon source to the growth medium. Ligands (NADPH, TMP, and folinic acid) were obtained from Sigma and used without further purification. Equimolar complexes of DHFR.TMP, DHFR.NADPH, DHFR.folinic acid, DHFR.TMP.NADPH, and DHFR.folinic acid.NADPH were prepared by adding excess ligands to 1−4 mM solutions of enzyme in 50 mM potassium phosphate and 100 mM KCl, pH* = 6.5 (the pH* values being meter readings, unadjusted for deuterium isotope effects). The samples were taken up either in D2O or in 95% H2O/5% D2O. In all the above complexes the ligands are tightly bound, and their spectra are characteristic of slow exchange between bound and free forms.
1D NH residual dipolar couplings were measured in a ternary mixture of ∼5% (v/v) n-octylpenta(ethylene glycol) (C8E5), ∼1.5% (v/v) n-octanol, and NMR buffer.(19) The liquid crystalline medium gave a stable quadrupolar splitting of the D2O 2H signal of 29.9 Hz, with a final concentration of apo-DHFR of ∼0.21 mM. Precise measurements of one-bond 1H−15N coupling constants were obtained (see Figure S1 in Supporting Information) from a series of 1JNH-modulated 2D spectra.(20)
NMR Experiments
All NMR experiments were performed on Varian UNITY, UNITY plus, and INOVA spectrometers equipped with z-gradient triple-resonance probes operating at 500, 600, and 800 MHz (1H frequency). The spectra were recorded at various temperatures in the range 8−35 °C using unlabeled, 15N-labeled, or 13C/15N-labeled protein samples.
The 1H chemical shifts were referenced to DSS by converting shifts from values obtained using the 1H signal from either internal dioxane or from the water as reference; the 15N and 13C chemical shifts were referenced to liquid NH3 and TSP, respectively, using the γ ratio method.21,22 The 1H, 13C, and 15N chemical shift data have been deposited in the BioMagResDataBank (Deposition Numbers BMRB ID: BMRB ID: 17125, BMRB ID: 5396, BMRB ID: 17310 and BMRB ID: 17311).
The details of the procedures for spectral processing and analysis, signal assignment, distance restraints and hydrogen bond restraints determinations, structure calculations, structure analysis, and methods used for checking the quality of the structures are all described in the Supporting Information. The numbers and distributions of NOEs are given in Table S1 of the Supporting Information.
Cα Distance Difference Plots
Cα distance difference (ΔD) plots have been proposed as an unbiased way of comparing pairs of protein structures(23) and for identifying domains within the proteins.(24) In this method the distances between all pairs of Cα atoms in one protein structure are subtracted from the corresponding distances in a second related structure and the results presented as a 2-dimensional contour plot as shown in Figure 1. Each point in the plot has the value
where D1(i,j) is the distance between the Cα atoms in residues i and j in structure 1 and D2(i,j) is the corresponding distance in structure 2. The ΔD(i,j) values are then plotted on a square matrix with the residue numbers of the protein on the axes (i on one axis and j on the other). Such plots are symmetrical about the diagonal and for convenience it is usual to display the positive and negative ΔD(i,j) values separately, one in the triangle above the diagonal (negative values in Figure 1) and the others (positive values) below the diagonal.
Figure 1.

2D ΔD plots of the Cα distance differences (Å) between structures of (A) lc DHFR.TMP and apo DHFR, (B) lc DHFR.NADPH and apo DHFR, and (C) lc DHFR.TMP.NADPH and apo DHFR. The triangular space above the diagonal contains the negative ΔD values corresponding to shortening of distances between pairs of Cα atoms on formation of the complex. Blocks 1−7 are discussed in the text. The unshaded regions of the plots contain the Cα distance differences between the adenosine binding domain residues (38−88) and the major domain residues (2−37, 89−160). The data for the residues 15−31 (unfolded in apo DHFR) have been omitted from the figures.
We used contour plots to display the distance differences (ΔD(i,j) values) between pairs of corresponding Cα atoms (i, j) for the protein in two structures such as apo DHFR and the DHFR.TMP binary complex (and for other pairs of structures). The distances were measured from the structures using Insight and the contour plot program was an extension of EXCEL.
Structure Analysis Programs
The program DynDom(25) was used to determine changes in rotational angles about a twist axis for defined domains in pairs of related structures (see web platform http://fizz.cmp.uea.ac.uk/dyndom/dyndomMain.do). The ligand induced changes in rotational angles are averaged values from measurements on 25 pairs of structures for each comparison (apo DHFR/DHFR.TMP, apo DHFR/DHFR.NADPH, and apo DHFR/DHFR.TMP.NADPH).
Plots of Connolly(26) surfaces (solvent-excluded surfaces) were made using the Insight program. Calculations of solvent accessible surface areas (SASA) were made using the NACCESS program:(27) the SASA values for each ligand in the region of interligand contact were obtained by first calculating the total SASA values for the two bound ligands (TMP and NADPH) using the structure of the ternary complex (PDB ID: 1LUD or PDB ID: 1YHO) and then measuring the increase in SASA values after removal of the structure of each ligand in turn from the ternary complex structural data (PDB ID: 1LUD (L. casei) or PDB ID: 1YHO (human)).
ITC Experiments
Enthalpies of binding were measured for TMP, folinic acid and NADPH binding to lc DHFR in binary and ternary complexes at 25 °C using a MicroCal Omega VP-ITC (MicroCal Inc., Northampton, MA). The DHFR was dialyzed against buffer containing 50 mM potasium phosphate (pH 7.0) and 100 mM KCl. The ligand solutions were made with the same buffer.
Solutions of TMP (150−440 μM), (±)-folinic acid (70−1500 μM), and NADPH (120−240 μM) were titrated in 30 injections of 10 μL aliquots. The concentration of lc DHFR in the cell was 3.5−73 μM.
The ITC data were processed with the software MicroCal Origin version 5.0 provided by the manufacturer.
Results and Discussion
Structures of Apo L. casei DHFR and Its Complexes with TMP and NADPH
We have used NMR spectroscopy to determine the solution structures of apo lc DHFR and its binary complex with NADPH and TMP(12) (PDB ID: 2L28, PDB ID: 2HQP, and PDB ID: 2HM9). A summary of the restraints used in the structure calculations is given in Table S1 of the Supporting Information. These structures together with the structure of the ternary complex lc DHFR.TMP.NADPH (PBD ID: 1LUD)(10) are shown in Figures 2 and 3. The protein residues with NH protons within 4 Å (and 10 Å) of the bound ligands in the binary and ternary complexes are indicated in Table S2 (Supporting Information). The above results allowed us to define structural differences between the enzyme in its bound and unbound states for all four components of a system showing positive binding cooperativity.
Figure 2.

Disposition of the secondary structure elements in structures of apo lc DHFR and its complexes DHFR.NADPH, DHFR.TMP, and DHFR.NADPH.TMP obtained using the Kabsch_Sander program in Insight.(39)
Figure 3.

Families of calculated structures for apo lc DHFR (25 structures) and its DHFR.NADPH complex (30 structures).
Apo lc DHFR
The NMR data analysis led to a family of 25 structures for the apo lc DHFR with a backbone rmsd of 1.42 Å. Superimposition of the well-defined regions of lc DHFR (residues 1−12 and 37−161) gave a backbone rmsd of 0.63 ± 0.12 Å: superimposition of these regions of apo lc DHFR on the structures of its binary TMP and NADPH and ternary DHFR complexes gave backbone rmsd values of 1.91 ± 0.15, 1.43 ± 0.16, and 1.91 ± 0.18 Å, respectively. The overall core structure of apo lc DHFR comprising the 8-stranded β-sheet and three of the four α-helices is well-determined. However, the region involving residues 15−31 (corresponding to part of the long A−B loop and part of helix B) is partially unfolded as evidenced by the severe line broadening observed for their backbone NH and α-CH signals which prevented their detection for many residues. The 1H, 13Cα, and 15N chemical shifts for only 7 residues in the region 15−31 could be measured directly from their weak broad signals: no signals could be detected for the remaining residues because of exchange line broadening presumably arising from interconversions between different conformational states.
The 1H and 15N chemical shifts for residues that could not be directly detected in the HSQC spectra were obtained by titrating apo lc DHFR with p-aminobenzoyl-l-glutamate (PABG): this sharpens the signals and allows one to extrapolate the titration shifts to zero PABG concentration (see Tables S2 and S3, Supporting Information).(10) The lc DHFR.PABG complex and the folded form of apo lc DHFR are in fast exchange, and thus the extrapolated chemical shift values correspond to the folded form of apo DHFR. The simplest model for the exchange behavior in apo DHFR would involve interconversions between two different conformational states for the region containing the A−B loop and helix B. Chemical shift evidence (from the PABG titration) indicates that one of these states is the folded closed conformation of the A−B loop. Estimates of line broadening sufficient to prevent detection can be made using a two-site exchange analysis (using the in-house program MUSES(28)) assuming various interconversion rates, chemical shift differences, and fractional populations of the two forms. It is found, for example, that for a 50/50 mixture of folded and unfolded forms with shift differences in the range 100−500 Hz and with exchange rates of 100−300 s−1 contributions to the line widths of between 32 and 88 Hz would be expected. Such contributions when considered together with the usual line widths of NH signals (∼15 Hz) would make it difficult to directly observe the signals affected by the exchange. Calculated line widths for other ratios of conformers and even higher rates would also give fairly broad undetectable signals. An approximate estimate of the fraction of the folded form can be obtained from considering the intensities of signals from L12, D16, and D25 which give small signals at the shift values expected for the folded conformation (confirmed by the PABG titrations). These are the only residues in the loop region 12−25 that have clearly observable 1H15N HSQC signals in nonoverlapped regions and have relative intensities of 0.20 ± 0.05 compared to nonloop signals indicating that ∼20% of the loop is folded at 20 °C. This is an approximate value because of the difficulties of measuring intensities of broad signals in HSQC spectra. If 80% of the apo DHFR has an unfolded A−B loop and binds only weakly to ligands, this would effectively reduce the ligand binding constant to apo DHFR by a factor of 5 compared to fully folded DHFR. This loss in binding could be recovered after binding the first ligand, contributing −0.95 kcals mol−1 to the cooperativity (ΔG0 = −RT ln 5). Evidence for the presence of folded and unfolded forms of L. casei apo DHFR is also provided by kinetic experiments of inhibitors binding to apo DHFR reported by Dunn and co-workers.(29) Their measurements of the association rates revealed a two-step process, a “fast” step followed by a “slower” step: in the initial “fast” step ∼50% of the DHFR binds with a very rapid association rate at pH 6.5, consistent with the inhibitor binding to a folded form of the protein.(29)
It can be seen from Tables S2 and S3 that the 1H and 15N chemical shifts for helix B residues in apo DHFR extrapolated from the PABG titrations agree well with those for the DHFR.NADPH complex where helix B residues are remote from the ligand. This indicates that the folded structure of helix B remains essentially intact in the fraction of apo DHFR molecules in the folded form (only the folded fraction of apo lc DHFR is being monitored in the PABG titration experiment).
Because most of the helix B residues show broad or undetectable resonances, it was not possible to measure any NOEs in this region although some TALOS based ϕ/ψ dihedral angle information could be obtained for residues 30−32. Only two long-range restraints for helix B (Y29He−W5HN and D25Od1−H153He1) could be measured. Thus, helix B and its position in the structure are poorly defined as shown in Figure 3A (even though some additional helical ϕ/ψ dihedral angle constraints based on chemical shifts were included in the calculation).
For the A−B loop, the few NOEs that were measured (namely between I13 and K127, L12 and D125, K15 and D125, and L12 and E123) were those expected for a closed conformation.
Lc DHFR.NADPH Binary Complex
The family of 30 calculated conformers has a backbone rmsd of 0.50 ± 0.07 Å. Superimposition of the representative structure of the binary complex onto that of the lc DHFR.TMP.NADPH ternary complex gives a backbone rmsd of 1.46 ± 0.13 Å. In both the binary and ternary complexes, the NADPH is bound in an extended conformation along a groove on the surface of the protein. The A−B loop region of the NADPH binary complex is found in the closed conformation and has a more extensive network of hydrogen bonds than in the lc DHFR.TMP complex. This is because of its proximity to the nicotinamide part of the NADPH molecule. The nicotinamide ribose ring protons HO2 and HO3 are H-bonded to G17(O) and H18(N), respectively. The interactions of I13(O) with a 7-NH2 and the H2 protons of NADPH are important for cofactor binding. The antiparallel β-sheet within the A−B loop is stabilized by the H-bond L15(NH)−H18(O). The T126 carbonyl oxygen accepts H-bonds from both I13(NH) and G14(NH) protons and G14(O) is H-bonded to T126(NH)—unlike in many other complexes (such as in lc DHFR.MTX) where it binds D125(NH).
The positions of the nicotinamide and adenine rings in the bound cofactor are fairly well-defined. The nicotinamide carboxamide group is in the trans conformation which allows its O7 and 7-NH2 atoms to form hydrogen bonds with NH of A6 and O of I13.
Some hydrophobic contacts (evidenced by corresponding NOEs) are also observed between the nicotinamide ring protons and the side-chain protons of A6 and Hα protons of G99. The adenine ring exhibits a better-developed pattern of hydrophobic interactions with protein residues than does the nicotinamide ring. In particular, several NOEs are observed between adenine ring protons and side-chain protons of residues L62, T63, H64, and H77 in the adenine binding pocket.
The ribose rings are somewhat less well-defined than the aromatic moieties; however, the data firmly suggest that both of them are bound in the 3′-endo conformation. The nicotinamide ribose ring is located close to residues G17, R44, and S48 while the adenine ribose ring is near residues R43, Q101, and I102. The pyrophosphate linkage is the least well-defined part of the bound cofactor—it was refined at a very late stage of the calculations by identifying possible hydrogen bonds from oxygen atoms of phosphates to hydrogens of the backbone. Identified interactions involve NHs of T45, G99, A100, and Q101. In the same fashion the position of the 2′-phosphate group was refined; it forms hydrogen bonds with T63(NH) and with R43 and Q65 side chains. All protons that are postulated as serving as hydrogen bond donors were found to have high protection factors in H/D exchange experiments. Chemical shifts were also used to help in assigning hydrogen bonds (for example, Ala100 NH shows a 2.48 ppm increase in 1H shielding on NADPH binding). Additionally, titrations of lc DHFR.TMP with phosphate buffer show fast exchange shifting of signals from relevant NH protons involved in interactions with the coenzyme phosphate group.
The DHFR.NADPH complex is more stable than apo DHFR, reflecting the increased stability of its more folded structure (particularly in the loop and helix B regions). At the prevailing concentrations of unbound NADPH in the cell (estimated around 2−5 μM30,31) most of the DHFR (97 to 99%) will be complexed with NADPH. Any evolutionary processes aimed at increasing the stability of the enzyme (in terms of producing a more folded structure) will have operated on the DHFR.NADPH complex rather than on apo DHFR, thus offering an explanation for the lower stability of the partially unfolded apo DHFR.
Lc DHFR.TMP Binary and lc DHFR.TMP.NADPH Ternary Complexes
The NMR-determined structure of the ternary lc DHFR.TMP.NADPH complex has been described previously (PBD 1LUD),(10) and the representative structure from a family of 32 structures has been used for the structural comparisons in the present work. The structure of the lc DHFR.TMP binary complex was also published earlier (PBD 2HM9),(12) and further details of the structure are provided in the Supporting Information.
Comparisons of the Structures
It can be seen from Figure 2 that the structures of apo lc DHFR and its binary and ternary complexes are fairly similar in terms of their overall secondary structure composition and disposition. The most notable differences involve the A−B loop in apo lc DHFR. The A−B loop and the unfolded part of helix B of apo DHFR is refolded on forming the binary complexes with obvious implications for cooperative binding.
The A−B Loop (Residues 9−23)
Conformational changes involving the A−B loop could be important in controlling cooperative binding because of its proximity to important ligand binding sites. A−B loop conformations, classified by Sawaya and Kraut(7) as either “open”, “closed”, or “occluded” forms, have been extensively studied for many DHFR complexes by X-ray crystallography(7) and NMR spectroscopy.32−37
The structure of apo lc DHFR in solution has a partially unfolded A−B loop (see Figure 3A) whereas in its binary and ternary complexes with TMP and NADPH the A−B loop is in the closed conformation. This can be seen in Figure 4, which shows the A−B loops extracted from the superimposed structures in the binary and ternary complexes with TMP and NADPH. These A−B loops are compared with the same loop in the E. coli DHFR complexes (PDB ID: 1rh3 and PDB ID: 1rx5)(7) where it was previously found in the closed and occluded conformation, respectively. Although the lc DHFR complexes all show the closed conformation, there are substantial differences in the positions of this loop in the binary and ternary complexes. It can be seen in Figure 4 that in the lc DHFR.TMP complex the closed conformation A−B loop partially occupies the NADPH nicotinamide ribose binding site and could potentially displace any bound water molecules in this part of the NADPH binding site. However, the incoming NADPH on binding to the DHFR.TMP complex no longer needs to find the energy to organize the partially unfolded A−B loop into its required closed conformation, and this will contribute to the positive cooperative binding. The A−B loop in the lc DHFR.NADPH complex is also in the closed conformation and occupies part of the TMP trimethoxybenzyl ring binding site. Thus, in the case of each binary complex the binding of the first ligand has not created the perfect protein conformation required for binding the second ligand: the binding site for the second ligand, although approximately in place, is not exactly set up in the binary complexes but requires some further adjustment.
Figure 4.

A comparison of the A−B loop structures in apo L. casei DHFR and its complexes with TMP and NADPH: the A−B loop structures are also shown for E. coli DHFR complexes with NADPH.methotrexate (closed) and 5,10-diazatetrahydrofolate (occluded) (PDB ID: 1rh3 and PDB ID: 1rx5, respectively).(7) The adenine part of NADPH is not shown.
The A−B loops in several other DHFR structures have been extensively studied. For example, for E. coli DHFR the closed and occluded conformations of the A−B loop have been shown to interchange during the catalytic process from studies of NMR(35) and X-ray data.(7) In X-ray diffraction studies of the E. coli DHFR.folate complex where only the substrate site is occupied, the A−B loop was found in the occluded conformation while the A−B loop in the E. coli apo DHFR and its binary MTX.DHFR complex the loop was found to be disordered.7,13,38
Ligand-Induced Changes in Positions of Secondary Structure Elements
The conformation of each of the secondary structure elements in apo lc DHFR is very similar to that of the corresponding element in the complexes. This can be seen from comparing the structures of lc apo DHFR and its ternary complex DHFR.TMP.NADPH where superimposition of each individual helix results in fairly small rmsd values (for helices C, E, and F the RMSDs are 0.18, 0.12, and 0.11 Å, respectively). Helix B could not be included in this comparison because it was ill-defined in the apo structure: however, the extrapolated 1H chemical shifts for helix B residues obtained from the PABG titration of apo lc DHFR indicate that helix B in folded apo lc DHFR is structurally very similar to those in the binary and ternary complexes.
Structural similarities in the β-strands of apo lc DHFR and its ternary complex are also found from superimpositions of the β-strands. Thus, for the β-strands A, F, and H the rmsd values are 0.25, 0.26, and 0.4 Å, respectively, and for the β-sheet A, E, F, H in the major binding domain the rmsd is 0.51 Å. Comparisons of chemical shifts in apo lc DHFR and its complexes (see below) also point to the local structures of the secondary structure elements being very similar in all cases. Pairwise superimpositions of the apo DHFR loop elements onto corresponding loops in the different complexes result in larger rmsd values. The differences in positions of the helices in the various pairs of structures result not only from direct ligand interactions with helix residues but also from ligand-induced conformational adjustments in the loops that change the relative positions of the helices with respect to the β-sheet. Figure 2 shows the Kabsch−Sander(39) Insight structures of apo lc DHFR and its complexes with NADPH and TMP where the relatively modest changes in the positions of the helices with respect to the β-sheet are visualized. Distances between the Cα atoms of the helices and the Cα carbon of L4 of β-strand A in the various structures have been measured, and these give an indication of how the helices are repositioned in the different complexes. L4 was chosen as a reference point since it is in the ligand binding site and close to parts of both bound TMP and NADPH. In pairwise comparisons of the apo lc DHFR structure with those of its binary and ternary complexes, the largest movements for the end residues of the helices are observed upon formation of the binary complexes with NADPH and TMP (average movements 1.12 and 1.26 Å, respectively). Smaller movements accompany the binding of the second ligand to each of the binary complexes (average movements 0.73 and 0.46 Å, respectively).
Differences in Distances Spanning the Binding Sites
Table 1 contains the distances between Cα atoms of residues spanning the binding sites for apo lc DHFR and its complexes. For 70% of the measurements the binding of the ligands results in shortening of the distances between such residues. In some cases the distances in the binary complexes are shorter than in the ternary complex. The distance differences between apo lc DHFR and the binary complexes are nonadditive when compared with the corresponding values for the ternary complex. The nonadditivity of the displacements is consistent with cooperative binding behavior.
Table 1. Distances (in Å) between Cα Atoms for Residues Spanning the Ligand Binding Sites in L. casei DHFR and Its Complexes with TMP and NADPHa.
| Res 1 | Res 2 | apo DHFR | TMP | NADPH | ternary | TMP-apo DHFR | NADPH-apo DHFR | ternary-apo DHFR |
|---|---|---|---|---|---|---|---|---|
| R44 Cα | Q101 Cα | 10.09 | 9.22 | 9.40 | 9.29 | −0.87 | −0.69 | −0.80 |
| R43 Cα | Q65 Cα | 9.86 | 9.49 | 8.79 | 9.56 | −0.37 | −1.07 | −0.30 |
| R43 Cα | H64 Cα | 9.32 | 8.39 | 7.62 | 8.60 | −0.93 | −1.70 | −0.72 |
| H77 Cα | Q101 Cα | 12.06 | 12.91 | 12.74 | 13.12 | 0.85 | 0.68 | 1.06 |
| H64 Cα | A105 Cα | 12.59 | 10.05 | 12.73 | 13.40 | −2.55 | 0.14 | 0.81 |
| G14 Cα | T45 Cα | 11.58 | 11.87 | 10.54 | 9.59 | 0.29 | −1.04 | −1.99 |
| A6 Cα | T45 Cα | 15.92 | 13.35 | 14.02 | 13.88 | −2.57 | −1.90 | −2.04 |
| I13 Cα | A97 Cα | 11.33 | 11.99 | 10.47 | 10.95 | 0.66 | −0.86 | −0.38 |
| D26 Cα | A97 Cα | 13.57 | 13.55 | 11.95 | 13.03 | −0.02 | −1.61 | −0.54 |
| L4 Cα | L27 Cα | 11.82 | 11.17 | 11.27 | 11.18 | −0.66 | −0.56 | −0.64 |
| L27 Cα | F49 Cα | 13.42 | 11.01 | 12.26 | 11.21 | −2.41 | −1.16 | −2.21 |
Distances are the average values from all 25 structures in the apo DHFR family.
Cα−Cα Distance Differences
A comprehensive and unbiased way of examining differences between two structures is to measure differences in the distances between all Cα atoms of corresponding residues in each of the different structures. The structures can then be conveniently compared by using the Cα distance difference (ΔD) two-dimensional (2D) contour plots introduced by Ooi and Nishikawa(23) (see Materials and Methods). Examination of the Cα ΔD values for all pairs of structures involving apo lc DHFR and its TMP and NADPH ternary and binary complexes reveals substantial differences for some pairs of residues. If we exclude residues in the A−B loop and helix B regions, then the majority of pairs of residues (86%) have Cα ΔD values less than 2 Å and only 1% have values greater than 4 Å.
We have calculated such 2D contour plots for all pairs of structures, and Figure 1A−C shows the three plots for apo lc DHFR and its binary and ternary complexes with TMP and NADPH. The triangular space above the diagonal contains negative ΔD values that correspond to shortening of distances between pairs of Cα atoms on formation of the complex. The triangular space below the diagonal contains positive ΔD values that correspond to bond lengthening on complex formation. There are roughly similar populations of negative and positive ΔD values seen in the upper and lower triangles, respectively, for all three complexes (see Figure 1). However, when these are weighted according to the magnitudes of the ΔD values, the results indicate an overall shrinkage of the protein upon complex formation.
The contours around the maxima and minima correspond to blocks of residues that all have similar ΔD values. Such blocks correspond to groups of residues that move together upon complex formation, and some are highlighted in boxes in Figure 1. Most of the blocks (whether highlighted or not) are found in the unshaded regions of Figure 1 which correspond to Cα ΔD values between pairs of atoms in two separate domains: the adenosine binding domain (residues 38−88) and the major domain (2−37, 89−160). Residues within the blocks move together when the two domains are displaced with respect to each other. Thus, block 1 in Figure 1A corresponds to shortening of Cα−Cα distances between residues 2−14 in the major domain and residues 42−49 (helix C) in the adenosine binding domain on forming the DHFR.TMP binary complex. Likewise, blocks 2−5 in Figure 1A correspond to distance shortening between residues in the major domain and residues in the adenosine binding domain. Most of the other nonhighlighted blocks of residues can be similarly assigned to distance shortening between residues in these two domains (for example, the region involving residues 45−75 and 122−155).
There are also some Cα distance lengthenings upon complex formation. The lower triangular spaces (positive ΔD values) in the 2D plots feature a strip of contours (block 6) connecting residues 77−79 with residues 90−160. This indicates that many of the latter residues move away from residues 77−79 on complex formation. Block 7 also shows distance lengthening (between residues 36−40 and many other residues). A few blocks show large differences between the complexes. For example, block 4 indicates distance shortening on complex formation with TMP (Figure 1A), no distance changes on formation of complex with NADPH, and distance lengthening on formation of the ternary complex. Thus, although most regions are perturbed in a generally similar way by the different ligands, there are some differences.
There are two main conclusions from these structure comparisons. (i) The preponderance of blocks indicating Cα−Cα distance shortening and lengthening between the two domains (unshaded regions of Figure 1) clearly indicates that the ligand binding is affecting the relative positions of the two domains. (ii) The most striking feature of the Cα ΔD 2D contour plots for all the DHFR complexes shown in Figure 1 is their general similarity. Thus, the binding of TMP or NADPH each causes generally similar overall changes in conformation, and furthermore these changes are approaching the final conformation of the ternary complex. Both ligands are each capable of moving the protein conformation toward that of the ternary complex. This is as would be expected for ligand induced conformational changes contributing to positive cooperative binding. The ΔD 2D contour plot for the binary lc DHFR.TMP and ternary lc DHFR.TMP.NADPH complexes (not shown) indicate that the relative position of the two domains is further adjusted when the coenzyme binds to the lc DHFR.TMP complex. Clearly organizing the conformation of the binding sites for the two ligands is a complex process that involves more than a simple progressive tightening of the structure as each ligand binds.
The nonadditivity of the displacements caused by each ligand and the fact that the conformational changes induced by binding the first ligand are in general approaching the final conformational state of the ternary complex are both consistent with positive cooperative binding behavior.
Domain Orientations
We have used the DynDom program(25) to characterize the rotation angles between the major and adenosine binding domains for pairs of structures for apo lc DHFR and its three NADPH/TMP complexes. For all pairs of lc DHFR structures it was necessary to define the two domains explicitly (major domain residues (1−12 and 89−160) and adenosine binding domain residues (38−88) because the program was unable to identify these domains directly in our structures. The rotation angles are defined about an interdomain screw axis determined by DynDom: the determined rotation angles showed differences between apo lc DHFR and its two binary complexes (10.0 ± 1.3° and 11.4 ± 1.4° for the NADPH and TMP binary complexes, respectively) and 16.2 ± 2.1° for the ternary DHFR.NADPH.TMP structure (see Figure S2, Supporting Information). The screw axes for the different pairs of structures are quite similar (the angles between the screw axis and the line joining the centers of mass of the two domains fall in the range 105°−114° for the different pairs of structures). While the screw axes identified by DynDom for all pairs of structures are similar to each other, they are too far (>3 Å) from the hinge residues to be effective hinge axes between the two domains.(25) However, it is clear that there are domain reorientations accompanying ligand binding and that the rotation angle changes seen on forming the binary complex constitute a substantial part of the larger angle change observed between the structures of apo DHFR and its ternary complex as expected for positive cooperative binding of the two ligands.
Earlier, Bystroff and Kraut(13) examined X-ray structures of E. coli apo DHFR and its complexes DHFR.NADP+, DHFR.Folate.NADP+, and DHFR.Methotexate. The positive cooperativity for such complexes involving NADP+ is at least 10-fold smaller than that observed for TMP and NADPH. We find that while the overall pattern of the changes in domain rotation angles and Cα−Cα distance differences for L. casei apo DHFR and its TMP and NADPH complexes in solution are similar to those observed in the X-ray studies on E. coli DHFR,(13) the magnitudes of the effects are substantially larger for the L. casei DHFR complexes. There are also some differences in the extent of the disordered regions. The partially unfolded region found in the solution structure of L. casei DHFR (residues 15−31) is larger than the disordered region seen in the crystal structure of E. coli apo DHFR (residues16−24) and extends beyond the A−B loop into helix B. Formation of the L. casei DHFR binary complexes in solution results in this unfolded region being refolded to its ordered form, whereas in the crystal structures of the E. coli DHFR binary complexes the A−B loop region remains disordered only becoming ordered on forming the ternary complex. Clearly refolding of DHFR on forming the binary complex, as seen in our NMR solution studies, will make a contribution to the positive binding cooperativity (up to −0.95 kcal mol−1). It would be useful to obtain a better estimate of the relative contribution to the cooperativity from the refolding. One possibility might be to engineer a mutant that has a refolded A−B loop/helix B such that the protein binds both ligands more tightly in the binary complex while showing lower positive cooperativity on forming the ternary complex. However, such an approach would not be straightforward because of the complexity of the possible changes in ligand protein interactions following such mutations.(40) Li and co-workers(41) have prepared an E. coli DHFR mutant with the A−B loop residues 16−19 replaced by a glycine residue that was shown to have reduced flexibility in the loop compared to in wild type.(36) This mutant provided useful insights into the possible role of protein flexibility in the catalytic process, but it was noted that its binding constants for NADPH and FH2 are lowered by a factor of 10.
Hydrophobic Binding from Interligand Contact in the Ternary Complex
Hydrophobic effects from nonpolar contacts between the two ligands in a ternary complex of DHFR have been suggested as possible contributors to cooperative binding.(42) We have examined the interligand contact in the lc DHFR.TMP.NADPH complex where the nicotinamide C4 carbon is located close to the TMP C7 carbon: the C4−C7 separation measured as the average value for the family of structures is 3.5 ± 1 Å (close to the unstrained difference of 3.7 Å(43)). The unoccupied reduced nicotinamide ring site in the lc DHFR.TMP binary complex is likely to contain bound water molecules, and displacement of such ordered water molecules on binding NADPH would make a favorable entropic contribution to the binding. Connolly(26) plots shown in Figure 5 indicate that for the two binary complexes there are solvent accessible areas (marked by asterisks) that are rendered inaccessible to solvent when the ternary complex is formed (Figure 5B). Values for the solvent accessible surface area (SASA) of bound NADPH (30.4 Å2) and TMP (24.2 Å2) in the region of interligand contact in the ternary complex were calculated using the NACCESS program.(27) These values would provide contributions to the cooperative binding of −1.4 kcal mol−1 for NADPH and −1.1 kcal mol−1 for TMP (based on using a value of 47 cal mol−1 per Å2).(44) While these entropic hydrophobic effects make a substantial contribution to the overall cooperativity, they do not account for the full effect of around −2.9 kcal mol−1.
Figure 5.

Connolly(26) plots showing the solvent accessible surface areas around the ligands for complexes of lc DHFR: (A) DHFR.TMP; (B) DHFR.TMP.NADPH; (C) DHFR.NADPH. The regions marked with asterisks in the two binary complexes are seen to be absent in the ternary complex. The radius of the solvent molecule used in determining the SASAs was 1.4 Å.
The interligand contributions of −1.4 and −1.1 kcal mol−1 are probably upper estimates because there is still some debate over the exact magnitude of the hydrophobic effect.(45) Earlier values of 25−30 cal mol−1 per Å2 based on measurements of solubilities and vapor pressures of nonpolar solutes46−48 were revised by Sharp and co-workers(44) to 47 cal mol−1 per Å2 based on measurements of surface tension hydrocarbon−water interfaces. More recent work has suggested lower estimates based on examining the thermodynamic parameters of protein−ligand complexes (12 cal mol−1 per Å2):(49) however, the values from such studies can be influenced by difficulties in allowing for entropic contributions related to refolding the apo protein on forming the ligand binding site.
Freisheim and Matthews(42) have raised the possibility that differences in interligand interactions in DHFR.TMP.NADPH complexes formed using the mammalian enzyme and bacterial enzymes might provide an explanation for the much lower positive cooperativity seen in the ligand binding to mammalian DHFR (a value of 4.7 was reported for SR-1 rodent lymphoma DHFR(6)). We have explored this by using the structure of the DHFR.TMP.NADPH complex formed with human DHFR(11) to determine SASAs of the bound ligands. The NACCESS calculations on the ternary complex of human DHFR (PBD 1YHO)(11) give estimates of (SASA) of bound NADPH (25.5 Å2) and TMP (16.2 Å2) in the region of interligand contact corresponding to binding energy contributions of −1.2 and −0.8 kcal mol−1, respectively. While these hydrophobic contributions are smaller than those from interligand contact in the lc DHFR ternary complex, they cannot explain the difference in observed binding cooperativity between the human and bacterial DHFRs in their complexes with TMP and NADPH.
Chemical Shift Studies
The 1H/15N/13C chemical shifts at 15 °C measured with respect to the corresponding random coil shifts (corrected for sequence effects) for apo lc DHFR and its TMP.NADPH ternary complex are shown as histogram plots in Figure S3 (Supporting Information) for all nuclei that are more than 4 Å from the ligand. Very similar chemical shifts are observed for corresponding nuclei in apo lc DHFR and its ternary complex indicating that the loops, α-helices, and β-strands generally have similar local structures. Thus, the chemical shifts indicate that there is no major change in overall local structure in regions around these residues when the complexes are formed. At the same time, some modest long-range ligand induced differences in chemical shifts are noted for example in the F−G (119−127) and F−F (106−111) loops. The chemical shift agreement between apo lc DHFR and its complexes is even better if we consider only those residues which are more than 10 Å from the ligand (98 remote residues for TMP and 77 for NADPH).
Is There Evidence for Hydrogen Bond Shortening on Complex Formation?
Previously, we have used measurements of NH proton exchange rates to show that ligand binding to lc DHFR results in a general increase in the protection factors for the amide NH protons, reflecting an overall tightening of the protein structure even in regions well-removed from the ligand binding site: the tightest binding ligands usually causing the greatest protection.(50) The tightening of the structure increases further on formation of the ternary complex in the case of positive cooperative binding (such as with TMP and NADPH). However, for complexes showing negative cooperativity (such as with folinic acid and NADPH), the NHs of some residues have decreased protection factors, indicating a loosening of the structure.(50) Williams and co-workers have postulated that on binding ligands under conditions of positive cooperativity there would be a shrinking of the protein structure while for negative cooperativity there would be an expansion of the structure of the protein−ligand complex.15,16
Could the observed ligand-induced tightening or loosening of the complex structure be related to changes in hydrogen bond lengths? Correlations of protein amide NH 1H chemical shifts with hydrogen bond lengths measured from X-ray structures51−53 and studies of pressure-induced amide 1H chemical shifts54,55 have provided estimates for the expected deshielding changes of 0.38 ± 0.17 ppm per 0.1 Å shortening of the hydrogen bond. On the basis of this correlation, changes in NH 1H shielding on ligand binding (1H Δδ) values can be used to detect any substantial shortenings of hydrogen bonds that occur on formation of a protein ligand complex. Clearly this approach can only be adopted for NH protons in residues remote from the binding site where direct ligand-induced shielding effects of the ligands can be neglected. Figure 6 provides the 1H Δδ values for amide NH protons in such remote secondary structure regions (>10 Å from ligand)(10) for two lc DHFR complexes. In the case of DHFR.TMP.NADPH (Figure 6A), none of the ligand-induced 1H chemical shifts are greater than 0.2 ppm, and for the relatively few observed positive 1H Δδ values (which could result from H-bond shortening) only one of the values is >0.1 ppm. Similar results are observed for all the binary complexes and for the ternary complex DHFR.folinic acid.NADPH (Figure 6B) where the ligands are bound more weakly than in the binary complexes (negative cooperativity). Thus, in both cases there is no evidence for any systematic ligand-induced hydrogen bond shortening of greater than 0.03 Å (i.e., corresponding to a positive 1H Δδ value of 0.1 ppm) for the DHFR.TMP.NADPH complex or lengthening of hydrogen bonds for the folinic acid.NADPH.DHFR complex. The effects of any hydrogen bond shortening/lengthening on the chemical shifts are either very small or masked by shifts of opposite sign arising from long-range conformational effects. In fact, the ligand-induced NH shifts for residues in the remote secondary structure regions are remarkably similar in the two ternary complexes despite the protection factors in the DHFR.TMP.NADPH complex being much larger than those in the DHFR.folinic acid.NADPH complex and the Kd values for NADPH in the two complexes differing by a factor of ∼70 000 (see Table S2, Supporting Information).
Figure 6.

Histograms of the ligand-induced 1H chemical shifts at 15 °C (Δδ ppm values) for the hydrogen-bonded amide NH protons(50) in helical (blue) and sheet (pink) residues in regions remote from the ligand (>10 Å from ligand) for the lc DHFR.TMP.NADPH and DHFR.folinic acid.NADPH complexes. Errors on the chemical shifts are ±0.01 ppm.
Similar findings on hydrogen bond shortening have been reported in recent studies of ligand binding effects on protein NH exchange rates in phosphoglycerate kinase.(56)
Cordier and co-workers have used novel measurements of 13C−15N scalar couplings across hydrogen bonds to estimate changes in hydrogen-bond lengths of between 0.02 and 0.12 Å accompanying ligand binding in the c-Src SH3 domain.(57) Their larger values appear to be associated with hydrogen bonds near the ends of short β-strands: our results relate to very stable backbone to backbone hydrogen bonds within an 8-stranded β-sheet and well removed from the binding ligands.
In the absence of any substantial changes in hydrogen bond lengths in DHFR on complex formation, a likely explanation for the structure tightening observed on formation of the complexes is a reduction in the populations of partially unfolded forms of the protein (that is, an additional component of protein refolding).
Binding Constants and Thermodynamic Data
The binding constants,5,14,58,59 ITC-derived enthalpies, and the calculated −TΔS values for complexes of lc DHFR with NADPH, TMP, and folinic acid are presented in Table 2. It can be seen that the formation of the binary complexes at 25 °C is enthalpy driven in all cases: large enthalpic contributions overcome the large unfavorable entropic contributions to the binding. The latter is consistent with the requirement to refold the protein particularly in the loop region 14−23 and in part of helix B (24−31) in order to allow important protein−ligand interactions to take place. Reduced unfavorable entropic contributions are seen at lower temperatures (see footnote in Table 2). These thermodynamic data are generally similar to those seen previously for cooperative binding of antifolate ligands to DHFRs from other sources in extensive studies by Briand and co-workers, who also implicated protein refolding in the large unfavorable entropic contributions and first pointed out its potential involvement in positive cooperative binding.60,61
Table 2. Binding Constants a (M−1) and Thermodynamic Data (kcal mol−1) for Ligands Binding to L. casei DHFR at 25 °C.
| ligand | binding to | Ka | refs | ΔG0 | ΔH0b | −TΔS0c |
|---|---|---|---|---|---|---|
| TMP | E | 2.0 (±0.1) × 107 | (5, 58) | −10.0 | −22.2 ± 0.7 | 12.2 |
| NADPH | E | 1.0 (±0.1) × 108 | (29) | −10.9 | −27.1 ± 1.5 | 16.2 |
| NADPH | E + TMP | 1.35 (±0.10) × 1010 | (5, 59) | −13.8 | −12.2 ± 0.7 | −1.6 |
| Folinic | E | 1.3 (±0.6) × 108 | (14) | −11.1 | −24.2 ± 0.7 | 13.1 |
| NADPH | E + Folinic | 2.1 (±0.5) × 105 | (14) | −7.3 | 0 ± 0.5d | −7.3 |
Literature values of the Ka values: it was not possible to measure good Ka values for the tightly binding ligands using ITC data. However, for (NADPH + DHFR.folinic acid) the ITC derived Ka value is 2.2 × 105 M−1, in good agreement with literature value.(14)
ΔH0 values measured from ITC experiments (at least three measurements for each complex).
−TΔS0 values at 10 °C for lc DHFR + TMP (2.0 kcal mol−1) and for lc DHFR + NADPH (7.5 kcal mol−1) were estimated from ITC ΔH0 values measured at 10 °C and assuming Ka values to be similar to those at 25 °C.
ΔH0 value estimated by interpolation of ITC data at 10 °C (3.0), 15 °C (2.2), 20 °C (1.0), and 30 °C (−2.1 kcal mol−1).
On forming the ternary complex, for example by binding NADPH to the lc DHFR.TMP complex, the enthalpic contributions are reduced but are compensated for by favorable entropic contributions which result in the NADPH binding more tightly in the ternary complex (positive cooperativity). A similar pattern will be expected for TMP binding to the lc DHFR.NADPH complex. These favorable entropy contributions arise mainly from hydrophobic effects related to reductions in the number of ordered water molecules and possibly also from vibrational entropic changes.
If we consider the energy changes associated with NADPH forming its binary and ternary complexes with TMP/DHFR at 25 °C, we find that (ΔG0Ter − ΔG0Bin) = −2.9, (ΔH0Ter − ΔH0Bin) = 14.9, and −(TΔS0Ter − TΔS0Bin) = −17.8 kcal mol−1. In forming the ternary complex from the binary complex where the refolding of the A−B loop and helix B has already taken place, the binding can be achieved with lower enthalpic interactions and with favorable entropic contributions. The enthalpy changes accompanying formation of the ternary complex oppose positive cooperativity whereas the entropy changes strongly support it: this indicates that entropy changes are a major factor driving the positive cooperativity in forming the lc DHFR.TMP.NADPH complex. Subramanian and Kaufman also identified entropically driven positive cooperativity in chicken liver DHFR.MTX.NADPH from thermodynamic data measurements.(62)
The energy changes associated with NADPH forming its binary lc DHFR complex and its ternary complex with DHFR and folinic acid are given by (ΔG0Ter − ΔG0Bin) = 3.60, (ΔH0Ter − ΔH0Bin) = 27.1, and −(TΔS0Ter − TΔS0Bin) = −23.5 kcal mol−1. Thus, there is a large reduction in the enthalpic contribution and a favorable increase in −TΔS when NADPH forms the ternary complex with folinic acid compared with the values for formation of the NADPH binary complex.
A possible model for the decreased binding in the ternary complex (negative cooperativity) would be one where parts of the NADPH and folinic acid molecules cannot simultaneously occupy the binding sites used in their binary complexes. In earlier NMR studies of the lc DHFR.folinic acid.NADPH complex(14) we postulated the presence of two bound states for the complex in order to explain anomalous off-rates. More recently, an X-ray study of 5,10-dideazatetrahydrofolate in its complex with E. coli DHFR found that the C7 position of the THF analogue is within 1.8 Å of the expected position of the C4 of bound NADPH:(63) the unfavorable steric interaction from such close proximity was proposed as the cause of the increase in rate of THF release by cofactor binding. Similar conclusions were reached from crystallographic studies of folinic acid complexed with E. coli DHFR.(64)
Conclusions
Origins of Positive Binding Cooperativity in L. casei DHFR.TMP.NADPH Complexes
The main difference in structure between apo lc DHFR and its complexes involves the A−B loop and part of helix B (residues 15−31) which are partially unfolded in apo DHFR but folded in the closed conformation in the complexes. Because this region contains several residues that interact directly with TMP and NADPH, the refolding of this part of the structure by the first ligand could certainly assist the binding of the second ligand and is a likely contributor to the positive cooperative binding. For example, a population of 80% unfolded apo DHFR with very weak ligand binding would lead to a contribution to cooperativity of up to −0.95 kcal mol−1.
In addition to the refolding in the binary complexes, there are some adjustments of the rest of the structure which reposition the helices and loops without major changes in their local conformations. The DynDom studies indicate that the reorientations of the major and adenosine binding domains observed in the binary complexes are approaching their final orientations in the ternary complex. The Cα−Cα ΔD studies indicate that while the binding site for the second ligand is not perfectly organized in the binary complexes, it is much closer to the final ternary conformation than that found in the apo lc DHFR structure: these conformational changes will also contribute to the cooperativity. On formation of the ternary complex there is an additional substantial contribution to cooperative binding from the hydrophobic effects related to the close proximity of parts of the two ligands: the removal of ordered bound water molecules on the surface of the ligands caused by interligand proximity results in favorable entropic binding contributions of up to −1.4 kcal mol−1 for NADPH and −1.1 kcal mol−1 for TMP.
In summary, the large positive binding cooperativity (−2.9 kcal mol−1) observed in forming the lc DHFR.TMP.NADPH complex results from a (i) significant contributions from both the refolding of the partially unfolded A−B loop and helix B in apo lc DHFR (up to −0.95 kcal mol−1) and also from favorable conformational changes induced by binding the first ligand that are required to facilitate tighter binding of the second ligand and (ii) an equally important favorable entropic contribution (up to −1.4 kcal mol−1) from hydrophobic effects related to interligand proximities in the ternary complex where ordered water are displaced from the surface of the bound ligands by the close proximity of the NADPH reduced nicotinamide ring to parts of trimethoprim.
It should be noted that similar favorable entropic contributions from interligand proximity are also present in the ternary complex of human DHFR.TMP.NADPH. Thus, the observed difference of cooperative binding in the human and bacterial DHFR complexes is not because of differences in hydrophobic effects related to interligand proximity.
Acknowledgments
The NMR measurements were carried out at the MRC Biomedical NMR Centre, Mill Hill, London. We thank Tom Frenkiel for help with the NMR measurements and Geoff Kelly, who carried out the experiments on the aligned samples used for the RDC measurements, John McCormick for expert technical support, John Bouquiere for computing support, Katrin Rittinger for assistance with the ITC measurements, and Nigel Birdsall for helpful discussions.
Glossary
Abbreviations
- Δδ
change in shielding on ligand binding
- ΔD
Cα−Cα distance differences
- DHFR
dihydrofolate reductase
- DHF
7,8-dihydrofolate
- ec
Escherichia coli, E. coli
- folinic acid
5-formyl-5,6,7,8-tetrahydrofolic acid
- HSQC
heteronuclear single quantum coherence spectroscopy
- ITC
isothermal titration calorimetry
- lc
Lactobacillus casei, L. casei
- NMR
nuclear magnetic resonance
- PABG
p-aminobenzoyl-l-glutamate
- rmsd
root-mean-square deviation
- SASA
solvent accessible surface area
- THF
5,6,7,8-tetrahydrofolate
- TMP
trimethoprim, 2,4-diamino-5-(3,4,5-trimethoxybenzyl)pyrimidine.
Supporting Information Available
Details of the structure calculations, domain orientations, and 1H, 13C, and 15N chemical shift data. This material is available free of charge via the Internet at http://pubs.acs.org.
The research was supported by the Medical Research Council, U.K. (J.F. and B.B.), the Wellcome Trust (Collaborative Research Initiative Grant 060140 (J.F. and V.I.P.)), the Russian Foundation for Basic Research (V.I.P), the Russian Ministry of Education and Science (Contract 16.740.11.0445, V.I.P.), and a research fellowship from the Cajamurcia Foundation (E.N.P).
Accession numbers in the Protein Data Bank for the structures of apo lc DHFR and its binary complex with NADPH and TMP are PDB ID: 2L28, PDB ID: 2HQP, and PDB ID: 2HM9, respectively. The 1H, 13C, and 15N chemical shift data have been deposited in the BioMagResDataBank (Deposition Numbers BMRB ID: BMRB ID: 17125, BMRB ID: 5396, BMRB ID: 17310 and BMRB ID: 17311).
Supplementary Material
References
- Blakley R. L.Dihydrofolate Reductase (1985) in Folates and Pterins (Blakley R. L., Benkovic S. J., Eds.) Vol. 1, Chapter 5, pp 191−253, J. Wiley, New York. [Google Scholar]
- Matthews D. A.; Bolin J. T.; Burridge J. M.; Filman D. J.; Volz K. W.; Kaufman B. T.; Beddell C. R.; Champness J. N.; Stammers D. K.; Kraut J. (1985) Refined crystal structures of Escherichia coli and chicken liver dihydrofolate reductase containing bound trimethoprim. J. Biol. Chem. 260, 381–391. [PubMed] [Google Scholar]
- Andrews J.; Fierke C. A.; Birdsall B.; Ostler G.; Feeney J.; Roberts G. C. K.; Benkovic S. J. (1989) A kinetic study of wild type and mutant dihydrofolate reductases. Biochemistry 28, 5743–5750. [DOI] [PubMed] [Google Scholar]
- Dauber-Osguthorpe P.; Roberts V. A.; Osguthorpe D. J.; Wolff J.; Genest M.; Hagler A. T. (1988) Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins 4, 31–47. [DOI] [PubMed] [Google Scholar]
- Birdsall B.; Burgen A. S. V.; Roberts G. C. K. (1980) Binding of coenzyme analogs to Lactobacillus casei dihydrofolate reductase: binary and ternary complexes. Biochemistry 19, 3723–3731. [DOI] [PubMed] [Google Scholar]
- Baccanari D. P.; Daluge S.; King R. W. (1982) Inhibition of dihydrofolate reductase: effect of reduced nicotinamide adenine dinucleotide phosphate on the selectivity and affinity of diaminobenzylpyrimidines. Biochemistry 21, 5068–5075. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. (1997) Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 36, 586–603. [DOI] [PubMed] [Google Scholar]
- Feeney J. (2000) NMR studies of ligand binding to dihydrofolate reductase. Angew. Chem., Int. Ed. 39, 290–312. [DOI] [PubMed] [Google Scholar]
- Martorell G.; Gradwell M. J.; Birdsall B.; Bauer C. J.; Frenkiel T. A.; Cheung H. T. A.; Feeney J. (1994) Solution structure of bound trimethoprim in its complex with Lactobacillus casei dihydrofolate reductase. Biochemistry 33, 12416–12426. [DOI] [PubMed] [Google Scholar]
- Polshakov V. I.; Smirnov E. G.; Birdsall B.; Kelly G.; Feeney J. (2002) NMR-based solution structure of the complex of Lactobacillus casei dihydrofolate reductase with trimethoprim and NADPH. J. Biomol. NMR 24, 67–70. [DOI] [PubMed] [Google Scholar]
- Kovalevskaya N. V.; Smurnyy Y. D.; Polshakov V. I.; Birdsall B.; Bradbury A. F.; Frenkiel T.; Feeney J. (2005) Solution structure of human dihydrofolate reductase in its complex with trimethoprim and NADPH. J. Biomol. NMR 33, 69–72. [DOI] [PubMed] [Google Scholar]
- Kovalevskaya N. V.; Smurnyi E. D.; Birdsall M.; Feeney J.; Polshakov V. I. (2007) Structural Factors Determine the Selectivity of Antibacterial Drug Trimethoprim Binding to Dihydrofolate Reductase. Khimiko-farmatsevticheskii Zhurnal 41, 8−11. English translation in (2007). Pharm. Chem. J. 41, 350–353. [Google Scholar]
- Bystroff C.; Kraut J. (1991) Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry 30, 2227–2239. [DOI] [PubMed] [Google Scholar]
- Birdsall B.; Hyde E. I.; Burgen A. S. V.; Roberts G. C. K.; Feeney J. (1981) Negative cooperativity between folinic acid and coenzyme in their binding to L. casei dihydrofolate reductase. Biochemistry 20, 7186–7195. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Stephens E.; Zhou M. (2003) Ligand binding energy and catalytic efficiency from improved packing with receptors and enzymes. J. Mol. Biol. 329, 389–399. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Stephens E.; O’Brien D. P.; Zhou M. (2004) Understanding non-covalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew. Chem., Int. Ed. 43, 6596–6616. [DOI] [PubMed] [Google Scholar]
- Dann J. G.; Ostler G.; Bjur R. A.; King R. W.; Scudder P.; Turner P. C.; Roberts G. C. K.; Burgen A. S. V. (1976) Large-scale purification and characterization of dihydrofolate reductase from a methotrexate-resistant strain of Lactobacillus casei. Biochem. J. 157, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews J.; Clore G. M.; Davies R. W.; Gronenborn A. M.; Kalderon D.; Papadopoulos P. C.; Schafer S.; Sims P. F.; Stancombe R. (1985) Nucleotide sequence of the dihydrofolate reductase gene of methotrexate-resistant Lactobacillus casei. Gene 35, 217–222. [DOI] [PubMed] [Google Scholar]
- Rückert M.; Otting G. (2000) Alignment of biological macromolecules in novel non-ionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 122, 7793–7797. [Google Scholar]
- Tjandra N.; Grzesiek S.; Bax A. (1996) Magnetic field dependence of nitrogen-proton J splitting in 15N-enriched human ubiquitin resulting from relaxation interference and residual dipolar coupling. J. Am. Chem. Soc. 118, 6264–6272. [Google Scholar]
- Live D. H.; Davies D. G.; Agosta W. C.; Cowburn D. (1984) Long range hydrogen bond mediated effects in peptides: nitrogen-15 NMR study of gramicidin S in water and organic solvents. J. Am. Chem. Soc. 106, 1939–1941. [Google Scholar]
- Wishart D. S.; Bigam C. G.; Yao J.; Abildgaard F.; Dyson H. J.; Oldfield E.; Wright P. E. (2004) The amide proton NMR chemical shift and hydrogen-bonded structure of peptides and polypeptides in the solid state as studied by high-frequency solid-state 1H NMR. Biochemistry 43, 16046–16055.15609999 [Google Scholar]; Erratum in: (2005) Biochemistry 44, 5948. [Google Scholar]
- Ooi T., Nishikawa K. (1973) in Conformation of Biological Macromolecules and Polymers (Bergman E., Pullman B., Eds.) p 173, Academic Press, New York. [Google Scholar]
- Go M. (1983) Modular structural units, exons, and function in chicken lysozyme. Proc. Natl. Acad. Sci. U.S.A. 80, 1964–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward S.; Kitao A.; Berendsen H. J. C. (1997) Model-free methods of analyzing domain motions in proteins from simulation: A comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins: Struct., Funct., Genet. 27, 425–437. [DOI] [PubMed] [Google Scholar]
- Connolly M. L. (1992) The molecular surface package. J. Mol. Graphics 11, 139–141. [DOI] [PubMed] [Google Scholar]
- Hubbard S. J., Thornton J. M. NACCESS Computer Program, Department of Biochemistry and Molecular Biology, University College, London.
- Polshakov V. I.; Birdsall B.; Frenkiel T. A.; Gargaro A.; Feeney J. (1999) Structure and dynamics in solution of the complex of Lactobacillus casei dihydrofolate reductase with the new lipophilic antifolate drug trimetrexate. Protein Sci. 8, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S. M. J.; Bachelor J. G.; King R. W. (1978) Kinetics and ligand binding to dihydrofolate reductase binary complex formation with NADPH and coenzyme analogues. Biochemistry 17, 2356–2364. [DOI] [PubMed] [Google Scholar]
- Veech R. L.; Eggleston L. V.; Krebs H. A. (1969) The redox state of free nicotinamide−adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 115, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman H. N.; Gaetani G. F.; Clemons E. H. (1986) NADP-binding proteins causing reduced availability and sigmoid release of NADP+ in human erythrocytes. J. Biol. Chem. 261, 4039–4045. [PubMed] [Google Scholar]
- Hammond S. J.; Birdsall B.; Searle M. S.; Roberts G. C. K.; Feeney J. (1986) Dihydrofolate reductase: 1H resonance assignments and coenzyme-induced conformational changes. J. Mol. Biol. 188, 81–97. [DOI] [PubMed] [Google Scholar]
- Gargaro A. R.; Soteriou A.; Frenkiel T. A.; Bauer C. J.; Birdsall B.; Polshakov V. I.; Barsukov I. L.; Roberts G. C. K.; Feeney J. (1998) Solution Structure of the Complex of Lactobacillus casei Dihydrofolate Reductase with Methotrexate. J. Mol. Biol. 277, 119–134. [DOI] [PubMed] [Google Scholar]
- Osborne M. J.; Venkitakrishnan R. P.; Dyson H. J.; Wright P. E. (2003) Diagnostic chemical shift markers for loop conformation and substrate and cofactor binding in dihydrofolate reductase complexes. Protein Sci. 12, 2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitakrishnan R. P.; Zaborowski E.; McElheny D.; Benkovic S. J.; Dyson H. J.; Wright P. E. (2004) Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry 43, 16046–55. [DOI] [PubMed] [Google Scholar]
- Falzone C. J.; Wright P. E.; Benkovic S. J. (1994) Dynamics of a flexible loop in Dihydrofolate Reductase from Escherichia coli and its implication for catalysis. Biochemistry 33, 439–442. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. (2006) The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- Matthews D. A.; Alden R. A.; Bolin J. T.; Freer S. T.; Hamlin R.; Xuong N.; Kraut J.; Poe M.; Williams M.; Hoogsteen K. (1977) Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate. Science 197, 452–455. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymer 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Wagner C. R.; Benkovic S. J. (1990) Site directed mutagenesis: a tool for enzyme mechanism dissection. Trends Biotechnol. 8, 263–270. [DOI] [PubMed] [Google Scholar]
- Li L.; Falzone C. J.; Wright P. E.; Benkovic S. J. (1992) Functional role of a mobile loop of Escherichia coli dihydrofolate reductase in transition-state stabilisation. Biochemistry 31, 7826–7833. [DOI] [PubMed] [Google Scholar]
- Freisheim J. H., Matthews D. A. (1984) The comparative biochemistry of dihydrofolate reductase, in Folate Antagonists as Therapeutic Agents (Sirotna F. M., Burchall J. J., Ensminger W. D., Montgomery J. A., Eds.) Vol. 1, p 69, Academic Press, New York. [Google Scholar]
- Chothia C. (1975) Structural invariants in protein folding. Nature 254, 304–308. [DOI] [PubMed] [Google Scholar]
- Sharp K. A.; Nicholls A.; Fine R. F.; Honig B. (1991) Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science 252, 106–109. [DOI] [PubMed] [Google Scholar]
- S. Chakravarty S.; Bhinge A.; Varadarajan R. (2002) A Procedure for Detection and Quantitation of Cavity Volumes in Proteins. Application to measure the strength of the hydrophobic driving force in protein folding. J. Biol. Chem. 277, 31345–31353. [DOI] [PubMed] [Google Scholar]
- Chothia C. (1974) Hydrophobic bonding and accessible surface area in proteins. Nature 248, 338–339. [DOI] [PubMed] [Google Scholar]
- Richards F. M. (1977) Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 6, 151–176. [DOI] [PubMed] [Google Scholar]
- Tanford C. (1980) The Hydrophobic Effect, 2nd ed., John Wiley and Sons, New York. [Google Scholar]
- Olsson T. S. G.; Williams M. A.; Pitt W. R.; Ladbury J. E. (2008) The thermodynamics of protein-ligand interactions and solvation: insights for ligand design. J. Mol. Biol. 384, 1002–1017. [DOI] [PubMed] [Google Scholar]
- Polshakov V. I.; Birdsall B.; Feeney J. (2006) Effects of cooperative ligand binding on protein amide NH hydrogen exchange. J. Mol. Biol. 356, 886–903. [DOI] [PubMed] [Google Scholar]
- Wagner G.; Pardi A.; Wuthrich K. (1983) Hydrogen bond length and proton NMR chemical shifts in proteins. J. Am. Chem. Soc. 105, 5948–5949. [Google Scholar]
- Pardi A.; Wagner G.; Wuthrich K. (1983) Protein conformation and proton nuclear magnetic resonance chemical shifts. Eur. J. Biochem. 137, 445–454. [DOI] [PubMed] [Google Scholar]
- Yamauchi K.; Kuroki F.; Fujii K.; Ando I. (2000) The amide proton NMR chemical shift and hydrogen-bonded structure of peptides and polypeptides in the solid state as studied by high-frequency solid-state 1H NMR. Chem. Phys. Lett. 324, 435–439. [Google Scholar]
- Asakura T.; Taoka K.; Demura M.; Williamson M. P. (1995) The relationship between amide proton chemical shifts and secondary structure in proteins. J. Biomol. NMR 6, 227–236. [DOI] [PubMed] [Google Scholar]
- Li H.; Yamada H.; Akasaka K. (1998) Effect of Pressure on Individual Hydrogen Bonds in Proteins. Basic Pancreatic Trypsin Inhibitor. Biochemistry 37, 1167–1173. [DOI] [PubMed] [Google Scholar]
- Marston J. P.; Cliff M. J.; Reed M. A. C.; Blackburn G. M.; Hounslow A. M.; Craven C. J.; Waltho J. P. (2010) Structural Tightening and Interdomain Communication in the Catalytic Cycle of Phosphoglycerate Kinase. J. Mol. Biol. 396, 345–360. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Wang C.; Grzesiek S.; Nicholson L. K. (2000) Ligand-induced strain in the c-Src SH3 domain detected by NMR. J. Mol. Biol. 304, 497–505. [DOI] [PubMed] [Google Scholar]
- Hood K.; Roberts G. C. K. (1978) Ultraviolet difference spectroscopic studies of substrate and inhibitor binding to Lactobacillus casei dihydrofolate reductase. Biochem. J. 171, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde E. I.; Birdsall B.; Roberts G. C. K.; Feeney J.; Burgen A. S. V. (1980) Proton nuclear magnetic resonance saturation transfer studies of coenzyme binding to Lactobacillus casei dihydrofolate reductase. Biochemistry 19, 3746–3754. [DOI] [PubMed] [Google Scholar]
- Gilli R. M.; Sari J. C.; Lopez C. L.; Odile S. R.; Briand C. M. (1990) Comparative thermodynamic study of the interaction of some antifolates with dihydrofolate reductase. Biochim. Biophys. Acta 1040, 245–250. [DOI] [PubMed] [Google Scholar]
- Sasso S. P.; Gilli R. M.; Sari J. C.; Rimet O. S.; Briand C. M. (1994) Thermodynamic study of dihydrofolate reductase inhibitor selectivity. Biochim. Biophys. Acta 1207, 74–79. [DOI] [PubMed] [Google Scholar]
- Subramanian S.; Kaufman B. T. (1978) Interaction of methotrexate, folates, and pyridine nucleotides with dihydrofolate reductase: Calorimetric and spectroscopic binding studies. Proc. Natl. Acad. Sci. U.S.A. 75, 3201–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes V. M.; Sawaya M. R.; Brown K. A.; Kraut J. (1995) Isomorphous Crystal Structures of Escherichia coli Dihydrofolate Reductase Complexed with Folate, 5-Deazafolate, and 5,10-Dideazatetrahydrofolate: Mechanistic Implications. Biochemistry 34, 2710–2723. [DOI] [PubMed] [Google Scholar]
- Lee H.; Reyes V. M.; Kraut J. (1996) Crystal Structures of Escherichia coli Dihydrofolate Reductase Complexed with 5-Formyltetrahydrofolate (Folinic Acid) in Two Space Groups: Evidence for Enolization of Pteridine O4. Biochemistry 35, 7012–7020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.