

Abstract
Epithelial-mesenchymal interactions guide tooth development through its early stages and establish the morphology of the dentin surface upon which enamel will be deposited. Starting with the onset of amelogenesis beneath the future cusp tips, the shape of the enamel layer covering the crown is determined by five growth parameters: the (1) appositional growth rate, (2) duration of appositional growth (at the cusp tip), (3) ameloblast extension rate, (4) duration of ameloblast extension, and (5) spreading rate of appositional termination. Appositional growth occurs at a mineralization front along the ameloblast distal membrane in which amorphous calcium phosphate (ACP) ribbons form and lengthen. The ACP ribbons convert into hydroxyapatite crystallites as the ribbons elongate. Appositional growth involves a secretory cycle that is reflected in a series of incremental lines. A potentially important function of enamel proteins is to ensure alignment of successive mineral increments on the tips of enamel ribbons deposited in the previous cycle, causing the crystallites to lengthen with each cycle. Enamel hardens in a maturation process that involves mineral deposition onto the sides of existing crystallites until they interlock with adjacent crystallites. Neutralization of acidity generated by hydroxyapatite formation is a key part of the mechanism. Here we review the growth parameters that determine the shape of the enamel crown as well as the mechanisms of enamel appositional growth and maturation.
Keywords: appositional growth, amelogenin, enamelin, ameloblastin, tooth
Introduction
Early tooth development is regulated by a series of epithelial-mesenchymal interactions between cells that have migrated from the cranial neural crest and the oral epithelium along the future alveolar ridge (Lumsden, 1988; Chai et al., 2000). The initiation of tooth formation involves the synthesis and secretion, by the oral epithelium, of diffusible growth factors (Thesleff and Sharpe, 1997; Cobourne and Sharpe, 2003; Tucker and Sharpe, 2004) that induce the expression of transcription factors in the underlying mesenchyme (Bei, 2009a,b; Tummers and Thesleff, 2009). A series of reciprocal interactions between the two opposing tissues orchestrates the development of tooth organs that, even before the onset of mineralization, have established the basic shape of the dental crown. Tooth development progresses through initiation, bud, cap, and bell histological stages (Nanci, 2008b). Important signaling centers that guide development are the primary (cap stage) enamel knot (Thesleff and Jernvall, 1997) and secondary enamel knots, which are associated with the inner enamel epithelium at the developing cusp tips (bell stage) (Thesleff et al., 2001; Matalova et al., 2005).
The importance of signaling to the early developmental process is highlighted by the types of genetic defects that disturb it. Genetic alterations that affect early developmental processes involve transcription factors, signaling molecules, and their receptors, and lead to familial tooth agenesis (MSX1, PAX9, AXIN2, EDA) or supernumerary teeth (RUNX2, APC) (Kere et al., 1996; Vastardis et al., 1996; Bayes et al., 1998; Stockton et al., 2000; Lammi et al., 2004; Nieminen, 2009; Wang et al., 2009).
The interface between the epithelium and mesenchyme during tooth development ultimately specifies the outer dentin surface. In the developing crown, the outer dentin surface becomes the dentino-enamel junction (DEJ). The cervical limit of the enamel crown is established at the point where the inner and outer enamel epithelia fuse to form Hertwig’s epithelial root sheath (HERS). From that point on, the interface between epithelium and mesenchyme becomes the outer surface of dentin along the root, which is covered by cementum. In this review, we focus on the later events in amelogenesis: the formation of the enamel crown on the outer dentin surface, starting with the onset of biomineralization at the future cusp tips. We discuss five growth parameters that determine the shape of the enamel crown, the mechanism of appositional growth that establishes the thickness of the enamel layer, and the mechanism of enamel maturation that hardens enamel.
Part I. Growth Parameters that Determine the Shape of the Enamel Crown
Incremental Lines in Teeth
Enamel and dentin form by accretionary modes of growth that preserve within the hard tissues short- and long-period lines of incremental growth. Dental structures are not remodeled by cycles of resorption and deposition, so growth lines formed during tooth development are permanent. In dental enamel, there are two regularly occurring incremental markers: daily cross-striations and long-period striae of Retzius (SR lines). These lines correspond to what was the enamel surface at precise points in time during the secretory stage of amelogenesis. Striae of Retzius are prominent cross-striations that occur every 7 to 11 days in humans (Reid and Ferrell, 2006). In a given individual, the striae of Retzius are the same number of days apart, but the interval between striae varies in different people. One hypothesis suggests that striae of Retzius are caused by the imperfect synchronization of two circadian rhythms. Periodically, an incremental deposit is delayed for the remainder of the daily cycle to restore synchrony between the two rhythms (Newman and Poole, 1974). Systemic disturbances (fever) and birth (neonatal line) cause highly accentuated striae of Retzius in addition to those caused by body rhythms (Kodaka et al., 1996). The striae of Retzius terminate at the enamel surface in shallow furrows, called perikymata, that run horizontally around the enamel crown (Fig. 1A) (Risnes, 1985, 1998). The distance between perikymata decreases near the cervical margin (CM) (Fig. 1B). Between the striae of Retzius are less distinct lines known as cross-striations. Cross-striations demarcate the amount of enamel deposited by ameloblasts in a single day (FitzGerald, 1998). Measuring the distance between adjacent cross-striations can be used to calculate the daily rate of enamel deposition by ameloblasts. The average rate is approximately 4 µm/day in humans (Risnes, 1986), 6 µm/day in mice, and 12-13 µm/day in rats (Smith, 1998). In primates, the distance between adjacent cross-striations tends to increase from the DEJ toward the outer enamel surface and to decrease from the cusp tip to the CM (Beynon et al., 1991; Lacruz and Bromage, 2006). The physical basis for cross-striations is unknown. Explanations include variations in prism thickness secondary to changes in the proportion of the matrix secreted at the walls vs. the tips of the Tomes’ processes, and variations in carbonate content and crystallinity of the mineral (Shellis, 1998).
Figure 1.
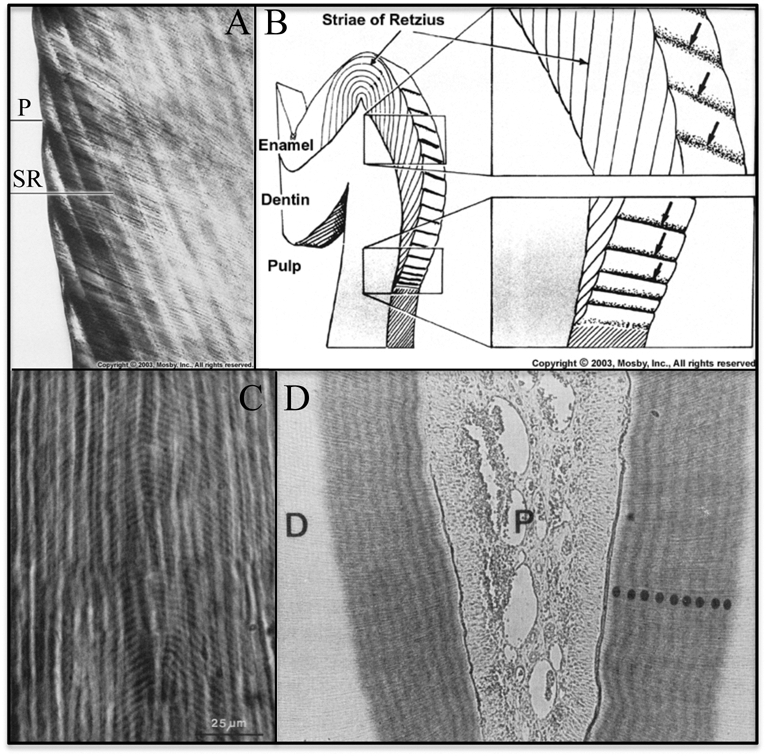
Incremental lines in teeth. (A) Ground section of enamel showing how perikymata (P) are surface manifestations of the striae of Retzius (SR) (Nanci, 2008a). (B) Line drawing showing the striae of Retzius in a molar (Nanci, 2008a). The striae of Retzius are long-period (6- to 11-day) growth lines that extend from the DEJ to the enamel surface. They show where the enamel surface was at one day during development. There are no perikymata at the cusp tip, because the Retzius lines run continuously in an elliptical arc from the DEJ on one side of the cusp tip to the DEJ on the other side of the cusp tip without breaking the surface. (C) Ground section of dentin (Dean, 1998) showing short-period growth lines (von Ebner’s lines: the curved, periodically repeating, inverted V’s in image). (D) Autoradiograph of rat dentin section showing 9 densely labeled circumpulpal bands  after infusion with labeled proline for 10 days, suggesting daily fluctuations in the secretion of collagen (Ohtsuka et al., 1998).
after infusion with labeled proline for 10 days, suggesting daily fluctuations in the secretion of collagen (Ohtsuka et al., 1998).
Incremental lines are also observed in dentin (Fig. 1C). The daily short-period lines in dentin are termed von Ebner’s lines. The spacing of short-period lines increases from about 2 µm early in dentin formation to a maximum of 4 µm (Risnes, 1986; Dean, 1998). Long-period lines in dentin are called contour lines of Owen, and are analogous to the striae of Retzius in enamel. There is a 1:1 relationship between long-period lines in enamel and dentin, so it appears that the same disturbance causes both (Dean and Scandrett, 1996).
The numbers of cross-striations in enamel and von Ebner’s lines in dentin correspond to the number of days between sequentially administered dyes and suggest that circadian rhythms are important in their formation (Dean, 1989, 1998; Shellis, 1998). Dentin collagen is deposited in a daily cycle (Fig. 1D). Twice as much collagen is secreted by rat odontoblasts during the daylight 12 hrs as during the nighttime 12 hrs (Ohtsuka et al., 1998). In rat incisors, variations in the rate of production and secretion of enamel proteins between early morning and late afternoon suggest that enamel protein secretion is under circadian control (Fig. 2).
Figure 2.
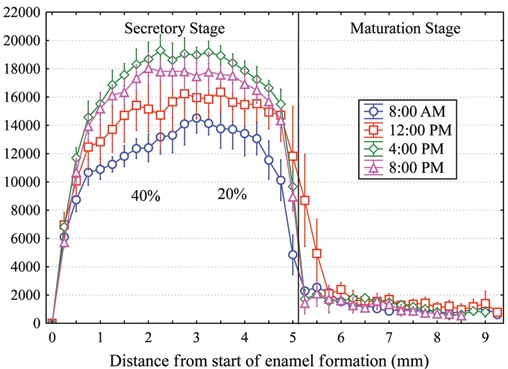
Daily variation in ameloblast secretion of proteins containing methionine. Graph of means ± 95% confidence interval illustrating the total amount of newly synthesized proteins released into developing enamel on rat mandibular incisors by 1 hr after a single intravenous injection of 3H-methionine administered at different times of the day. Substantially greater amounts of secretory activity for enamel proteins occur in the late afternoon (4:00 p.m., diamonds) compared with early morning (8:00 a.m., circles) throughout the secretory stage. These differences are noticeably larger (up to 40%) for inner enamel formation (distance, 0.5-3.0 mm) than for outer enamel formation (20%). Each datapoint represents mean counts of sections from 6 incisors per time-point. Technical information about how animals were injected, tissues were processed, and quantitative data were obtained by computerized image analysis has been described previously (Smith and Nanci, 1996).
Growth Parameters that Determine the Enamel Crown Shape
Once early developmental processes, based upon epithelial-mesenchymal interactions, establish the position of the DEJ, the shape of the enamel crown is determined by five growth parameters, which are potentially important points of biological control. The five growth parameters are: the (1) appositional growth rate, (2) duration of appositional growth (at the cusp tip), (3) ameloblast extension rate, (4) duration of ameloblast extension, and (5) spreading rate of appositional termination (Fig. 3). Incremental lines in enamel facilitate the measurement of these growth parameters.
Figure 3.
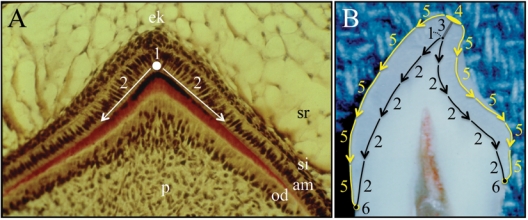
Pattern of ameloblast differentiation during crown formation. (A) Section through the developing primate cusp tip. Pre-ameloblasts first differentiate from inner enamel epithelia on the dentin surface covering the pulp horn (1) beneath what will become the cusp tip. From this beginning, a wave of ameloblast differentiation moves through the inner enamel epithelia down the slope of the mineralized dentin surface (2). Key: am, ameloblasts; od, odontoblasts; ek, enamel knot; p, pulp; si, stratum intermedium; sr, stellate reticulum. (B) Incisor split open to show the growth parameters that determine the shape of the enamel crown once earlier developmental processes establish the dentin surface. (1) Ameloblasts differentiate beneath the future cusp tip and deposit the first increment of enamel. (2) A wave of ameloblast differentiation extends down the slope of the dentin surface and ends when it reaches its limit, where the inner enamel epithelium previously fused with the outer enamel epithelium to form Hertwig’s epithelial root sheath. (3) Enamel mineral builds up in daily increments (appositional growth) during the secretory stage of amelogenesis. (4) Ameloblasts at the cusp tip end the secretory stage (appositional growth) and transition into maturation-stage ameloblasts. (5) A wave of ameloblast re-organization that terminates the secretory stage and transitions the ameloblasts into the maturation stage moves down the enamel surface. (6) Ameloblast termination reaches the last secretory ameloblasts at the cervical margin, and the shape of the enamel crown is established. After this point, the entire crown is in maturation stage, which involves the removal of residual enamel proteins and the growth of existing enamel crystallites in width and thickness.
Appositional Growth Rate
The final thickness of the enamel layer is determined by the amount of appositional growth (Smith, 1998; Smith et al., 2005). “Appositional growth” has varied meanings in different contexts. The appositional growth rate is the increase in thickness of the enamel layer perpendicular to the DEJ per day. Appositional growth rate varies with location, so this parameter is a function rather than a constant. Enamel rods and the oriented crystallites in them are deposited at an angle to the DEJ, so the actual daily increase in the total length of enamel rods (as determined by measuring the spacing between adjacent cross-striations) is roughly 15% greater than the appositional growth rate perpendicular to the DEJ (Risnes, 1986). The final enamel surface area is larger than the dentin surface it covers, and the enamel rods (each deposited by a single ameloblast) do not thicken. Depositing enamel rods at oblique angles to the perpendicular along with the net movement of secretory ameloblasts in the cuspal direction accommodates coverage of the expanding enamel surface (Radlanski and Renz, 2004).
Appositional growth is the product of pre-ameloblasts and secretory ameloblasts. (A secretory ameloblast has a Tomes’ process that organizes enamel crystallites into rods; pre-ameloblasts are secretory ameloblasts that have not yet formed a Tomes’ process.) On the dentin horn (the dentin surface beneath the future cusp tip), the first epithelial cells differentiate into pre-ameloblasts, which initiate enamel formation and form a thin layer of aprismatic enamel on the dentin surface. The signals driving this differentiation come from the enamel knot and the underlying odontoblasts (Thesleff and Jernvall, 1997; Thesleff et al., 2001). Enamel mineral deposition takes place in the extracellular space along the ameloblast (distal) cell membrane, and is associated with the secretion of enamel matrix proteins (amelogenin, ameloblastin, enamelin) and enamelysin, a proteolytic enzyme that cleaves enamel matrix proteins (Fincham et al., 1999). Enamel proteins form a mineralization front and induce the simultaneous production of thousands of oriented enamel ribbons near the plasma membrane of each ameloblast. As ameloblasts secrete enamel proteins and extend the mineral ribbons, they retreat from the existing enamel surface, thus increasing the thickness of the enamel extracellular space. Unlike collagen-based mineralization processes, which are two-step processes (secretion of an organic matrix followed later by mineralization of the matrix), secretory-stage enamel formation occurs in a single step, with secretion of the organic matrix and mineralization of the matrix being coupled.
Duration of Appositional Growth
The duration of appositional growth is the number of days during which ameloblasts are engaged in appositional growth. Near the end of the secretory stage (starting at the cusp tips), ameloblasts retract their Tomes’ processes, lay down a final layer of aprismatic enamel, and then undergo a transition that ends the secretory stage, and with it, appositional growth. The final enamel thickness is the product of the appositional growth rate times the duration of appositional growth. The important regulatory points for the duration of appositional growth are at the cusp tips, where the secretory stage first concludes and the transition to maturation begins.
Ameloblast Extension Rate
From its starting point on the dentin horn where the enamel knot stimulates differentiation of inner enamel epithelia into the first pre-ameloblasts, the differentiation of adjacent epithelial cells into pre-ameloblasts spreads down the slopes of the crown in all directions (Shellis, 1998). This “wave” of ameloblast differentiation continues until it stops at the CM, where the crown ends and the root begins. Ameloblasts are about 5 to 6 microns in diameter, and the rate at which successive rows of epithelial cells become pre-ameloblasts corresponds to the ameloblast extension rate. The extension rate slows substantially from 20 to 30 µm/day at the cusp tip to 3 to 6 µm/day near the CM (Birch and Dean, 2009), so the striae of Retzius (which are separated by a constant number of days) are closer together at the DEJ nearer to the CM than they are at the cusp tip (Fig. 1B).
Duration of Ameloblast Extension
Expansion of the ameloblast layer along the dentin surface concludes at the CM, where the wave of ameloblast extension ceases. The CM coincides with the locus where the inner and outer enamel organ epithelia fuse to form HERS. How far the enamel crown extends along the dentin surface is a function of the rate at which inner enamel epithelia differentiate into pre-ameloblasts (the extension rate) times the amount of time this “wave” of differentiation (duration of ameloblast extension) continues to spread down the dentin surface toward what will become the cervical margin, which is at or near the cemento-enamel junction or CEJ.
Spreading Rate of Appositional Termination
This is analogous to the ameloblast extension rate, but instead of starting, the ameloblasts are ending the appositional growth phase of amelogenesis. We hypothesize that at a cusp tip the same cohort of ameloblasts that was previously induced to become the first pre-ameloblasts on the dentin horn, after having deposited enamel for a set period of time, terminates appositional enamel growth. Then a “wave” of ameloblast transition to maturation stage spreads from this cohort down the cusp slope in all directions. Whereas the ameloblast extension rate is the rate at which the wave of pre-ameloblast differentiation spreads along the DEJ, the spreading rate of appositional termination (for simplicity, the termination rate) is the rate at which the wave of ameloblast transition (secretory-stage termination) spreads along the outer enamel surface (OES) to the CM.
Regulation of Enamel Crown Morphogenesis
Early tooth development maps out the shape of the dentin surface (in a process driven by epithelial-mesenchymal interactions), and then the five growth parameters listed above determine the shape of the enamel crown. The five growth parameters are not constants, but can vary from crown to crown and during the formation of a single crown. Little is known about how these parameters are regulated, but it is certain to be complex, since altering one parameter affects others (more on this in the next section). The thickness of enamel at the cusp tip is equal to the rate of appositional growth times its duration. The rate of appositional growth is a function of the daily increment of mineral deposition that lengthens the enamel rod and the angle that the long axis of the rod deviates from perpendicular to the DEJ. The average distances between cross-striations in human teeth are about 2.5 µm at the DEJ and 6.5 µm at the enamel surface (Birch and Dean, 2009). The duration of appositional growth at the cusp tip is determined by factors that cause secretory ameloblasts at the cusp tip to transition into maturation. This transition occurs when the enamel has reached its final thickness and ameloblasts are too distant from odontoblasts to be regulated by epithelial-mesenchymal interactions. We hypothesize that a molecular counter may help regulate the duration of appositional growth at the cusp tip.
The position of the cervical margin, which affects crown height, is determined by factors that cause the cervical loop (CL; the growth center that causes the crown to grow down toward the latent root) to transition into HERS (the growth center that causes the root to grow longer). HERS is a bicellular layer comprised of inner and outer enamel epithelia, whereas in the cervical loop these cells are separated by stellate reticulum (star-shaped cells separated by large intercellular spaces) (Sasaki, 1990; Luan et al., 2006). It has been proposed that the conversion of CL into HERS may be associated with the down-regulation of growth factors that sustain proliferation of the intervening stellate reticulum, such as epidermal growth factor (Fujiwara et al., 2009).
Enamel thickness decreases from the cusp tip to the cervical margin. Since variations in the appositional growth rate are relatively minor (Beynon et al., 1991; Lacruz and Bromage, 2006), the duration of appositional growth must be less at the CM than at the cusp tip. For this to occur, the wave of ameloblast termination must move faster down the outer enamel surface than the wave of ameloblast extension moves down the outer dentin surface, so that ameloblasts from the cusp tip to the cervical margin spend progressively less time in the secretory stage. Variations in the termination rate (as determined by measuring the distances between perikymata) appear to be an important parameter in the evolution of crown morphology in hominids (Dean and Reid, 2001).
Summary
Early tooth formation establishes the interface between odontogenic epithelium and mesenchyme, which dictates the position of the (latent) dentin surface upon which enamel forms. Given the architecture of the latent dentin surface, the shape of the enamel crown results from the interplay of five growth parameters. A better understanding of how variations in the five growth parameters affect crown morphology and how these parameters are regulated would provide insights into the mechanisms underlying the development and evolution of crown shape in mammals. How simultaneous changes in one or more of the five growth parameters affect enamel crown morphology can be difficult to imagine, so computer simulations are being developed for this purpose. In Fig. 4, we show two runs of a simple two-dimensional model that depicts enamel apposition on a single cusp using different extension and termination rates. Computer simulations can be used to generate several models that illustrate how changes in the five growth parameters might explain observed variations in crown morphology between two related species. Then, analyses of incremental lines in teeth from the two species could provide the data necessary to choose among the alternative models. In addition, computer simulations can lead to new hypotheses. In the simple model presented here, the most realistic cervical margin shapes are generated by conditions in which propagation of the wave of extension is stopped by the “passing” wave of termination. This suggests that the signals causing the transition of secretory ameloblasts into maturation ameloblasts might participate in the formation of HERS.
Figure 4.
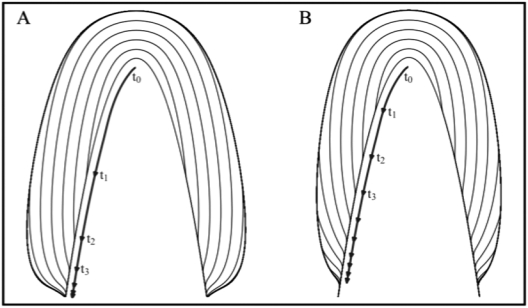
Models depicting enamel apposition while varying the extension and termination rates. An inverted parabola populated with virtual ameloblasts represents the hypothetical DEJ. Ameloblast activation sweeps from the horn of the DEJ down the length of the curve, with the rate of extension decreasing exponentially down the slope of the tooth. Once activated, cells move along vectors normal to the DEJ at a fixed rate. Black lines represent the positions of the secretory front at evenly spaced time intervals (t0, t1, t2, . . .) and can be thought of as virtual striae of Retzius. Bold lines with arrows show how the wave of activation moves down the DEJ. The appositional growth rate and duration of appositional growth (at the cusp tip) are identical in both models. Extension ends when the wave of termination reaches the wave of activation. (A) In this simulation, a wave of ameloblast activation moves down the crown at a rate based on an exponential decay equation with a decay constant of 0.025. The wave of appositional termination moves down the crown at a constant rate that is rapid relative to the rate at which the wave of activation advances. (B) In this simulation, a lower decay constant of 0.01 was used for activation, resulting in a slower advance of the wave. Termination of apposition moves down the crown at a rate defined by an exponential decay equation with a decay constant of 0.02.
Part II. the Mechanism of Appositional Growth
During tooth development, the earliest odontoblasts and ameloblasts differentiate at the DEJ under the future cusp tip. Odontoblasts differentiate into columnar cells and secrete mantle predentin, which is rich in type I collagen and matrix vesicles. The basal lamina associated with the inner enamel epithelium disintegrates and is penetrated by slender processes from the overlying epithelial cells (Reith, 1967; Kallenbach, 1976). Disruption of the basal lamina occurs after the odontoblasts have begun to produce predentin, but before the onset of calcification, and is associated with a major up-regulation in the production and secretion of enamel matrix proteins and Mmp-20 (Inai et al., 1991). Initial mineralization occurs in the predentin matrix, perhaps within matrix vesicles (Katchburian, 1973). Mineral is first deposited subjacent to the DEJ and progresses toward the DEJ to the terminal ends of the collagen fibrils (Arsenault and Robinson, 1989). Finger-like projections of the ameloblast plasma membrane penetrate the fenestrated basal lamina, extend to the dentin surface, and deposit enamel proteins (Ronnholm, 1962). Then numerous enamel mineral ribbons measuring 10-15 nm in width and 1-2 nm in thickness originate suddenly at the DEJ (Daculsi and Kerebel, 1978; Kerebel et al., 1979; Weiss et al., 1981; Cuisinier et al., 1992). The initial enamel ribbons are distinct from those of dentin (Diekwisch et al., 1995). They are uniform in size, parallel (highly oriented in the long axis direction), evenly spaced, and randomly oriented with respect to their widths or the long axes of their cross-sections (Nylen et al., 1963). The distance between crystallites is approximately 20 nm (Diekwisch et al., 1995). The growing tip of an enamel crystallite has a cross-section of 15 x 1.5 nm. The dimensions are the same in the earliest enamel ribbons and the elongating tips of enamel ribbons throughout the secretory stage. In developing human primary teeth, the density at the surface is 1240 crystallites/mm2. Mature erupted teeth have 558 crystallites/mm2 at the tooth surface (Kerebel et al., 1979), the decreasing number being due to crystallite fusions.
While the exact nature of the mineral inside the early enamel ribbons is controversial, electron diffraction evidence supports the interpretation that enamel ribbons elongate at the mineralization front by adding successive increments of amorphous calcium phosphate (ACP) (Landis et al., 1988; Beniash et al., 2009). The addition of new mineral appears to be directly on the tips of existing mineral ribbons, elongating them. It is difficult to estimate their actual lengths, but based upon the long lengths of crystallites isolated from enamel, it is likely that “enamel crystals extend without interruption from their beginning at the dentino-enamel junction to their ends at the enamel surface” (Daculsi et al., 1984). The mineral near the growing tips of the enamel ribbons is not crystalline (amorphous calcium phosphate), but the deeper, more developed, part of the ribbons is (hydroxyapatite). Thus, the initial shape of an enamel ribbon is not intrinsic to the mineral itself, but may be molded by the space available for it. Enamel matrix proteins (amelogenin, ameloblastin, and enamelin) are secreted at the mineralization front where the mineral ribbons grow in length (Fig. 5) and are necessary for the formation of enamel ribbons. The onset of enamel mineral deposition correlates with the first release of ameloblastin, enamelin, and enamelysin and the massive up-regulation of amelogenin. In the Enam null and Ambn mutant mice, the mineralization front fails, and no enamel forms (Fig. 6) (Fukumoto et al., 2004; Hu et al., 2008; Wazen et al., 2009). Only a thin layer of aprismatic enamel (~20 µm, instead of more than 100 µm) is deposited on dental crowns in AmelX null mice (Gibson et al., 2001; Prakash et al., 2005).
Figure 5.
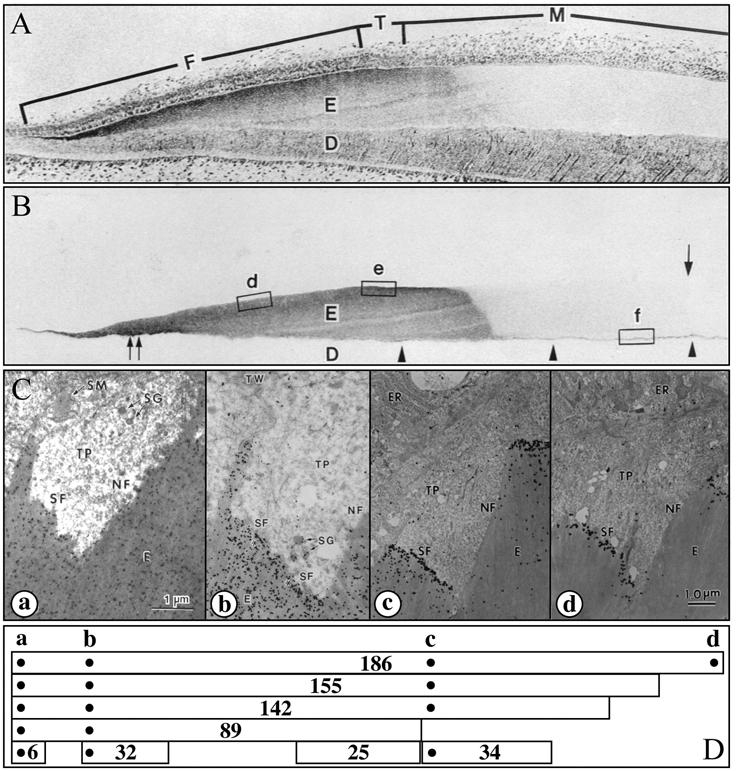
Enamelin localization in developing enamel. (A,B) Consecutive sections of a developing porcine incisor. The top section is stained with toluidine blue; the second section is immunostained with the 32-kDa enamelin antibody (Uchida et al., 1991a). The 32-kDa enamelin signal is observed throughout the secretory stage in the enamel matrix, from the DEJ to the surface. However, because enamelin is extensively processed by proteases and the C-terminal parts are removed from the matrix, its distribution depends upon which part of the protein is recognized by the antibody. (C,D) Transmission electron microscopy (TEM) showing immunogold staining patterns of forming enamel near the ameloblast Tomes’ processes using affinity-purified anti-peptide antibodies raised against the enamelin (a) N-terminus (Dohi et al., 1998), (b) 32-kDa cleavage product (Uchida et al., 1991a), (c) 34-kDa cleavage product (Hu et al., 1997a), and (d) C-terminus (Hu et al., 1997a). Note that each antibody shows a different localization pattern. Below the TEMs are diagrams showing enamelin (186 kDa) and known cleavage products (155, 142, 89, 34, 32, 25, and 6 kDa) and the positions of the sequences used to make antibodies (dots) (Hu and Yamakoshi, 2003). An important observation is that the enamelin C-terminus is found only at the mineralization front (d).
Figure 6.
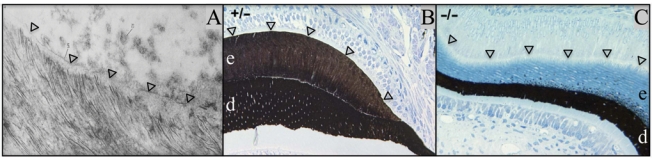
The secretory-stage mineralization front. Arrowheads mark the mineralization front. (A) TEM of developing human tooth showing enamel crystallites extending from a layer of enamel proteins along the secretory surface of the ameloblast distal membrane (Ronnholm, 1962). [Note: In Fig. 5C part d, we show that intact enamelin (containing the C-terminus) is found only along the mineralization front.] (B,C) Von Kossa (which turns mineralized tissues dark) -stained sections of developing mouse teeth (dentin, d; enamel, e). (B) Enamelin heterozygous (+/–) mouse section showing that both the dentin and enamel layers are mineralized. (C) Enamelin null mouse (–/–) showing that, without enamelin, the mineralization front fails and does not stimulate enamel mineralization. Only small, punctate foci of mineralization are detected within the enamel layer near the dentino-enamel junction, despite there being a thick accumulation of organic material (light blue) in the enamel layer (Hu et al., 2008).
Besides enamel proteins, normal appositional growth requires the secretion of enamelysin (Mmp-20, matrix metalloproteinase 20) (Bartlett et al., 1996). Enamel proteins are processed by enamelysin, so that intact enamel proteins are found only in the superficial layer of the developing enamel (Uchida et al., 1991b; Hu et al., 1997b; Murakami et al., 1997), with intact enamelin being restricted to the mineralization front (Hu et al., 1997a). Enamel ribbons form normally in Mmp20 null mice, and the ribbons convert into hydroxyapatite, but the enamel layer as a whole is thinner, less hard, and the organization of rod and interrod enamel is disturbed (Caterina et al., 2002; Bartlett et al., 2004). Without enamelysin, appositional growth apparently cannot be sustained.
In normal enamel formation, as the enamel ribbons continue to lengthen at the mineralization front, they also grow progressively thicker and wider with depth. The cross-sectional geometry of the deeper part of a ribbon closer to the DEJ is a flattened hexagon with sharp outlines that are characteristic of crystallites (Nylen et al., 1963; Kallenbach, 1990; Miake et al., 1993; Nanci, 2003). Growth on the sides of enamel crystallites initially increases their width and later their thickness. While the width-to-thickness ratio of the earliest crystallites is nearly 10, this ratio falls to 2.6 for the enamel crystallites of erupted teeth (Daculsi and Kerebel, 1978). Enamel crystallites continue to thicken after they come into contact with adjacent crystallites, causing their final cross-sectional geometries to be irregular. The cross-sections of mature enamel crystallites average 26 x 68 nm (Warshawsky and Nanci, 1982; Daculsi et al., 1984).
Appositional growth involves the deposition of thousands of similarly shaped ribbons of ACP that lengthen near the ameloblast distal membrane at a mineralization front consisting of enamel proteins and enamelysin. The ACP ribbons convert into calcium hydroxyapatite (HAP) a short distance away from the enamel surface, while they are associated with enamel protein cleavage products. The bulk of enamel proteins that separate the mineral ribbons are amelogenins, which comprise over 90% of the matrix and self-assemble into spherical structures (Fincham et al., 1994) approximately the same width (~20 nm) as the space between crystallites. Amelogenins influence the conversion of ACP to HAP in vitro (Kwak et al., 2009). According to this model of appositional growth, the principal functions of enamel matrix proteins are: (1) to generate a mineralization front that partitions the space available for mineral into molds that shape the amorphous mineral into ribbons, (2) to ensure that successive mineral increments are deposited on the tips of ribbons deposited in the previous cycle, (3) to separate the mineral ribbons, and (4) to regulate the conversion of ACP into HAP, which presumably involves the absorption of the hydrogen ions released by HAP formation. Enamel proteins may also facilitate the movement and attachment of ameloblasts (Sonoda et al., 2009; Beyeler et al., 2010). Many other functions of enamel proteins are possible, since the mechanism of dental enamel formation at the molecular level is poorly understood.
The Classic Model of Appositional Growth
The mineralization front model for appositional growth of dental enamel differs radically from the classic view of enamel formation (Simmer and Fincham, 1995). The classic model was formulated when “amelogenin” and “enamelin” were terms for hypothetical classes of proteins, rather than specific genes and proteins (Termine et al., 1980), and preceded the first cloning of cDNAs for any of the enamel matrix proteins (Snead et al., 1983, 1985). As molecular data became available, it was found that amelogenin, enamelin, and ameloblastin are not acidic like the non-collagenous proteins of dentin and bone, but belong to the proline and glutamine group of secretory calcium-binding phosphoprotein (SCPP) proteins (Kawasaki and Weiss, 2008). The genes for enamel proteins evolved from a common ancestral gene and belong to a single class. The classic model holds that enamel mineral forms as crystalline material (octacalcium phosphate or hydroxyapatite) that is shaped into long ribbons by the selective binding of acidic beta-sheet secondary structures of enamel proteins to the sides of the crystallites (Fan et al., 2008). Proteolytic cleavage of the enamel proteins releases them from the sides of the crystallites, allowing the crystallites to grow in width and thickness (Sun et al., 2008). Another aspect of the classic model is that amelogenin buffers calcium ion concentration to control the degree of saturation of enamel fluid so as to favor the formation of hydroxyapatite over other calcium phosphate solid phases (Aoba and Moreno, 1987). The classic theory is based upon ideas about biomineralization that, for the most part, can be supported in principle by in vitro studies of crystal growth. Although this model is still accepted by many, we believe that it is inconsistent with many observations of how enamel is observed to form in vivo. Even if one function of enamel proteins is to inhibit mineral deposition on the sides of enamel crystallites, it remains unclear how protein inhibitors can distinguish between the sides of hydroxyapatite crystals (which have identical faces) to selectively inhibit first crystallite thickening and then crystallite widening. Furthermore, enamel crystallites grow in width and thickness during the secretory stage, when enamel proteins are abundant, and during the early maturation stage prior to the removal of significant amounts of matrix protein (Smith, 1998). Large increases in crystallite width and thickness even occur in the Mmp20 (Caterina et al., 2002) and Klk4 (Simmer et al., 2009) knockout mice, where there is no significant extracellular proteolytic activity, and enamel proteins persist in the matrix. The classic theory ignores the mineralization front, which in vivo studies suggest is inherent to the mechanism of enamel appositional growth. When the specialized enamel proteins enamelin or ameloblastin are missing or defective, the mineralization front fails to form, and the enamel layer is virtually absent (Fukumoto et al., 2004; Hu et al., 2008). The mineralization front gives shape to the mineral ribbons before they are crystalline (Beniash et al., 2009).
Regulation of Appositional Growth
The thickness of the enamel layer covering dentin is determined by the amount of appositional growth. Appositional growth starts with the differentiation of inner enamel epithelium into pre-ameloblasts and ends with the transition of secretory ameloblasts into maturation ameloblasts. The amount of time ameloblasts spend in appositional growth (secretory stage) varies, and the enamel thickness varies accordingly. The developmental signals that regulate the onset and termination of the secretory stage are largely unexplored, but these steps are associated with changes in the expression of extracellular matrix molecules. At the onset of the secretory stage, amelogenin, enamelin, ameloblastin, and Mmp-20 expressions are initiated. The transition to maturation initiates kallikrein 4, amelotin, and odam (apin) expression (Hu et al., 2000; Moffatt et al., 2006b, 2008), reformation of a glycoprotein-rich basal lamina on the surface of the enamel (Sawada and Inoue, 2001; Al Kawas and Warshawsky, 2008), and some ameloblast apoptosis (Joseph et al., 1994; Bronckers et al., 2000; Kondo et al., 2001). Perhaps continued characterization of the transcription factors that regulate amelogenin gene expression will provide insights into the developmental mechanisms that mediate the initial differentiation and the later transition of ameloblasts (Adeleke-Stainback et al., 1995; Gibson, 1999; Zhou and Snead, 2000; Xu et al., 2009; Xu et al., 2006, 2007a,b). A rat wct (whitish chalk-like teeth) mutant exhibits an arrest of enamel formation at ameloblast transition (Masuyama et al., 2005; Osawa et al., 2007).
Appositional growth appears to involve the repetition of a secretory cycle that results in the formation of long- and short-period incremental lines. A potentially important function of enamel proteins is to ensure proper alignment of each ribbon addition with the tip of the previously deposited mineral increment, so that the crystallites grow to great lengths. The incremental growth of enamel suggests that clock genes (Cermakian and Boivin, 2009) could be important. Molecular clocks and counters potentially play a role in determining the onset of ameloblast termination at the cusp tip, which could occur a set number of days after pre-ameloblast differentiation. After the onset of termination is initiated, the wave of differentiation appears to be propagated by successive horizontal bands of ameloblasts parallel to the cervical loop and would not depend upon a timing mechanism.
On their course from the DEJ to the enamel surface, ameloblasts move relative to each other, resulting in decussating rod patterns and Hunter-Schraeger bands (optical phenomenon produced by changes in direction between adjacent groups of enamel rods) (Hanaizumi et al., 1996). As ameloblasts retreat, some migrate cuspally from the cervical loop (to maintain coverage of the radially expanding enamel surface). Ameloblasts in a circumcoronal loop may have differentiated and begun secretion at different times during tooth development, yet all transition to maturation stage simultaneously and form a circumcoronal perikyma that is a horizontal closed circle (Risnes, 1985). In the absence of experimental data concerning the mechanisms that terminate appositional growth, we hypothesize that a clock/counter mechanism might dictate when ameloblasts at the cusp tip enter transition, but this is followed by a propagated signal spreading from these cells that pushes successive rows of ameloblasts into transition.
A Role for Molecular Clocks in Enamel Formation?
The existence of daily lines in enamel suggests that circadian rhythms play a role in its formation. Circadian rhythms in mammals are regulated globally by the master clock in the suprachiasmatic nucleus (SCN), and locally by clock cells that control tissue-specific rhythmic outputs (Amir et al., 2004). Circadian oscillations are generated by a set of genes encoding transcription factors that form a transcriptional autoregulatory feedback loop (Siepka et al., 2007). Lamellar bone, like dentin and enamel, forms incrementally, mirroring the long-period rhythm represented by the striae of Retzius in enamel (Bromage et al., 2009). The expression of clock genes in osteoblasts (bone-forming cells) is regulated by the sympathetic nervous system and the hormone leptin (Fu et al., 2005). Mice with mutations in circadian genes, such as Period (Per1 and Per2) and Cryptochrome (Cry1), show increased numbers of osteoblasts and develop high bone mass (Fu et al., 2006), suggesting that clock genes inhibit bone formation by preventing osteoblast proliferation (Fu et al., 2005). Lesions of the SCN in the brain cause a loss of circadian increments in dentin (Ohtsuka-Isoya et al., 2001). Clock encodes a transcription factor that is essential for circadian rhythms (King et al., 1997) and localizes within the nucleus of differentiating ameloblasts and odontoblasts (Fig. 7). What these molecules are doing is unknown.
Figure 7.
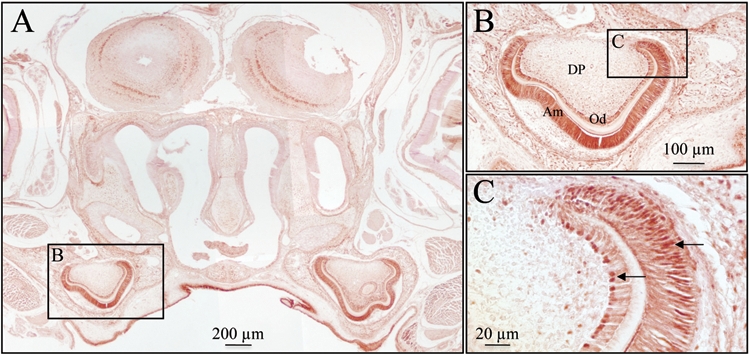
Immunohistochemistry of clock protein in a post-natal day 4 mouse. (A) Clock protein expression was detected in the developing first molars. (B,C) Higher magnifications showing that the nuclei (arrows) of ameloblasts (AM) and odontoblasts (OD) have strong clock expression relative to the dental pulp (DP) cells. Samples were fixed at 4°C for 4 hrs in 4% formalin, washed (3x) with rinse buffer (2 mM MgCl2 and 0.1% Nonidet P40 in PBS), demineralized in EDTA for 2 wks, embedded in paraffin, and sectioned. The sections (~4 µm) were rehydrated, blocked, treated for antigen retrieval in 10 mM sodium citrate for 30 min by being microwaved prior to incubation with an anti-clock primary antibody (dilution 1:100; Calbiochem, San Diego, CA, USA) for 1 hr at room temperature, followed by incubations with a biotinylated secondary antibody (1:200, Vector Laboratories, Burlingame, CA, USA) and a horseradish peroxidase-streptavidin conjugate (1:200, Zymed, San Francisco, CA, USA). Signal was detected by means of the DAB Plus Substrate kit (Zymed).
Part III. Mechanisms of Enamel Maturation
Enamel maturation involves the deposition of ions onto the sides of enamel crystallites. The final hardness of dental enamel is dependent upon the growth in width and thickness of the enamel ribbons so that adjacent crystallites come into contact and interlock. Once the secretory stage is complete, no additional crystallites form, and there is no further lengthening of existing crystallites. Enamel crystallites start to mature during the secretory stage as the ribbons grow in length, so the cross-sectional dimensions of secretory-stage crystallites increase progressively from the mineralization front to the DEJ (Kerebel et al., 1979). During the maturation stage, when more than 60% of enamel mineral deposition takes place, ions are deposited on the sides of existing crystallites at the expense of matrix protein remnants and fluid, which exit the enamel layer from the surface, presumably facilitated in whole or in part by maturation-stage ameloblasts (Smith, 1998).
After undergoing a brief post-secretory transition, ameloblasts develop into maturation (or modulating) ameloblasts. These cells modulate between two forms: ruffle-ended ameloblasts (RA), which have tight distal and loose proximal junctional complexes with a striated border toward the enamel surface, and smooth-ended ameloblasts (SA), which have disassociated distal and tight proximal junctional complexes and unmodified distal membranes (Warshawsky and Smith, 1974; Josephsen and Fejerskov, 1977). In rats, a modulation cycle lasts for about 8 hrs, with about 75% of the time being spent in the ruffle-ended phase. A remarkable finding is that the pH of the enamel matrix covered by RA is acidic, dropping to as low as pH = 6, while enamel covered by SA has a nearly physiologic pH (pH = 7.2) (Sasaki et al., 1991; Smith et al., 1996).
During the maturation stage, residual enamel proteins are degraded by Klk4 and, to a lesser extent, by Mmp-20 (Bartlett and Simmer, 1999; Lu et al., 2008). Genetic defects in human MMP20 and KLK4 cause hypomaturation forms of amelogenesis imperfecta (Hart et al., 2004; Kim et al., 2005), and the Mmp20 (Caterina et al., 2002) and Klk4 (Simmer et al., 2009) null mice exhibit defective enamel. Although enamel proteins are retained in Klk4 null mice, the enamel crystallites are able to grow considerably in width and thickness. The enamel layer hardens, for the most part, but there is a weakness near the DEJ, and the enamel layer abrades away when the teeth erupt into function (Simmer et al., 2009).
While degradation and re-absorption of the organic matrix are important, the principal activity in the maturation stage is the regulated movement of ions into and out of the matrix. Essentially, ameloblasts move calcium, phosphate, and bicarbonate into the matrix and remove water. The bicarbonate is needed to neutralize the hydrogen ions that are generated by hydroxyapatite formation. Some of the critical molecular participants involved in pH regulation have been identified by the characterization of null mice with enamel defects and by human genetic studies of kindreds with inherited enamel defects. Next we present a simplified model for the regulation of pH during the maturation stages that is analogous to mechanisms used in salivary glands, and differs somewhat from other explanations (Lacruz et al., 2010).
Neutralizing Protons Released during Hydroxyapatite Formation
A simple formula for the formation of hydroxyapatite from its constituent ions is:
The formula is misleading, because it ignores the ionic species that are actually available at physiological pH to produce calcium hydroxyapatite. The anionic components (6PO43- + 20H-) are rare at physiological pH, and are abundant only in their protonated forms. At pH = 7.2, the phosphate is about half HPO42- and half H2PO4-, so an average of 9 hydrogen ions are released to generate the 6PO43- ions required to deposit a single unit cell of hydroxyapatite (3HPO42- + 3H2PO4- ⇔ 6PO43- + 9H+). The hydroxyls are generated by the dissociation of water, which releases two more hydrogen ions per unit cell of hydroxyapatite (2H2O ⇔ 20H- + 2H+). The net effect is that 11 H+ ions are generated for every unit cell of hydroxyapatite that forms at pH 7.2.
During the maturation stage of amelogenesis, hydrogen ions released by hydroxyapatite formation are neutralized by bicarbonate (Simmer and Fincham, 1995; Smith, 1998; Lacruz et al., 2009), by a mechanism similar to the way that striated duct cells excrete bicarbonate into saliva (Fig. 8). Bicarbonate is generated by carbonic anhydrase II (CA2), which is strongly expressed by ameloblasts starting in the transition stage (Toyosawa et al., 1996), as well as by striated duct cells in mature salivary glands (Redman et al., 1979). Using a zinc ion at the active site, carbonic anhydrase combines CO2 generated by aerobic metabolism with H2O to generate HCO3- (bicarbonate) + H+. The bicarbonate ion is exchanged across the ameloblast plasma membrane for a chloride ion (Cl-) by anion exchanger 2 (AE2). Anion exchanger 2 is expressed specifically by maturation-stage ameloblasts and is required for normal enamel maturation (Lyaruu et al., 2008; Bronckers et al., 2009). Since the amount of bicarbonate transported is necessarily high, a chloride channel may be necessary to support HCO3-/Cl- exchange. In salivary glands, this role is filled by the cystic fibrosis transmembrane conductance regulator (CFTR), which co-localizes in the apical membrane of the striated duct cells with AE2 and cGMP-dependent protein kinase II (cGKII), which activates CFTR (Kulaksiz et al., 2002). CFTR null mice display hypomaturation enamel defects (Wright et al., 1996), which allows them to be distinguished from their heterozygous or wild-type littermates at an early age (Grubb and Boucher, 1999). CFTR null mice show defective regulation of pH during the maturation stage (Sui et al., 2003). They have enamel with normal thickness and prism structure, but there is reduced mineral volume (hypomineralization), enamel proteins are retained, and the chalky enamel readily chips away from the incisal edges following eruption into function.
Figure 8.
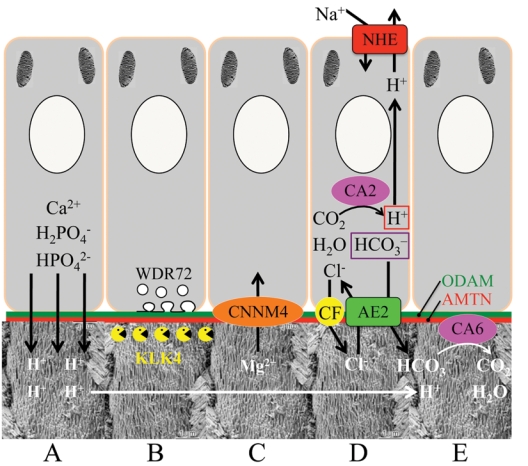
Major activities of maturation stage ameloblasts. (A) Calcium (Ca2+) and phosphate (H2PO4- and HPO42-) ions are transported and add to the width and thickness of existing calcium hydroxyapatite crystals generating hydrogen ions (H+). (B) Enamel proteins are cleaved by kallikrein (KLK4) and reabsorbed into the cells, possibly with the assistance of WDR72. (C) Magnesium ions (Mg2+) are potentially removed from the matrix by CNNM4. (D) Carbonic anhydrase II (CA2) catalyzes the combination of carbon dioxide (CO2) and water (H2O) to form a bicarbonate ion (HCO3-) and a hydrogen ion. The H+ is removed from the cell, possibly by the action of a sodium (Na+) and hydrogen ion exchanger (NHE) on the proximal membrane. The bicarbonate ion is transported into the matrix by exchanging it for a chloride ion (Cl-) by anion exchanger 2 (AE2). The cystic fibrosis transmembrane regulator protein may facilitate this exchange by transporting Cl- out of the cell and into the matrix. (E) Carbonic anhydrase VI (CA6) catalyzes the combination of bicarbonate and a hydrogen ion generated by hydroxyapatite formation to form carbon dioxide and water. ODAM and amelotin (AMTN) are components of the basal lamina along the distal membrane of ameloblasts throughout the maturation stage. No attempt has been made to distinguish between the activities of ruffle-ended and smooth ended ameloblasts. The proximal side of the ameloblast is at the top; the distal side is at the bottom. The enamel rod images are from Nanci (2003).
The hydrogen ions generated by the intracellular production of bicarbonate must exit the proximal side of maturation ameloblasts, where they can be neutralized and carried away by the circulation. (If the hydrogen ions generated by intracellular bicarbonate formation transported out the distal membrane along with the bicarbonate ions, there would be a futile cycling of the bicarbonate and H+ back into CO2 and water.) By analogy to salivary glands, this would be accomplished by a Na+/H+ exchanger (NHE1) (Park et al., 2001), although no work has been performed to localize such an exchanger in ameloblasts. Once in the enamel matrix, bicarbonate is rapidly combined with hydrogen ions generated by mineral formation, catalyzed by carbonic anhydrase VI (CA6), the secreted form of carbonic anhydrase that is expressed by maturation-stage ameloblasts (Fig. 9) (Moffatt et al., 2006a; Smith, 2006). Carbonic anhydrase VI is secreted by salivary glands along with bicarbonate and becomes part of the acquired enamel pellicle of erupted teeth, where it neutralizes acid generated by cariogenic bacteria as a part of the caries prevention mechanism of saliva (Kivela et al., 1999).
Figure 9.
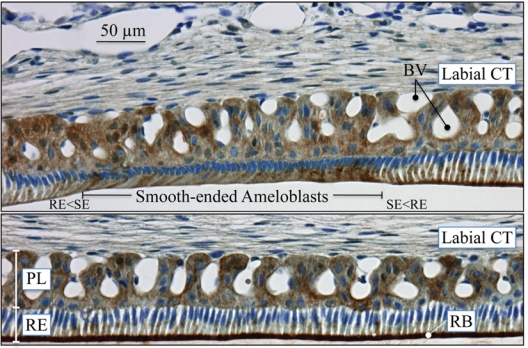
Immunohistochemistry of carbonic anhydrase VI distribution in maturation-stage ameloblasts of the rat incisor. CA6 localizes primarily to membrane invaginations along the distal surface of ruffle-ended ameloblasts (bottom panel). Strong reactions for CA6 are also evident in some papillary layer cells, especially in areas near blood vessels. The maxillary incisors of 100 g male rats were perfused via the vascular system for 20 min with 4% paraformaldehyde + 0.1% glutaraldehyde in 0.8 M sodium cacodylate buffer + 0.05% calcium chloride, pH 7.2. The jaws were decalcified for 3 wks in disodium EDTA, washed, then processed for embedding in paraffin. The sections (~5 µm) were treated for antigen retrieval in 10 mM sodium citrate for 15 min by being microwaved prior to immunolocalization with the anti-CA6 antibody, which was custom-made by Affinity BioReagents (Thermo Fisher Scientific, Inc., Rockford, IL, USA). The CA6 antibody is an affinity-purified chicken anti-rat IgY (egg yolk) antibody raised against the peptide CGGERQSPIDVKRREVHFSS, from near the rat CA6 N-terminus (aa 41–60). The CA6 antibody was incubated at 1:1000 dilution overnight at 4°C. The secondary rabbit anti-chicken antibody was incubated for 30 min on the section and then revealed with an immunoperoxidase kit (Vector Laboratories, Burlingame, CA, USA). The slide was counterstained with toluidine blue. Key: BV, blood vessels; CT, connective tissue; PL, papillary layer; RB, ruffled border; RE, ruffle-ended ameloblasts; SE, smooth-ended ameloblasts; transition zones between ruffled and smooth-ended ameloblasts, RE<SE and SE<RE.
Genetic Diseases Causing Inherited Enamel Defects
The genes associated with isolated enamel defects encode proteins that are specialized for ameloblast activities. FAM83H (Kim et al., 2008) and WDR72 (El-Sayed et al., 2009) are associated with the secretion or re-absorption of enamel proteins. AMELX (Lagerström et al., 1991), ENAM (Rajpar et al., 2001), MMP20 (Kim et al., 2005), and KLK4 (Hart et al., 2004) encode proteins that are secreted into the enamel matrix. Ameloblasts also rely on a toolbox of genes that are necessary for other processes and are associated with enamel defects in syndromes. Junctional epidermolysis bullosa (JEB) is a heterogeneous group of inherited skin disorders associated with trauma-induced blistering that sometimes includes enamel hypoplasia as part of the phenotype. Two components of the basal lamina associated with the inner enamel epithelium that is degraded by pre-ameloblasts are laminin 5 and type 17 collagen (Varki et al., 2006), and defects in the genes encoding components of these proteins (LAMB3 and COL17A1) can cause JEB (McGrath et al., 1996; Buchroithner et al., 2004; Almaani et al., 2009). Defects in CNNM4, a putative magnesium ion transporter, cause cone-rod dystrophy and amelogenesis imperfecta (Parry et al., 2009; Polok et al., 2009), suggesting that removal of magnesium from the enamel matrix is an important function. Identification of the genes involved in inherited enamel defects is shedding light on ameloblast functional components that are critical for dental enamel formation.
Conclusions
Dental enamel formation is a complicated process that cannot, at present, be reproduced in the laboratory. In recent years, there have been great advances in our knowledge of the genes and proteins that are critical for amelogenesis. In Part I we described five growth parameters that determine the shape of the enamel crown once developmental processes have determined the morphology of the latent dentin surface upon which it grows. Future studies seeking to understand tooth morphogenesis might focus on the regulation of these parameters. In Part II, we described our understanding of the mechanism of appositional growth. We did not attempt to review all possible mechanisms, but emphasized the importance of the mineralization front in establishing crystal morphology and organization. We believe that the key to understanding the molecular mechanism of dental enamel formation is to understand what is happening at the mineralization front. In Part III, we discussed how enamel hardens by the maturation of enamel crystals, and presented a model for how maturation-stage ameloblasts neutralize acidity, which is a normal by-product of hydroxyapatite formation. We summarized some of the recent genetic findings that identified unexpected molecular participants in enamel formation, and showed that much must be discovered before the molecular mechanisms of dental enamel formation are understood.
Acknowledgments
USPHS Research Grants DE011301, DE015846, DE018878, DE019622, and DE019775 (NIDCR/NIH) supported this investigation. All authors declare that there are no conflicting interests.
References
- Adeleke-Stainback P, Chen E, Collier P, Yuan ZA, Piddington R, Decker S, et al. (1995). Analysis of the regulatory region of the bovine X-chromosomal amelogenin gene. Connect Tissue Res 32:115-118 [DOI] [PubMed] [Google Scholar]
- Al Kawas S, Warshawsky H. (2008). Ultrastructure and composition of basement membrane separating mature ameloblasts from enamel. Arch Oral Biol 53:310-317 [DOI] [PubMed] [Google Scholar]
- Almaani N, Liu L, Dopping-Hepenstal PJ, Lovell PA, Lai-Cheong JE, Graham RM, et al. (2009). Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol 160:1094-1097 [DOI] [PubMed] [Google Scholar]
- Amir S, Lamont EW, Robinson B, Stewart J. (2004). A circadian rhythm in the expression of PERIOD2 protein reveals a novel SCN-controlled oscillator in the oval nucleus of the bed nucleus of the stria terminalis. J Neurosci 24:781-790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoba T, Moreno EC. (1987). The enamel fluid in the early secretory stage of porcine amelogenesis: chemical composition and saturation with respect to enamel mineral. Calcif Tissue Int 41:86-94 [DOI] [PubMed] [Google Scholar]
- Arsenault AL, Robinson BW. (1989). The dentino-enamel junction: a structural and microanalytical study of early mineralization. Calcif Tissue Int 45:111-121 [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP. (1999). Proteinases in developing dental enamel. Crit Rev Oral Biol Med 10:425-441 [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP, Xue J, Margolis HC, Moreno EC. (1996). Molecular cloning and mRNA tissue distribution of a novel matrix metalloproteinase isolated from porcine enamel organ. Gene 183:123-128 [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Beniash E, Lee DH, Smith CE. (2004). Decreased mineral content in MMP-20 null mouse enamel is prominent during the maturation stage. J Dent Res 83:909-913 [DOI] [PubMed] [Google Scholar]
- Bayes M, Hartung AJ, Ezer S, Pispa J, Thesleff I, Srivastava AK, et al. (1998). The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats. Hum Mol Gen 7:1661-1669 [DOI] [PubMed] [Google Scholar]
- Bei M. (2009a). Molecular genetics of ameloblast cell lineage. J Exp Zool B Mol Dev Evol 312(B):437-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei M. (2009b). Molecular genetics of tooth development. Curr Opin Genet Dev 19:504-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beniash E, Metzler RA, Lam RS, Gilbert PU. (2009). Transient amorphous calcium phosphate in forming enamel. J Struct Biol 166:133-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyeler M, Schild C, Lutz R, Chiquet M, Trueb B. (2010). Identification of a fibronectin interaction site in the extracellular matrix protein ameloblastin. Exp Cell Res 316:1202-1212 [DOI] [PubMed] [Google Scholar]
- Beynon AD, Dean MC, Reid DJ. (1991). On thick and thin enamel in hominoids. Am J Phys Anthropol 86:295-309 [Google Scholar]
- Birch W, Dean C. (2009). Rates of enamel formation in human deciduous teeth. Front Oral Biol 13:116-120 [DOI] [PubMed] [Google Scholar]
- Bromage TG, Lacruz RS, Hogg R, Goldman HM, McFarlin SC, Warshaw J, et al. (2009). Lamellar bone is an incremental tissue reconciling enamel rhythms, body size, and organismal life history. Calcif Tissue Int 84:388-404 [DOI] [PubMed] [Google Scholar]
- Bronckers AL, Goei SW, Dumont E, Lyaruu DM, Woltgens JH, van Heerde WL, et al. (2000). In situ detection of apoptosis in dental and periodontal tissues of the adult mouse using annexin-V-biotin. Histochem Cell Biol 113:293-301 [DOI] [PubMed] [Google Scholar]
- Bronckers AL, Lyaruu DM, Jansen ID, Medina JF, Kellokumpu S, Hoeben KA, et al. (2009). Localization and function of the anion exchanger Ae2 in developing teeth and orofacial bone in rodents. J Exp Zoolog B Mol Dev Evol 312(B):375-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchroithner B, Klausegger A, Ebschner U, Anton-Lamprecht I, Pohla-Gubo G, Lanschuetzer CM, et al. (2004). Analysis of the LAMB3 gene in a junctional epidermolysis bullosa patient reveals exonic splicing and allele-specific nonsense-mediated mRNA decay. Lab Invest 84:1279-1288 [DOI] [PubMed] [Google Scholar]
- Caterina JJ, Skobe Z, Shi J, Ding Y, Simmer JP, Birkedal-Hansen H, et al. (2002). Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 277:49598-49604 [DOI] [PubMed] [Google Scholar]
- Cermakian N, Boivin DB. (2009). The regulation of central and peripheral circadian clocks in humans. Obes Rev 10(Suppl 2):25-36 [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, et al. (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127:1671-1679 [DOI] [PubMed] [Google Scholar]
- Cobourne MT, Sharpe PT. (2003). Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Arch Oral Biol 48:1-14 [DOI] [PubMed] [Google Scholar]
- Cuisinier FJ, Steuer P, Senger B, Voegel JC, Frank RM. (1992). Human amelogenesis. I: High resolution electron microscopy study of ribbon-like crystals. Calcif Tissue Int 51:259-268 [DOI] [PubMed] [Google Scholar]
- Daculsi G, Kerebel B. (1978). High-resolution electron microscope study of human enamel crystallites: size, shape, and growth. J Ultrastruct Res 65:163-172 [DOI] [PubMed] [Google Scholar]
- Daculsi G, Menanteau J, Kerebel LM, Mitre D. (1984). Length and shape of enamel crystals. Calcif Tissue Int 36:550-555 [DOI] [PubMed] [Google Scholar]
- Dean MC. (1989). The developing dentition and tooth structure in hominoids. Folia Primatol (Basel) 53:160-176 [DOI] [PubMed] [Google Scholar]
- Dean MC. (1998). Comparative observations on the spacing of short-period (von Ebner’s) lines in dentine. Arch Oral Biol 43:1009-1021 [DOI] [PubMed] [Google Scholar]
- Dean MC, Reid DJ. (2001). Perikymata spacing and distribution on hominid anterior teeth. Am J Phys Anthropol 116:209-215 [DOI] [PubMed] [Google Scholar]
- Dean MC, Scandrett AE. (1996). The relation between long-period incremental markings in dentine and daily cross-striations in enamel in human teeth. Arch Oral Biol 41:233-241 [DOI] [PubMed] [Google Scholar]
- Diekwisch TG, Berman BJ, Genter S, Slavkin HC. (1995). Initial enamel crystals are not spatially associated with mineralized dentin. Cell Tissue Res 279:149-167 [DOI] [PubMed] [Google Scholar]
- Dohi N, Murakami C, Tanabe T, Yamakoshi Y, Fukae M, Yamamoto Y, et al. (1998). Immunocytochemical and immunochemical study of enamelins, using antibodies against porcine 89-kDa enamelin and its N-terminal synthetic peptide, in porcine tooth germs. Cell Tissue Res 293:313-325 [DOI] [PubMed] [Google Scholar]
- El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, et al. (2009). Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet 85:699-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan D, Lakshminarayanan R, Moradian-Oldak J. (2008). The 32kDa enamelin undergoes conformational transitions upon calcium binding. J Struct Biol 163:109-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham AG, Moradian-Oldak J, Simmer JP, Sarte P, Lau EC, Diekwisch T, et al. (1994). Self-assembly of a recombinant amelogenin protein generates supramolecular structures. J Struct Biol 112:103-109 [DOI] [PubMed] [Google Scholar]
- Fincham AG, Moradian-Oldak J, Simmer JP. (1999). The structural biology of the developing dental enamel matrix. J Struct Biol 126:270-299 [DOI] [PubMed] [Google Scholar]
- FitzGerald CM. (1998). Do enamel microstructures have regular time dependency? Conclusions from the literature and a large-scale study. J Hum Evol 35:371-386 [DOI] [PubMed] [Google Scholar]
- Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. (2005). The molecular clock mediates leptin-regulated bone formation. Cell 122:803-815 [DOI] [PubMed] [Google Scholar]
- Fu L, Patel MS, Karsenty G. (2006). The circadian modulation of leptin-controlled bone formation. Prog Brain Res 153:177-188 [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Akimoto T, Otsu K, Kagiya T, Ishizeki K, Harada H. (2009). Reduction of Egf signaling decides transition from crown to root in the development of mouse molars. J Exp Zool B Mol Dev Evol 312(B):486-494 [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G, et al. (2004). Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol 167:973-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CW. (1999). Regulation of amelogenin gene expression. Crit Rev Eukaryot Gene Expr 9:45-57 [PubMed] [Google Scholar]
- Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen E, Thyagarajan T, et al. (2001). Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 276:31871-31875 [DOI] [PubMed] [Google Scholar]
- Grubb BR, Boucher RC. (1999). Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev 79(1 Suppl):S193-S214 [DOI] [PubMed] [Google Scholar]
- Hanaizumi Y, Maeda T, Takano Y. (1996). Three-dimensional arrangement of enamel prisms and their relation to the formation of Hunter-Schreger bands in dog tooth. Cell Tissue Res 286:103-114 [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al. (2004). Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 41:545-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CC, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. (1997a). Sheathlin: cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel sheath proteins. J Dent Res 76:648-657 [DOI] [PubMed] [Google Scholar]
- Hu CC, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. (1997b). Cloning and characterization of porcine enamelin mRNAs. J Dent Res 76:1720-1729 [DOI] [PubMed] [Google Scholar]
- Hu JC, Yamakoshi Y. (2003). Enamelin and autosomal-dominant amelogenesis imperfecta. Crit Rev Oral Biol Med 14:387-398 [DOI] [PubMed] [Google Scholar]
- Hu JC, Zhang C, Sun X, Yang Y, Cao X, Ryu O, et al. (2000). Characterization of the mouse and human PRSS17 genes, their relationship to other serine proteases, and the expression of PRSS17 in developing mouse incisors. Gene 251:1-8 [DOI] [PubMed] [Google Scholar]
- Hu JC, Hu Y, Smith CE, McKee MD, Wright JT, Yamakoshi Y, et al. (2008). Enamel defects and ameloblast-specific expression in Enam knock-out/lacz knock-in mice. J Biol Chem 283:10858-10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inai T, Kukita T, Ohsaki Y, Nagata K, Kukita A, Kurisu K. (1991). Immunohistochemical demonstration of amelogenin penetration toward the dental pulp in the early stages of ameloblast development in rat molar tooth germs. Anat Rec 229:259-270 [DOI] [PubMed] [Google Scholar]
- Joseph BK, Savage NW, Young WG, Waters MJ. (1994). Insulin-like growth factor-I receptor in the cell biology of the ameloblast: an immunohistochemical study on the rat incisor. Epithelial Cell Biol 3:47-53 [PubMed] [Google Scholar]
- Josephsen K, Fejerskov O. (1977). Ameloblast modulation in the maturation zone of the rat incisor enamel organ. A light and electron microscopic study. J Anat 124(Pt 1):45-70 [PMC free article] [PubMed] [Google Scholar]
- Kallenbach E. (1976). Fine structure of differentiating ameloblasts in the kitten. Am J Anat 145:283-317 [DOI] [PubMed] [Google Scholar]
- Kallenbach E. (1990). Evidence that apatite crystals of rat incisor enamel have hexagonal cross sections. Anat Rec 228:132-136 [DOI] [PubMed] [Google Scholar]
- Katchburian E. (1973). Membrane-bound bodies as initiators of mineralization of dentine. J Anat 116(Pt 2):285-302 [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K, Weiss KM. (2008). SCPP gene evolution and the dental mineralization continuum. J Dent Res 87:520-531 [DOI] [PubMed] [Google Scholar]
- Kere J, Srivastava A, Montonen O, Zonana J, Thomas N, Ferguson B, et al. (1996). X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nature Genet 13:409-416 [DOI] [PubMed] [Google Scholar]
- Kerebel B, Daculsi G, Kerebel LM. (1979). Ultrastructural studies of enamel crystallites. J Dent Res 58(Spec Iss B):844-851 [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al. (2005). MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 42:271-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, Lee MH, et al. (2008). FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet 82:489-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DP, Vitaterna MH, Chang AM, Dove WF, Pinto LH, Turek FW, et al. (1997). The mouse clock mutation behaves as an antimorph and maps within the W19H deletion, distal of Kit. Genetics 146:1049-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivela J, Parkkila S, Parkkila AK, Leinonen J, Rajaniemi H. (1999). Salivary carbonic anhydrase isoenzyme VI. J Physiol 520(Pt 2):315-320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaka T, Sano T, Higashi S. (1996). Structural and calcification patterns of the neonatal line in the enamel of human deciduous teeth. Scanning Microsc 10:737-743 [PubMed] [Google Scholar]
- Kondo S, Tamura Y, Bawden JW, Tanase S. (2001). The immunohistochemical localization of Bax and Bcl-2 and their relation to apoptosis during amelogenesis in developing rat molars. Arch Oral Biol 46:557-568 [DOI] [PubMed] [Google Scholar]
- Kulaksiz H, Rehberg E, Stremmel W, Cetin Y. (2002). Guanylin and functional coupling proteins in the human salivary glands and gland tumors: expression, cellular localization, and target membrane domains. Am J Pathol 161:655-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak SY, Wiedemann-Bidlack FB, Beniash E, Yamakoshi Y, Simmer JP, Litman A, et al. (2009). Role of 20-kDa amelogenin (P148) phosphorylation in calcium phosphate formation in vitro. J Biol Chem 284:18972-18979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Bromage TG. (2006). Appositional enamel growth in molars of South African fossil hominids. J Anat 209:13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Nanci A, Kurtz I, Wright JT, Paine ML. (2010). Regulation of pH during amelogenesis. Calcif Tissue Int 86:91-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, et al. (1991). A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 10:971-975 [DOI] [PubMed] [Google Scholar]
- Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. (2004). Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 74:1043-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis WJ, Burke GY, Neuringer JR, Paine MC, Nanci A, Bai P, et al. (1988). Earliest enamel deposits of the rat incisor examined by electron microscopy, electron diffraction, and electron probe microanalysis. Anat Rec 220:233-238 [DOI] [PubMed] [Google Scholar]
- Lu Y, Papagerakis P, Yamakoshi Y, Hu JC, Bartlett JD, Simmer JP. (2008). Functions of KLK4 and MMP-20 in dental enamel formation. Biol Chem 389:695-700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan X, Ito Y, Diekwisch TG. (2006). Evolution and development of Hertwig’s epithelial root sheath. Dev Dyn 235:1167-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden AG. (1988). Spatial organization of the epithelium and the role of neural crest cells in the initiation of the mammalian tooth germ. Development 103(Suppl):155-169 [DOI] [PubMed] [Google Scholar]
- Lyaruu DM, Bronckers AL, Mulder L, Mardones P, Medina JF, Kellokumpu S, et al. (2008). The anion exchanger Ae2 is required for enamel maturation in mouse teeth. Matrix Biol 27:119-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuyama T, Miyajima K, Ohshima H, Osawa M, Yokoi N, Oikawa T, et al. (2005). A novel autosomal-recessive mutation, whitish chalk-like teeth, resembling amelogenesis imperfecta, maps to rat chromosome 14 corresponding to human 4q21. Eur J Oral Sci 113:451-456 [DOI] [PubMed] [Google Scholar]
- Matalova E, Antonarakis GS, Sharpe PT, Tucker AS. (2005). Cell lineage of primary and secondary enamel knots. Dev Dyn 233:754-759 [DOI] [PubMed] [Google Scholar]
- McGrath JA, Gatalica B, Li K, Dunnill MG, McMillan JR, Christiano AM, et al. (1996). Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol 148:1787-1796 [PMC free article] [PubMed] [Google Scholar]
- Miake Y, Shimoda S, Fukae M, Aoba T. (1993). Epitaxial overgrowth of apatite crystals on the thin-ribbon precursor at early stages of porcine enamel mineralization. Calcif Tissue Int 53:249-256 [DOI] [PubMed] [Google Scholar]
- Moffatt P, Smith CE, Sooknanan R, St-Arnaud R, Nanci A. (2006a). Identification of secreted and membrane proteins in the rat incisor enamel organ using a signal-trap screening approach. Eur J Oral Sci 114(Suppl 1):139-146 [DOI] [PubMed] [Google Scholar]
- Moffatt P, Smith CE, St-Arnaud R, Simmons D, Wright JT, Nanci A. (2006b). Cloning of rat amelotin and localization of the protein to the basal lamina of maturation stage ameloblasts and junctional epithelium. Biochem J 399:37-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt P, Smith CE, St-Arnaud R, Nanci A. (2008). Characterization of Apin, a secreted protein highly expressed in tooth-associated epithelia. J Cell Biochem 103:941-956 [DOI] [PubMed] [Google Scholar]
- Murakami C, Dohi N, Fukae M, Tanabe T, Yamakoshi Y, Wakida K, et al. (1997). Immunochemical and immunohistochemical study of 27 and 29 kDa calcium binding proteins and related proteins in the porcine tooth germ. Histochem Cell Biol 107:485-494 [DOI] [PubMed] [Google Scholar]
- Nanci A. (2003). Enamel: composition, formation, and structure. In: Ten Cate’s oral histology development, structure, and function. Nanci A, editor. St. Louis, MO, USA: Mosby, pp. 145-191 [Google Scholar]
- Nanci A. (2008a). Development of the tooth and its supporting tissues. In: Ten Cate’s oral histology development, structure, and function. 7th ed. Nanci A, editor. St. Louis, MO, USA: Mosby, pp. 79-107 [Google Scholar]
- Nanci A. (2008b). Enamel: composition, formation, and structure. In: Ten Cate’s oral histology development, structure, and function. 7th ed. Nanci A, editor. St. Louis, MO, USA: Mosby, pp. 141-190 [Google Scholar]
- Newman HN, Poole DF. (1974). Observations with scanning and transmission electron microscopy on the structure of human surface enamel. Arch Oral Biol 19:1135-1143 [DOI] [PubMed] [Google Scholar]
- Nieminen P. (2009). Genetic basis of tooth agenesis. J Exp Zool B Mol Dev Evol 312(B):320-342 [DOI] [PubMed] [Google Scholar]
- Nylen MU, Eanes ED, Omnell KA. (1963). Crystal growth in rat enamel. J Cell Biol 18:109-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka M, Saeki S, Igarashi K, Shinoda H. (1998). Circadian rhythms in the incorporation and secretion of 3H-proline by odontoblasts in relation to incremental lines in rat dentin. J Dent Res 77:1889-1895 [DOI] [PubMed] [Google Scholar]
- Ohtsuka-Isoya M, Hayashi H, Shinoda H. (2001). Effect of suprachiasmatic nucleus lesion on circadian dentin increment in rats. Am J Physiol Regul Integr Comp Physiol 280:R1364-R1370 [DOI] [PubMed] [Google Scholar]
- Osawa M, Kenmotsu S, Masuyama T, Taniguchi K, Uchida T, Saito C, et al. (2007). Rat wct mutation prevents differentiation of maturation-stage ameloblasts resulting in hypo-mineralization in incisor teeth. Histochem Cell Biol 128:183-193 [DOI] [PubMed] [Google Scholar]
- Park K, Evans RL, Watson GE, Nehrke K, Richardson L, Bell SM, et al. (2001). Defective fluid secretion and NaCl absorption in the parotid glands of Na+/H+ exchanger-deficient mice. J Biol Chem 276:27042-27050 [DOI] [PubMed] [Google Scholar]
- Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H, et al. (2009). Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet 84:266-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polok B, Escher P, Ambresin A, Chouery E, Bolay S, Meunier I, et al. (2009). Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am J Hum Genet 84:259-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash SK, Gibson CW, Wright JT, Boyd C, Cormier T, Sierra R, et al. (2005). Tooth enamel defects in mice with a deletion at the Arhgap6/AmelX locus. Calcif Tissue Int 77:23-29 [DOI] [PubMed] [Google Scholar]
- Radlanski RJ, Renz H. (2004). A possible interdependency between the wavy path of enamel rods, distances of Retzius lines, and mitotic activity at the cervical loop in human teeth: a hypothesis. Med Hypotheses 62:945-949 [DOI] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet 10:1673-1677 [DOI] [PubMed] [Google Scholar]
- Redman RS, Cohen IM, Greer RO., Jr (1979). Focal odontodysgenesis of the maxillary second premolars in a child. Oral Surg Oral Med Oral Pathol 47:349-353 [DOI] [PubMed] [Google Scholar]
- Reid DJ, Ferrell RJ. (2006). The relationship between number of striae of Retzius and their periodicity in imbricational enamel formation. J Hum Evol 50:195-202 [DOI] [PubMed] [Google Scholar]
- Reith EJ. (1967). The early stage of amelogenesis as observed in molar teeth of young rats. J Ultrastruct Res 17:503-526 [DOI] [PubMed] [Google Scholar]
- Risnes S. (1985). Circumferential continuity of perikymata in human dental enamel investigated by scanning electron microscopy. Scand J Dent Res 93:185-191 [DOI] [PubMed] [Google Scholar]
- Risnes S. (1986). Enamel apposition rate and the prism periodicity in human teeth. Scand J Dent Res 94:394-404 [DOI] [PubMed] [Google Scholar]
- Risnes S. (1998). Growth tracks in dental enamel. J Hum Evol 35:331-350 [DOI] [PubMed] [Google Scholar]
- Ronnholm E. (1962). The amelogenesis of human teeth as revealed by electron mircoscopy I. The fine structure of the ameloblasts. J Ultrastruct Res 6:229-248 [DOI] [PubMed] [Google Scholar]
- Sasaki S, Takagi T, Suzuki M. (1991). Cyclical changes in pH in bovine developing enamel as sequential bands. Arch Oral Biol 36:227-231 [DOI] [PubMed] [Google Scholar]
- Sasaki T. (1990). Cell biology of tooth enamel formation. Functional electron microscopic monographs. Monogr Oral Sci 14:1-199 [PubMed] [Google Scholar]
- Sawada T, Inoue S. (2001). Ultrastructure and composition of basement membranes in the tooth. Int Rev Cytol 207:151-194 [DOI] [PubMed] [Google Scholar]
- Shellis RP. (1998). Utilization of periodic markings in enamel to obtain information on tooth growth. J Hum Evol 35:387-400 [DOI] [PubMed] [Google Scholar]
- Siepka SM, Yoo SH, Park J, Lee C, Takahashi JS. (2007). Genetics and neurobiology of circadian clocks in mammals. Cold Spring Harb Symp Quant Biol 72:251-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer JP, Fincham AG. (1995). Molecular mechanisms of dental enamel formation. Crit Rev Oral Biol Med 6:84-108 [DOI] [PubMed] [Google Scholar]
- Simmer JP, Hu Y, Lertlam R, Yamakoshi Y, Hu JC. (2009). Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. J Biol Chem 284:19110-19121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE. (1998). Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med 9:128-161 [DOI] [PubMed] [Google Scholar]
- Smith CE, Nanci A. (1996). Protein dynamics of amelogenesis. Anat Rec 245:186-207 [DOI] [PubMed] [Google Scholar]
- Smith CE, Issid M, Margolis HC, Moreno EC. (1996). Developmental changes in the pH of enamel fluid and its effects on matrix-resident proteinases. Adv Dent Res 10:159-169 [DOI] [PubMed] [Google Scholar]
- Smith CE, Chong DL, Bartlett JD, Margolis HC. (2005). Mineral acquisition rates in developing enamel on maxillary and mandibular incisors of rats and mice: implications to extracellular acid loading as apatite crystals mature. J Bone Miner Res 20:240-249 [DOI] [PubMed] [Google Scholar]
- Smith TM. (2006). Experimental determination of the periodicity of incremental features in enamel. J Anat 208:99-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead ML, Zeichner-David M, Chandra T, Robson KJ, Woo SL, Slavkin HC. (1983). Construction and identification of mouse amelogenin cDNA clones. Proc Natl Acad Sci USA 80:7254-7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead ML, Lau EC, Zeichner-David M, Fincham AG, Woo SL, Slavkin HC. (1985). DNA sequence for cloned cDNA for murine amelogenin reveal the amino acid sequence for enamel-specific protein. Biochem Biophys Res Commun 129:812-818 [DOI] [PubMed] [Google Scholar]
- Sonoda A, Iwamoto T, Nakamura T, Fukumoto E, Yoshizaki K, Yamada A, et al. (2009). Critical role of heparin binding domains of ameloblastin for dental epithelium cell adhesion and ameloblastoma proliferation. J Biol Chem 284:27176-27184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton DW, Das P, Goldenberg M, D’Souza RN, Patel PI. (2000). Mutation of PAX9 is associated with oligodontia. Nat Genet 24:18-19 [DOI] [PubMed] [Google Scholar]
- Sui W, Boyd C, Wright JT. (2003). Altered pH regulation during enamel development in the cystic fibrosis mouse incisor. J Dent Res 82:388-392 [DOI] [PubMed] [Google Scholar]
- Sun Z, Fan D, Fan Y, Du C, Moradian-Oldak J. (2008). Enamel proteases reduce amelogenin-apatite binding. J Dent Res 87:1133-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termine JD, Belcourt AB, Christner PJ, Conn KM, Nylen MU. (1980). Properties of dissociatively extracted fetal tooth matrix proteins. I. Principal molecular species in developing bovine enamel. J Biol Chem 255:9760-9768 [PubMed] [Google Scholar]
- Thesleff I, Jernvall J. (1997). The enamel knot: a putative signaling center regulating tooth development. Cold Spring Harb Symp Quant Biol 62:257-267 [PubMed] [Google Scholar]
- Thesleff I, Sharpe P. (1997). Signaling networks regulating dental development. Mech Dev 67:111-123 [DOI] [PubMed] [Google Scholar]
- Thesleff I, Keranen S, Jernvall J. (2001). Enamel knots as signaling centers linking tooth morphogenesis and odontoblast differentiation. Adv Dent Res 15:14-18 [DOI] [PubMed] [Google Scholar]
- Toyosawa S, Ogawa Y, Inagaki T, Ijuhin N. (1996). Immunohistochemical localization of carbonic anhydrase isozyme II in rat incisor epithelial cells at various stages of amelogenesis. Cell Tissue Res 285:217-225 [DOI] [PubMed] [Google Scholar]
- Tucker A, Sharpe P. (2004). The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet 5:499-508 [DOI] [PubMed] [Google Scholar]
- Tummers M, Thesleff I. (2009). The importance of signal pathway modulation in all aspects of tooth development. J Exp Zool B Mol Dev Evol 312(B):309-319 [DOI] [PubMed] [Google Scholar]
- Uchida T, Tanabe T, Fukae M, Shimizu M. (1991a). Immunocytochemical and immunochemical detection of a 32 kDa nonamelogenin and related proteins in porcine tooth germs. Arch Histol Cytol 54:527-538 [DOI] [PubMed] [Google Scholar]
- Uchida T, Tanabe T, Fukae M, Shimizu M, Yamada M, Miake K, et al. (1991b). Immunochemical and immunohistochemical studies, using antisera against porcine 25 kDa amelogenin, 89 kDa enamelin and the 13-17 kDa nonamelogenins, on immature enamel of the pig and rat. Histochemistry 96:129-138 [DOI] [PubMed] [Google Scholar]
- Varki R, Sadowski S, Pfendner E, Uitto J. (2006). Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet 43:641-652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. (1996). A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13:417-421 [DOI] [PubMed] [Google Scholar]
- Wang XP, O’Connell DJ, Lund JJ, Saadi I, Kuraguchi M, Turbe-Doan A, et al. (2009). Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development 136:1939-1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshawsky H, Smith CE. (1974). Morphological classification of rat incisor ameloblasts. Anat Rec 179:423-446 [DOI] [PubMed] [Google Scholar]
- Wazen RM, Moffatt P, Zalzal SF, Yamada Y, Nanci A. (2009). A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol 28:292-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MP, Voegel JC, Frank RM. (1981). Enamel crystallite growth: width and thickness study related to the possible presence of octocalcium phosphate during amelogenesis. J Ultrastruct Res 76:286-292 [DOI] [PubMed] [Google Scholar]
- Wright JT, Hall KI, Grubb BR. (1996). Enamel mineral composition of normal and cystic fibrosis transgenic mice. Adv Dent Res 10:270-274 [DOI] [PubMed] [Google Scholar]
- Xu L, Matsumoto A, Sasaki A, Harada H, Taniguchi A. (2009). Identification of a suppressor element in the amelogenin promoter. J Dent Res 89:246-251 [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhou YL, Luo W, Zhu QS, Levy D, MacDougald OA, et al. (2006). NF-Y and CCAAT/enhancer-binding protein alpha synergistically activate the mouse amelogenin gene. J Biol Chem 281:16090-16098 [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhou YL, Erickson RL, Macdougald OA, Snead ML. (2007a). Physical dissection of the CCAAT/enhancer-binding protein alpha in regulating the mouse amelogenin gene. Biochem Biophys Res Commun 354:56-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhou YL, Gonzalez FJ, Snead ML. (2007b). CCAAT/enhancer-binding protein delta (C/EBPdelta) maintains amelogenin expression in the absence of C/EBPalpha in vivo. J Biol Chem 282:29882-29889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshawsky H, Nanci A. (1982). Stereo electron microscopy of enamel crystallites. J Dent Res 61:1504-1514 [PubMed] [Google Scholar]
- Zhou YL, Snead ML. (2000). Identification of CCAAT/enhancer-binding protein alpha as a transactivator of the mouse amelogenin gene. J Biol Chem 275:12273-12280 [DOI] [PubMed] [Google Scholar]