

Abstract
BACKGROUND AND PURPOSE
γ-Secretase modulators represent a promising therapeutic approach for Alzheimer's disease (AD) because they selectively decrease amyloid β 42 (Aβ42), a particularly neurotoxic Aβ species that accumulates in plaques in the brains of patients with AD. In the present study, we describe the in vitro and in vivo pharmacological properties of a potent novel γ-secretase modulator, 2-(S)-(3,5-bis(4-(trifluoromethyl)phenyl)phenyl)-4-methylpentanoic acid (JNJ-40418677).
EXPERIMENTAL APPROACH
The potency and selectivity of JNJ-40418677 for Aβ reduction was investigated in human neuroblastoma cells, rat primary neurones and after treatment with single oral doses in non-transgenic mouse brains. To evaluate the effect of JNJ-40418677 on plaque formation, Tg2576 mice were treated from 6 until 13 months of age via the diet.
KEY RESULTS
JNJ-40418677 selectively reduced Aβ42 secretion in human neuroblastoma cells and rat primary neurones, but it did not inhibit Notch processing or formation of other amyloid precursor protein cleavage products. Oral treatment of non-transgenic mice with JNJ-40418677 resulted in an excellent brain penetration of the compound and a dose- and time-dependent decrease of brain Aβ42 levels. Chronic treatment of Tg2576 mice with JNJ-40418677 reduced brain Aβ levels, the area occupied by plaques and plaque number in a dose-dependent manner compared with transgenic vehicle-treated mice.
CONCLUSIONS AND IMPLICATIONS
JNJ-40418677 selectively decreased Aβ42 production, showed an excellent brain penetration after oral administration in mice and lowered brain Aβ burden in Tg2576 mice after chronic treatment. JNJ-40418677 therefore warrants further investigation as a potentially effective disease-modifying therapy for AD.
Keywords: Alzheimer's disease, amyloid β, γ-secretase modulator, plaque
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia in the elderly population. One of the major pathological hallmarks of AD is the extracellular deposition of amyloid β (Aβ) peptides in the form of plaques in the brain of patients. Aβ is formed by sequential proteolysis of amyloid precursor protein (APP) by β-secretase and γ-secretase (Selkoe, 2004). APP is cleaved by α-secretase (non-amyloidogenic pathway) or β-secretase (amyloidogenic pathway) within the extracellular domain resulting in the release of soluble APP fragments and the generation of membrane-bound carboxy-terminal fragments (CTF-α and CTF-β respectively). CTF-α and CTF-β can serve as a substrate for intramembrane cleavage by γ-secretase, producing an amino-terminally truncated Aβ fragment and full-length Aβ, respectively, and the APP intracellular domain (AICD).
Proteolysis of APP by γ-secretase is heterogeneous, leading to the formation of Aβ peptides of different length. The most abundant of these peptides is Aβ40. One of the longer forms, Aβ42, is particularly neurotoxic, has a high potential to aggregate and is the first and predominant species accumulating in plaques in AD brain (Iwatsubo et al., 1994).
Mutations in APP and presenilin (PS1 and PS2) genes that cause rare familial forms of AD typically result in a relative increase in the production of Aβ42. In transgenic mice, familial AD mutations markedly accelerate Aβ pathology leading to cognitive deficits, while treatment with Aβ-lowering agents can result in improvements in cognitive performance. These and other data from genetic, biochemical, pathological and pharmacological studies support the hypothesis that increased brain levels of Aβ42 caused by overproduction and/or failing clearance of this peptide are the initiating factor for the pathological cascade ultimately leading to dementia in AD (Hardy and Selkoe, 2002).
γ-Secretase is a particularly interesting target in the search for a disease-modifying therapy for AD because of its crucial function in Aβ production and the key role of Aβ in AD pathogenesis. γ-Secretase is a protein complex with several components, including the presenilin proteins that form its catalytic core. Besides APP, more than 30 other type I transmembrane proteins can serve as a substrate for γ-secretase and the pharmacologically most relevant alternative substrate is the Notch receptor (De Strooper, 2010). Signalling from this receptor plays a role in a variety of cell differentiation events from embryogenesis until adulthood. γ-Secretase mediates the final proteolytic cleavage of Notch to release the transcription modifier Notch intracellular domain (NICD).
While highly efficient in blocking Aβ production, γ-secretase inhibitors (GSIs) also inhibit Notch processing and downstream signalling leading to toxic side effects in the gastrointestinal and immune system (Hadland et al., 2001; Searfoss et al., 2003; Milano et al., 2004; Wong et al., 2004). An approach avoiding these side effects might be found in the modulation rather than the complete inhibition of γ-secretase activity. The phenomenon of γ-secretase modulation was first discovered for certain non-steroidal anti-inflammatory drugs (NSAIDs). A subset of NSAIDs, including flurbiprofen and its enantiomers, was shown to selectively lower Aβ42 levels in cell-based assays and transgenic mouse models independent of their cyclooxygenase (COX) inhibitory activity (Weggen et al., 2001; Morihara et al., 2002; Eriksen et al., 2003). These NSAIDs act as γ-secretase modulators (GSMs), that is, they modulate the preferred site of γ-secretase cleavage leading to a selective decrease in the production of Aβ42, the putative toxic Aβ species, and simultaneously they increase the production of shorter, more soluble and less toxic Aβ peptides like Aβ38 (Weggen et al., 2001; Kukar et al., 2005). Unlike classical inhibitors of γ-secretase, modulators selectively inhibit the amyloidogenic function of γ-secretase without interfering with the cleavage of other substrates like Notch (Weggen et al., 2001; 2003a;) and they do not cause an accumulation of APP-CTF (Weggen et al., 2001; Gasparini et al., 2004) or interfere with the production of AICD (Weggen et al., 2003a). GSMs might thus represent a safer approach towards an effective therapy for AD.
In general, NSAIDs have a rather low potency towards Aβ42 reduction and show relatively low brain penetration (Eriksen et al., 2003; Stock et al., 2006). Considerable efforts in recent years have been aimed at developing novel compounds with γ-secretase modulatory activity, both NSAID- and non-NSAID-derived, with improved pharmacodynamic and pharmacokinetic characteristics (Peretto and La Porta, 2008).
We describe here the characterization of a novel, NSAID-derived, orally active GSM, 2-(S)-(3,5-bis(4-(trifluoromethyl)phenyl)phenyl)-4-methylpentanoic acid (JNJ-40418677), with a strong potency towards Aβ42 inhibition and excellent brain penetration in mice. We show that JNJ-40418677 selectively decreased Aβ42 production in vitro and in vivo and that chronic JNJ-40418677 treatment reduced amyloid burden in transgenic APP mice. This compound therefore warrants further in vitro and in vivo investigation to explore its potential as a safe and effective disease-modifying treatment for AD.
Methods
Compound synthesis
2-(S)-(3,5-bis(4-(trifluoromethyl)phenyl)phenyl)-4-methylpentanoic acid (JNJ-40418677) (Figure 1) was synthesized as described previously (Ho, 2009).
Figure 1.
Chemical structure of JNJ-40418677.
Cellular Aβ assays
To evaluate the effect of JNJ-40418677 on Aβ secretion in vitro, human neuroblastoma SKNBE-2 cells transfected with wild-type (WT) APP were treated with JNJ-40418677 (0.2 nM–300 µM) for 16 h and rat primary cortical neurones (5 days in vitro) harvested from embryonic day 18 embryos were treated with JNJ-40418677 (0.2 nM–300 µM) for 48 h. Human or rodent Aβ42 in conditioned media from APP WT SKNBE-2 cells or rat primary neurones, respectively, was quantified in a sandwich enzyme-linked immunosorbent assay (elisa) utilizing Innotest®β-amyloid(1–42) plates (Innogenetics, Zwijnaarde, Belgium) and biotinylated 3D6 or 4G8 (Covance, Emeryville, CA, USA) followed by streptavidin-horseradish peroxidase (HRPO) (Jackson Immunoresearch Laboratories, West Grove, PA, USA) incubation for detection. Human or rodent Aβtotal in conditioned media from APP WT SKNBE-2 cells or rat primary neurones, respectively, was measured in a sandwich elisa utilizing species-specific monoclonal antibodies JRF/AbN/25 or JRF/rAb/2 (Mercken et al., 2000), respectively, as capture antibodies and biotinylated 4G8 followed by streptavidin-HRPO incubation for detection. More details on elisa procedures can be found in the section on brain Aβ measurements.
Cellular toxicity
Cellular toxicity was measured by assaying the cleavage of the tetrazolium salt WST-1 (Roche, Mannheim, Germany) by mitochondrial dehydrogenases in viable cells according to manufacturer's recommendations.
Cell-free γ-secretase modulation assay
A cell-free γ-secretase assay was performed to investigate whether JNJ-40418677 modulates γ-secretase activity directly. Recombinant substrate (hC99-Flag, 1 µM) was incubated with an enriched γ-secretase preparation (Winkler et al., 2009) from HeLa cells and 10 µM JNJ-40418677 or 1 µM N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT; Calbiochem, San Diego, CA, USA) at 37°C for 18 h. Reaction products were separated in a 12% Tris-Bicine-Urea gel and transferred to an Immobilon P membrane (Millipore, Billerica, MA, USA). The blot was probed with antibody 6E10 (Signet, Dedham, MA, USA) followed by IRDye 800 anti-mouse secondary antibody (LI-COR Biosciences, Lincoln, NE, USA) and analysed with the Odyssey infrared imaging system (LI-COR Biosciences).
In vitro APP processing assays
To evaluate the effect of JNJ-40418677 treatment on the formation of other APP cleavage products besides Aβ, APP-CTF and AICD formation was investigated in vitro. For analysis of APP-CTF, APP WT SKNBE-2 cells were treated with JNJ-40418677 (6 nM–20 µM) or the GSI DAPT (Calbiochem; 0.3 nM–3 µM), which is known to affect Notch processing (Dovey et al., 2001; Fraering et al., 2005), for 16 h. Cell lysates were separated in a NuPAGE® Novex 4–12% BisTris gel (Invitrogen, Carlsbad, CA, USA) and transferred to an Immobilon P membrane (Millipore). The blot was probed with rabbit anti-APP carboxy-terminus antibody (Sigma, St. Louis, MO, USA) followed by IRDye 800 anti-rabbit secondary antibody (LI-COR Biosciences) and analysed with the Odyssey infrared imaging system (LI-COR Biosciences). For analysis of AICD, recombinant substrate hC99-Flag (1 µM) was incubated with an enriched γ-secretase preparation (Winkler et al., 2009) from HEK293 cells and 10–100 µM JNJ-40418677 or 10 µM DAPT at 37°C for 18 h. Reaction products were separated in a NuPAGE® Novex 4–12% BisTris gel (Invitrogen), transferred to an Immobilon P membrane (Millipore) and fixed to the membrane by boiling in phosphate buffered saline (PBS) for 3 min. The blot was probed with polyclonal anti-APP carboxy-terminus antibody (AB5352, Chemicon, Temecula, CA, USA) followed by IRDye 800 anti-rabbit secondary antibody (LI-COR Biosciences) and analysed with the Odyssey infrared imaging system (LI-COR Biosciences).
Notch processing assays
Cell-based and cell-free assays were performed to investigate the possible effects of JNJ-40418677 treatment on the Notch processing activity of the γ-secretase complex. The Notch cell-based assay utilized co-culture of Chinese hamster ovary (CHO) cells with stable expression of mouse Notch2 (N2-CHO) and cells with stable expression of Notch ligand mouse Delta (DL-CHO). N2-CHO cells were transiently transfected with plasmids pCMV-Rluc and pTP1-Luc to express firefly and Renilla luciferase, respectively (Dual Glo Luciferase Assay; Promega, Fitchburg, WI, USA). DL-CHO cells were co-cultured with transfected N2-CHO cells and treated with JNJ-40418677 (5 nM–50 µM) or DAPT (Calbiochem; 0.5 nM–5 µM) for 16 h in the presence of 200 µg·mL−1 zeocine (Invitrogen). Subsequently, cells were lysed in Passive Lysis Buffer (Promega) and luciferase signals were obtained according to manufacturer's recommendations and read using the Envision 2101 Multilabel Reader (Perkin Elmer, Waltham, MA, USA). For the Notch cell-free assay, recombinant substrate mN99-Flag (0.794 µM) was incubated with an enriched γ-secretase preparation (Winkler et al., 2009) from HeLa cells and 0.1–100 µM JNJ-40418677 or 1 µM DAPT at 37°C for 18 h. Reaction products were separated in a NuPAGE® Novex 12% BisTris gel, transferred to an Immobilon P membrane (Millipore) and fixed to the membrane by boiling in PBS for 3 min. The blot was probed with monoclonal anti-Flag antibody (Sigma) followed by IRDye 800 anti-mouse secondary antibody (LI-COR Biosciences) and analysed with the Odyssey infrared imaging system (LI-COR Biosciences).
COX activity assay
COX-1/2 activity of JNJ-40418677 (0.7–60 µM) was assessed utilizing a colorimetric ovine COX inhibitor screening assay (Cayman, Tallinn, Estonia) according to the manufacturer's recommendations. Peroxidase activity of COX was assayed by measuring oxidized TMPD levels at 600 nm utilizing Envision 2101 Multilabel Reader (Perkin Elmer).
Animals and treatments
All animals were housed in an animal facility that is fully compliant with the European policy on the use of Laboratory Animals. Experimental protocols were approved by the Institutional Review Committee of Janssen Pharmaceutica (Beerse, Belgium) and meet the European and Belgian guidelines on animal experimentation. The effect of single oral doses of JNJ-40418677 on the in vivo production of Aβ in brain was examined in male non-transgenic CF-1 or CD-1 mice (Charles River, Sulzfeld, Germany) after treatment by gastric intubation. The duration of action of JNJ-40418677 was examined in a time-course experiment: six animals per time point (15 min–24 h after administration) were treated with 30 mg·kg−1 JNJ-40418677. For dose–response experiments, six animals per dose group (10, 30, 100 or 300 mg·kg−1 JNJ-40418677) were treated and killed 4 h after treatment. Animals were deprived of food overnight and during the experiment, water was available ad libitum. The effect of chronic JNJ-40418677 treatment on brain Aβ levels and plaque formation was investigated in hemizygous female Tg2576 mice (Hsiao et al., 1996). We chose to evaluate the activity of JNJ-40418677 in Tg2576 mice that express the Swedish double mutation (APP-Swe; K670N/M671L) that is located near the APP β-secretase cleavage site because we wanted to avoid the use of a transgenic mouse model with APP mutations around the γ-secretase cleavage site and the presence of PS mutations can modulate the Aβ42 response to GSM treatment (Weggen et al., 2003b; Czirr et al., 2007; Page et al., 2008). The APP-Swe mutation was shown to respond to GSM treatment in a similar way as APP WT (Weggen et al., 2003b). Tg2576 mice can be used as an animal model of brain amyloidosis and plaque deposition starts around the age of 9–10 months (Hsiao et al., 1996; Kawarabayashi et al., 2001; Terai et al., 2001). Starting at the age of 26–28 weeks (6 months), mice were treated via the diet with JNJ-40418677 for a period of 31 weeks (7 months). Four groups of 17 females each received an estimated daily dose of 0 (vehicle), 20, 60 or 120 mg·kg−1 JNJ-40418677. Food consumption and body weight were registered on a weekly basis and showed that the mean actual dose over the whole study was similar to the target dose (21, 61 and 123 mg·kg−1·day−1 respectively).
Tissue collection
Non-transgenic mice were killed by decapitation and blood was collected on EDTA by exsanguination. Blood samples were placed immediately on ice and plasma was obtained following centrifugation at 4°C for 10 min at ∼1900 ×g. Samples were stored at −18°C prior to pharmacokinetic analysis. Brains were excised and olfactory lobe and hindbrain were removed. Brain hemispheres were weighed, immediately frozen on dry ice and stored at −20°C or −80°C prior to pharmacokinetic (left hemisphere) or Aβ (right hemisphere) analysis, respectively. At the end of the chronic study, Tg2576 mice (age 57–59 weeks) were anaesthetized with an isoflurane/oxygen mixture and killed by exsanguination. Blood was collected on EDTA and plasma was obtained as described above. Brains were excised and the cerebellum and the cranial and caudal parts of the right hemisphere were frozen immediately on dry ice and stored at −80°C prior to APP/APP-CTF, Aβ and pharmacokinetic analysis respectively. The left brain hemisphere was fixed for immunohistochemistry in formalin-based FADE-4 fixative (HistoGeneX, Edegem, Belgium) at room temperature for a maximum of 72 h.
Pharmacokinetics
Brain samples were homogenized in demineralized water at 4°C (1/9 w·v−1 or +3 mL if tissue weight < 0.33 g) under dimmed light conditions. Unchanged parent compound was quantified in individual plasma and brain samples with a qualified liquid chromatography-mass spectrometry (MS)/MS method. Samples were subjected to a selective clean-up, followed by HPLC-MS/MS. HPLC separation was done using non-chiral reversed phase liquid chromatography. Subsequent MS/MS analysis was performed using triple quadrupole mass spectrometry in the Multiple Reaction Monitoring mode, optimized for JNJ-40418677. Samples were quantified against calibration curves prepared in the same matrix as the study samples. For each analytical batch, independent quality control samples prepared in the same matrix as the samples were analysed together with the study samples and calibration curve. All analytical batches were accepted based on calibration curve and quality control acceptance criteria in line with the current Food and Drug Administration guidelines.
Brain Aβ measurements
Non-transgenic mouse brains were thawed on ice in 8 × (w·v−1) 0.4% diethylamine in 50 mM NaCl buffer with a protease inhibitor cocktail (Complete® EDTA-free, Roche). Homogenization was done utilizing MP BIO FastPrep-24 with Lysing matrix D 1.4 mm ceramic beads (MP Biomedicals, Solon, OH, USA). Tubes were spun shortly at 4°C to pellet beads and cell debris and supernatant was spun at ∼220 000×g for 50 min at 4°C. Supernatant was collected and neutralized by addition of 1:10 volume 0.5 M Tris-HCl pH 6.8. Rodent Aβ38 and Aβ42 in brain extracts was quantified in a sandwich elisa with capture antibodies J&JPRDAβ38/5 and JRF/cAb42/26 that specifically recognize Aβ ending at amino acid 38 and 42, respectively, and detection with rodent-specific HRPO-labelled JRF/rAb/2 antibody (Mercken et al., 2000). Rodent Aβtotal was measured in a sandwich elisa with capture antibody JRF/rAb/2 and detection with biotinylated 4G8 and streptavidin-HRPO. Aβ from brains of Tg2576 mice was extracted using a two-step protocol, as described previously (Pype et al., 2003). Briefly, brain tissue was thawed in 1.2 mL ice-cold buffer A [50 mM Tris pH 8.0, 150 mM NaCl and a protease inhibitor cocktail (Complete® EDTA-free, Roche)] and homogenized by sonication on ice. Homogenates were centrifuged for 1 h at 128 000×g at 4°C. Supernatant was collected (‘soluble’ Aβ fraction) and the pellet was resuspended in 0.48 mL guanidine hydrochloride (GuHCl) buffer (50 mM Tris pH 8.0, 6 M GuHCl) and sonicated for 10 s. After 15 min incubation on ice, sonication was repeated for 5 s and the homogenate was diluted sixfold with ice-cold buffer A. Homogenates were centrifuged for 1 h at 128 000×g at 4°C and supernatant was collected (‘deposited’ Aβ fraction). Human Aβ1–38, Aβ1–40 and Aβ1–42 were quantified in a sandwich elisa with capture antibodies J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26, respectively, and detection with human-specific HRPO-labelled JRF/AbN/25 antibody (Mercken et al., 2000). Human Aβtotal was measured with capture antibody JRF/AbN/25 and detection with biotinylated 4G8 and streptavidin-HRPO.
Human (Anaspec, San Jose, CA, USA) and rodent (Bachem, Bubendorf, Switzerland) Aβ1–38, Aβ1–40 and Aβ1–42 standards were dissolved in DMSO at 0.1 mg·mL−1 and stored at −80°C. For use in elisa, standards were diluted in casein buffer (0.1% casein in PBS) down to 1 pg·mL−1. Ninety-six-well plates (half-area black plates; Costar, Bethesda, MD, USA) were coated with 1.5 µg·mL−1 capture antibodies in coating buffer (10 mM NaCl, 10 mM Tris-HCl, 10 mM NaN3, pH 8.6). After overnight coating at 4°C, plates were washed and blocked with casein buffer for 1–4 h at room temperature. For Tg2576 brain homogenates, soluble and deposited Aβ fractions were diluted 10-fold and 50- to 1600-fold, respectively, in casein buffer. The prediluted samples and standards were incubated overnight at 4°C together with HRPO-coupled detection antibodies for Aβ38, Aβ40 and Aβ42 assays or together with biotinylated 4G8 (Covance) for Aβtotal assays. After overnight incubation, plates were washed and Aβ38, Aβ40 and Aβ42 assays were developed with Quantablu substrate (Pierce, Rockford, IL, USA) according to the manufacturer's recommendations. For Aβtotal assays, incubation with streptavidin-HRPO (Jackson Immunoresearch Laboratories) for 1 h at room temperature was done before substrate addition.
APP and APP-CTF Western blot
Brain tissue was thawed in 8 × (w·v−1) ice-cold homogenization buffer [10 mM Tris, 250 mM sucrose, 1 mM EGTA and a protease inhibitor cocktail (Complete® EDTA-free, Roche)]. Homogenization was done utilizing MP BIO FastPrep-24 with Lysing matrix D 1.4 mm ceramic beads (MP Biomedicals). Tubes were spun shortly at 4°C to pellet beads and cell debris and supernatant was spun at ∼100 000×g for 1 h at 4°C. Supernatant was collected (soluble fraction) and the pellet (membrane fraction) was resuspended in homogenization buffer with 1% Triton X-100 and incubated for 1 h at 4°C. After this incubation step, homogenates were centrifuged for 30 min at ∼21 000×g at 4°C and supernatant was collected. Membrane fractions were separated in a NuPAGE® Novex 10% BisTris gel (Invitrogen) and transferred to a nitrocellulose membrane (Invitrogen). The blot was probed with JRF/Abtot/17-HRPO (monoclonal antibody directed against an N-terminal epitope of Aβ), C1/6.1 (monoclonal antibody directed against a C-terminal epitope of APP; kind gift from Dr Mathews, Nathan S. Kline Institute, Orangeburg) and vGlut1 (affinity purified polyclonal antibody, SYnaptic Systems, Goettingen, Germany) antibodies for detection of APP, APP-CTF and vGlut1 respectively. This was followed by incubation with sheep anti-mouse IgG-HRPO secondary antibody (GE Healthcare, Waukesha, WI, USA) for C1/6.1 and with donkey anti-rabbit IgG-HRPO secondary antibody (GE Healthcare) for vGlut1. Signals were generated utilizing SuperSignal West Dura (Thermo Fisher Scientific, Fremont, CA, USA) and analysed with Lumiscan imaging system (Syngene, Cambridge, UK).
Immunohistochemistry and image analysis
Sagittal sections of 6 µm thickness were prepared from FADE-4-fixed, paraffin-embedded Tg2576 mouse brains using a microtome (Leica, Wetzlar, Germany). Staining was performed after randomization of samples (Labvision Autostainer; Thermo Fisher Scientific). Briefly, after deparaffination and 70% formic acid epitope retrieval, sections were blocked for endogenous peroxidase. Subsequently, sections were incubated with primary antibody (4G8, JRF/AbN/25, J&JPRDAβ38/5, JRF/cAb40/28 or JRF/cAb42/26). Secondary anti-mouse antibody conjugated to HRPO was applied (Dako, Glostrup, Denmark) and 3,3′-diaminobenzidine (DAB; Dako) was utilized as a chromogen. Finally, all sections were coverslipped after being counterstained with haematoxylin (Dako).
Quantification of amyloid load was performed at five levels in the brain with a 250 µm interlevel distance. Automatic quantification was performed on slides that were digitalized using the Mirax Virtual Slide scanner (Carl Zeiss, Germany). Using Mirax Viewer software (Carl Zeiss), a snapshot was taken of each brain slide at a 1.5 × magnification. Cerebellum and olfactory lobe were not included in the analysis. Images were analysed using Axiovision Software (Carl Zeiss) with an automated antibody-specific measurement routine, which delineates both tissue area and immunoreactive regions. DAB thresholds were kept constant for analysis of different staining runs and a measure for DAB staining intensity was included to ensure that staining remained constant for a particular antibody between different staining runs. The percentage of immunopositive area versus total brain area and the number of immunopositive areas per mm2 brain tissue were determined as measures of plaque load.
Statistics
Statistical analysis was performed utilizing GraphPad Prism software (GraphPad Software, San Diego, CA, USA). Results are expressed as mean ± SEM. For acute studies in non-transgenic mice, one-way analysis of variance (anova) was used to detect a significant effect of treatment, followed by Dunnett adjusted post hoc comparisons with the vehicle group. For the chronic study, one-way anova on loge transformed data of brain Aβ levels and measures of plaque load was used to detect a significant effect of treatment, followed by Dunnett adjusted post hoc comparisons with the vehicle group. Transformation of the amyloid data was necessary to transform the skewed distribution of the vehicle group data into a normal distribution. The estimated mean treatment difference and limits of the 95% confidence interval (CI) were back transformed as 100 ×[1-exp(x)] resulting in Dunnett adjusted geometric mean reductions versus vehicle together with a 95% CI. P < 0.05 was set as a statistically significant level.
Results
JNJ-40418677 selectively inhibits Aβ42 secretion in vitro
The effect of JNJ-40418677 on Aβ secretion was evaluated in WT APP human neuroblastoma SKNBE-2 cells and in primary rat cortical neuronal cultures. JNJ-40418677 selectively inhibited Aβ42 secretion in cell culture supernatants of WT APP SKNBE-2 cells (mean IC50= 200 nM; n = 10) (Figure 2A) and primary rat cortical neuronal cultures (mean IC50= 185 nM; n = 37) (Figure 2B). In contrast to a classical GSI, JNJ-40418677 did not affect levels of total Aβ to a similar extent (IC50 > 10 µM for both WT APP SKNBE-2 and primary rat cortical neurones) (Figure 2A and B). This resulted in a selectivity difference between Aβ42 and Aβtotal inhibition in the cellular assays of at least 50 times. Reduction of total Aβ levels at high concentrations was typically associated with cellular toxicity as assessed by a WST-1 assay (Figure 2A and B).
Figure 2.
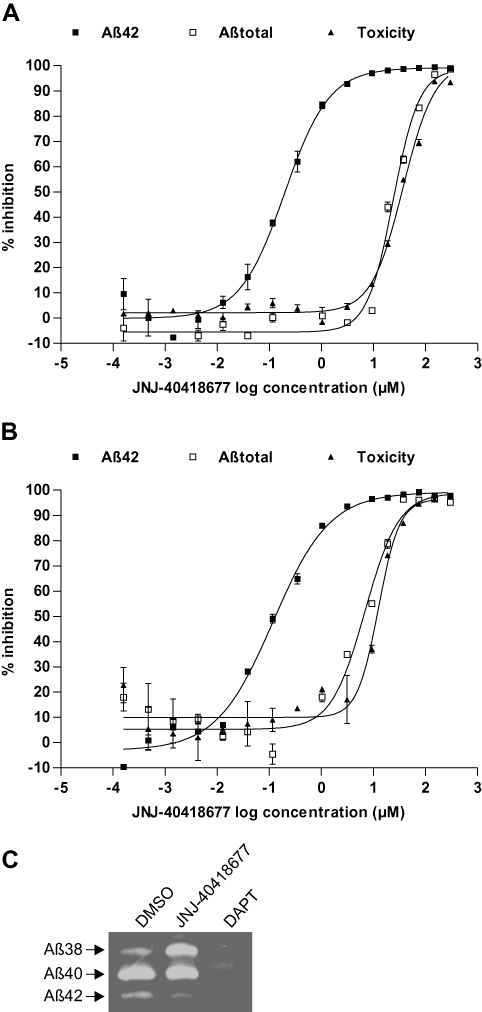
Selective inhibition of Aβ42 secretion after JNJ-40418677 treatment (0.2 nM–300 µM) in cell culture supernatants of APP WT SKNBE-2 cells (A) and primary rat cortical neuronal cultures (B). Levels of secreted Aβ42 and Aβtotal as measured with elisa and cellular toxicity as measured with a WST-1 assay are expressed as a percentage inhibition compared with values in vehicle-treated control cells (mean ± SEM). The mean IC50 value for Aβ42 inhibition is 200 and 185 nM for APP WT SKNBE-2 cells (n = 10) and primary rat cortical neurones (n = 37) respectively. (C) Selective decrease of Aβ42 and increase of Aβ38 after JNJ-40418677 (10 µM) treatment in a cell-free γ-secretase assay. Aβ40 levels remained unchanged. Recombinant CTF-β substrate was incubated with an enriched γ-secretase preparation as described, and Aβ38, Aβ40 and Aβ42 levels were analysed by Western blotting utilizing antibody 6E10. Treatment with 1 µM DAPT reduced the production of all Aβ species analysed.
To further investigate if JNJ-40418677 modulated γ-secretase activity directly, a cell-free γ-secretase assay was performed utilizing an enriched γ-secretase preparation known to contain all the γ-secretase subunits (Winkler et al., 2009). When this preparation and recombinantly expressed CTF-β substrate were incubated in the presence of JNJ-40418677, Aβ42 levels decreased while Aβ38 levels were concomitantly increased with no effect seen on levels of Aβ40 (Figure 2C). In contrast, GSI DAPT reduced the production of all Aβ species measured (Figure 2C).
JNJ-40418677 does not affect formation of other APP cleavage products, Notch processing or COX-1/2 activity
The model of γ-secretase modulation would predict that JNJ-40418677 treatment would not alter the formation of other APP cleavage products besides Aβ. Levels of full-length APP were not affected after either DAPT or JNJ-40418677 treatment of WT APP SKNBE-2 cells (Figure 3A). No changes in the steady state levels of CTF-α and CTF-β were observed after treatment of WT APP SKNBE-2 cells with JNJ-40418677 at concentrations much higher than those required for Aβ42 inhibition (up to 6 µM) (Figure 3A). Treatment with the GSI DAPT on the other hand, resulted in a clear build-up of γ-secretase substrates (CTF) with increasing concentrations, starting around 30 nM (Figure 3A). This corresponds to concentrations of DAPT needed for Aβ42 inhibition, as the IC50 for Aβ reduction is 40 nM for DAPT in WT APP SKNBE-2 cells.
Figure 3.
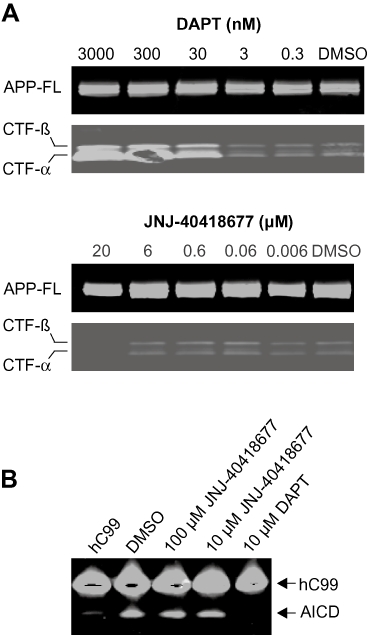
Effect of JNJ-40418677 treatment on the formation of other APP processing products. (A) No changes in the steady state levels of CTF-α and CTF-β were observed after treatment of APP WT SKNBE-2 cells with JNJ-40418677 up to a 6 µM concentration, while DAPT treatment caused a build-up of APP-CTF with increasing concentrations. Levels of full-length (FL) APP were not affected by either DAPT or JNJ-40418677 treatment. Cell lysates of APP WT SKNBE-2 cells treated with JNJ-40418677 or DAPT were analysed by Western blotting utilizing an anti-APP carboxy-terminus antibody. (B) JNJ-40418677 treatment at concentrations of 10 and 100 µM did not inhibit AICD production. A recombinant substrate hC99 was incubated with an enriched γ-secretase preparation (Winkler et al., 2009) from HEK293 cells as described, and reaction products were analysed by Western blotting with anti-APP carboxy-terminus antibody (B5352). 10 µM DAPT completely blocked AICD formation.
Production of the AICD fragment from a recombinant CTF-β substrate in a cell-free γ-secretase assay was also unaffected by JNJ-40418677 treatment at concentrations up to 100 µM, while AICD formation was completely abolished by treatment with 10 µM DAPT (Figure 3B).
To investigate the possible effects of JNJ-40418677 treatment on the Notch processing activity of the γ-secretase complex, cells expressing a Notch construct were treated with JNJ-40418677 at concentrations up to 10 µM. Even at the highest concentration tested, no effects on the activation of transcription by NICD were detected, while DAPT had an IC50 of 40 nM in the same experimental set-up (data not shown). Additionally, a recombinant Notch substrate was synthesized and examined in a cell-free assay using an enriched γ-secretase preparation. In agreement with the results of the cell-based assay, no inhibitory effects on NICD formation were observed after JNJ-40418677 treatment with concentrations up to 10 µM, which is 50-fold higher than the in vitro IC50 for lowering the levels of Aβ42 in cell-based assays, while treatment with 1 µM DAPT completely blocked NICD production (Figure 4).
Figure 4.

Notch processing was not affected by JNJ-40418677 treatment in a cell-free Notch assay. A recombinant substrate mN99-Flag was incubated with an enriched γ-secretase preparation (Winkler et al., 2009) from HeLa cells as described, and NICD levels were analysed by Western blotting utilizing an anti-Flag antibody. With up to 10 µM JNJ-40418677, no inhibition of Notch processing was observed, while NICD production was completely blocked by 1 µM DAPT.
By utilizing a COX-1/2 activity assay, JNJ-40418677 was shown to be devoid of any COX-1/2 inhibitory activity up to and including a concentration of 60 µM (data not shown).
Treatment with single oral doses of JNJ-40418677 lowers Aβ42 and increases Aβ38 levels in non-transgenic mouse brain
The effect of single oral doses of JNJ-40418677 on the in vivo production of Aβ was examined in non-transgenic mice. Aβ levels in brain extracts were measured with sandwich elisas that selectively detect rodent Aβ38, Aβ42 or total Aβ (Figure 5).
Figure 5.
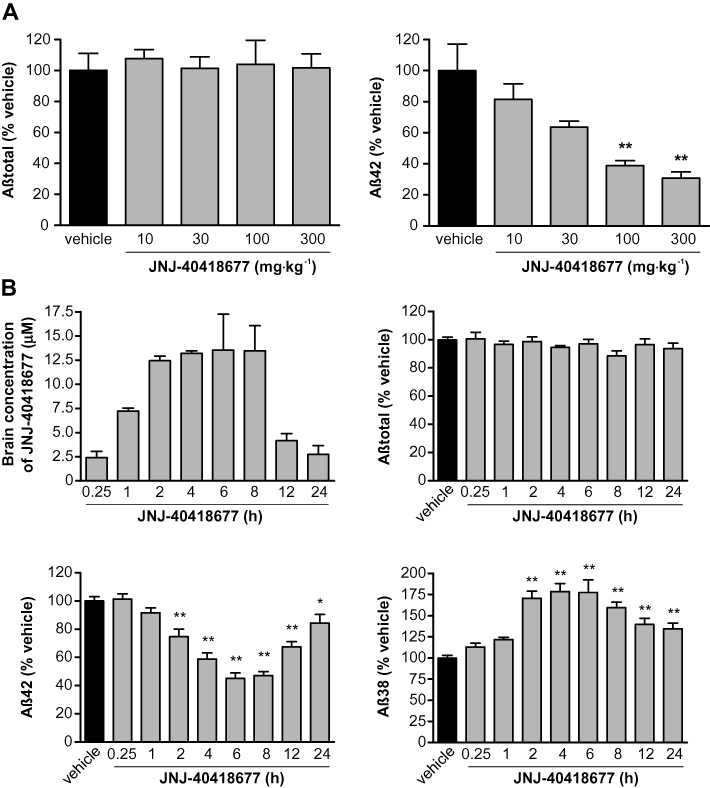
Effects of acute oral administration of JNJ-40418677 on Aβ levels in non-transgenic mouse brain as measured with differential elisa s. Mean (+SEM) brain Aβ levels after drug treatment are expressed as percentage of Aβ levels in brain of vehicle-treated mice. Mean (+SEM) brain JNJ-40418677 concentrations after drug treatment are expressed in µM. (A) Effect of JNJ-40418677 treatment on Aβtotal (left) and Aβ42 (right) levels in non-transgenic mouse brain 4 h after a single oral dose of 10, 30, 100 or 300 mg·kg−1 (n = 6 mice per data point). Brain Aβtotal levels were not affected at any dose, while Aβ42 levels were dose-dependently reduced. (B) Brain levels of JNJ-40418677 (top left) and effect of JNJ-40418677 treatment on Aβtotal (top right), Aβ42 (bottom left) and Aβ38 (bottom right) levels in non-transgenic mouse brain at different time points after a single oral dose of 30 mg·kg−1 (n = 6 mice per data point). Brain Aβ42 levels were significantly reduced compared with vehicle-treated mice, total Aβ levels were not affected and Aβ38 levels were significantly increased at time points between 2 and 24 h. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle.
Four hours after oral treatment of mice with various doses of JNJ-40418677, there was a significant effect of treatment on Aβ42 levels [F(4,22) = 8.58, P < 0.001] (Figure 5A). Aβ42 brain levels were reduced in a dose-dependent manner 4 h after JNJ-40418677 treatment to 82 ± 10%, 64 ± 4%, 39 ± 3% and 31 ± 4% of the levels in vehicle-treated mice for doses of 10, 30, 100 and 300 mg·kg−1 respectively (Figure 5A). JNJ-40418677 had no effect on the total Aβ levels in brain at any dose between 10 and 300 mg·kg−1[F(4,22) = 0.08, P > 0.05] (Figure 5A).
The duration of action of JNJ-40418677 was examined in a time-course experiment after a single oral dose of 30 mg·kg−1 in non-transgenic mice (Figure 5B). JNJ-40418677 had a significant effect on both Aβ42 and Aβ38 levels in brain [F(8,55) = 28.92 and 20.23, respectively, P < 0.0001 for both] and significantly reduced Aβ42 levels from 2 h after the start of treatment onwards (75 ± 5% of Aβ42 levels in vehicle-treated mice; P < 0.01). Aβ42 levels continued to decrease reaching a maximum reduction to 45 ± 4% of Aβ42 levels in vehicle-treated mice after 6 h (P < 0.01). Even at 24 h after administration, Aβ42 levels remained moderately reduced below vehicle values (84 ± 6% of Aβ42 levels in vehicle-treated mice; P < 0.05) (Figure 5B). Similarly, Aβ38 levels in brain were significantly increased from 2 to 24 h after JNJ-40418677 treatment (P < 0.01 for time points from 2 to 24 h) with a maximum 78 ± 15% increase compared with vehicle-treated mice at 6 h (Figure 5B). Total levels of Aβ in brain were not changed at any time point after JNJ-40418677 treatment [F(8,55) = 1.44, P > 0.05]. The extent of the increase in Aβ38 levels and reduction of Aβ42 levels in brain correlated with the concentration of the compound in the brain, except for the 1 h time point where a compound concentration that was higher than at 12 and 24 h was not accompanied by an effect on Aβ levels (Figure 5B). This can most probably be explained by the half-life of Aβ resulting in an effect on Aβ levels that occurs later than the rise in compound concentrations. Mean (±SEM) brain and plasma levels of JNJ-40418677 4 h after a single oral dose of 30 mg·kg−1 were 17 ± 1 µM and 17 ± 1 µM, respectively, indicative of good brain penetration.
Tolerance to chronic treatment with JNJ-40418677 in Tg2576 mice
The effects of chronic JNJ-40418677 treatment on plaque formation were investigated in Tg2576 mice. Female transgenic Tg2576 mice (n = 17 per group) were treated with JNJ-40418677 for a period of 7 months. Treatment was started at the age of 6 months, that is, before the appearance of plaques and mice received 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677 via a medicated diet.
Treatment with JNJ-40418677 for 7 months via medicated diet was well-tolerated in Tg2576 mice at doses up to 120 mg·kg−1·day−1. Eight transgenic mice were found dead or were killed before the completion of the treatment period. Their number was similar in vehicle- and JNJ-40418677-treated groups: two animals died or were killed prematurely in the vehicle group, one animal died in the 20 and 120 mg·kg−1·day−1 groups and four animals died in the 60 mg·kg−1·day−1 group. There were no adverse effects on body weight in any of the JNJ-40418677-treated groups over the treatment period.
Chronic JNJ-40418677 treatment reduces brain Aβ levels in Tg2576 mice
After 7 months of vehicle or JNJ-40418677 treatment via medicated diet, levels of Aβ in brain fractions of Tg2576 mice extracted with Tris buffered saline (‘soluble’ fraction) and GuHCl (‘deposited’ fraction) were measured with sandwich elisas that selectively detect human Aβ1–38, Aβ1–40, Aβ1–42 or total Aβ1–x (Figure 6A).
Figure 6.
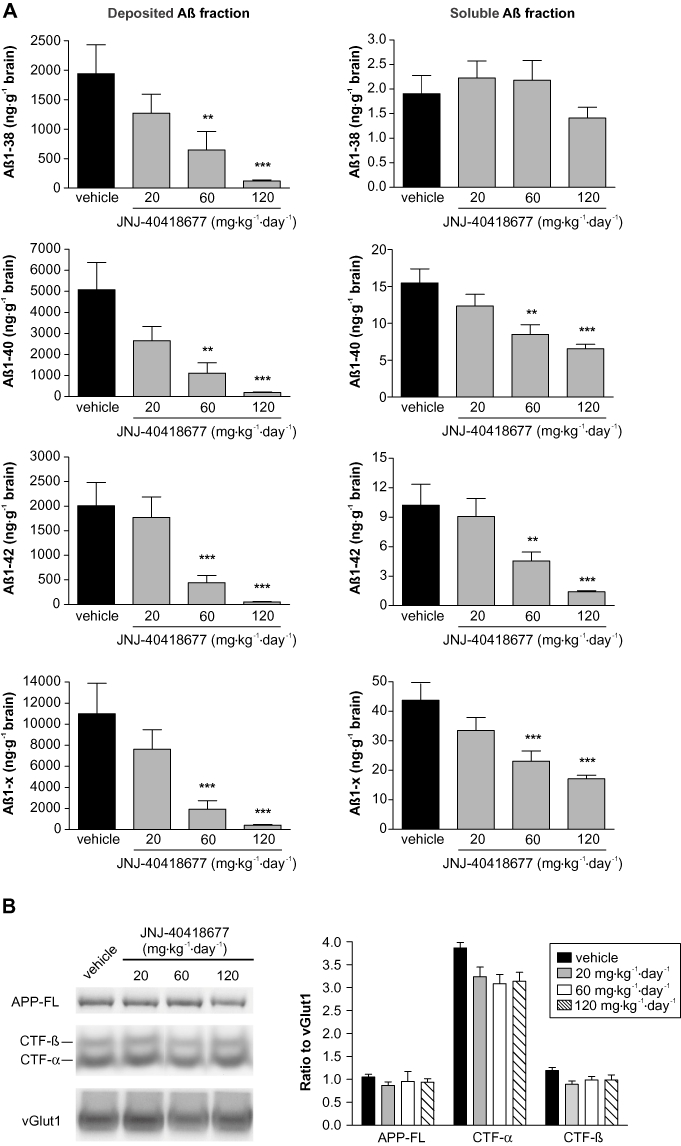
(A) Brain Aβ levels were reduced after chronic treatment of Tg2576 mice with JNJ-40418677 via a medicated diet. Female Tg2576 mice were treated from 6 to 13 months of age with 20 (n = 16), 60 (n = 13) or 120 mg·kg−1·day−1 (n = 16) JNJ-40418677 or vehicle (n = 15). Mean (+SEM) levels of human Aβ1–x, Aβ1–38, Aβ1–40 and Aβ1–42 in deposited (left histograms) and soluble (right histograms) brain fractions were measured by differential elisas and expressed as ng·g−1 brain. Chronic administration of JNJ-40418677 resulted in dose-dependent reductions of all Aβ species measured in soluble and deposited fractions, except for soluble Aβ1–38. (B) No changes in the steady state levels of full-length APP (APP-FL), CTF-α and CTF-β were observed after chronic treatment of Tg2576 mice with JNJ-40418677 via a medicated diet up to 120 mg·kg−1·day−1. Representative Western blots of membrane fractions of brains of Tg2576 mice treated with vehicle and various doses of JNJ-40418677 are shown. Antibodies JRF/Abtot/17 and C1/6.1 were utilized for detection of APP-FL and APP-CTF respectively. Data from densitometric analyses are expressed as ratio of APP-FL, CTF-α and CTF-β signals to loading control vGlut1. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle.
Chronic administration of JNJ-40418677 resulted in significant differences between treatment groups for all Aβ species measured in the deposited Aβ fraction [F(3,55) = 10.38 for Aβ1–38; F(3,56) = 16.91 for Aβ1–40; F(3,56) = 38.16 for Aβ1–42; F(3,56) = 26.40 for Aβ1–x; P < 0.0001 for all]. The effect was dose-dependent: compared with transgenic vehicle-treated animals, 20 mg·kg−1·day−1 JNJ-40418677 treatment tended to reduce all Aβ species in the deposited fraction but this was not significant, whereas 60 mg·kg−1·day−1 JNJ-40418677 treatment resulted in a significant reduction of deposited Aβ1–38, Aβ1–40, Aβ1–42 and Aβ1–x, and the reduction of these Aβ species was even more pronounced after 120 mg·kg−1·day−1 JNJ-40418677 treatment (Figure 6A). The mean percentages of reduction of Aβ levels in the deposited fraction compared with vehicle-treated transgenic mice are summarized in Table 1. All Aβ species measured in the deposited fraction (Aβ1–38, Aβ1–40, Aβ1–42 and Aβ1–x) were reduced to a similar extent after 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment (77–85% and 89–96% respectively).
Table 1.
Effect of chronic administration of JNJ-40418677 on soluble and deposited human Aβ1–38, Aβ1–40, Aβ1–42 and Aβ1–x levels in Tg2576 brain
| 20 mg·kg−1·day−1 JNJ-40418677 | 60 mg·kg−1·day−1 JNJ-40418677 | 120 mg·kg−1·day−1 JNJ-40418677 | |||||
|---|---|---|---|---|---|---|---|
| Peptide | Brain Aß fraction | Mean reduction (%) | 95% CI reduction (%) | Mean reduction (%) | 95% CI reduction (%) | Mean reduction (%) | 95% CI reduction (%) |
| Aß1–38 | Soluble | −13 | −100, 36 | −16 | −112, 37 | 26 | −31, 58 |
| Deposited | 20 | −134, 73 | 77 | 28, 93 | 89 | 66, 96 | |
| Aß1–40 | Soluble | 23 | −14, 48 | 47 | 20, 65 | 57 | 36, 71 |
| Deposited | 35 | −75, 76 | 81 | 45, 93 | 93 | 81, 97 | |
| Aß1–42 | Soluble | 18 | −45, 54 | 56 | 19, 76 | 84 | 71, 91 |
| Deposited | 0.3 | −144, 59 | 81 | 51, 93 | 96 | 92, 99 | |
| Aß1–x | Soluble | 26 | −8, 49 | 49 | 24, 65 | 59 | 40, 72 |
| Deposited | 15 | −111, 65 | 85 | 60, 94 | 94 | 85, 98 | |
Female Tg2576 mice were treated with 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677 via a medicated diet from 6 to 13 months of age.
Data are expressed as Dunnett adjusted geometric mean reduction (%) compared with vehicle-treated transgenic mice together with 95% CI (lower limit, upper limit).
Analysis of the soluble Aβ fraction in Tg2576 brains after chronic JNJ-40418677 treatment revealed significant differences between treatment groups for all Aβ species measured, except Aβ1–38 [F(3,56) = 1.51, P = 0.22 for Aβ1–38; F(3,56) = 10.62, P < 0.0001 for Aβ1–40; F(3,56) = 23.93, P < 0.0001 for Aβ1–42; F(3,56) = 12.87, P < 0.0001 for Aβ1–x]. The effect was dose-dependent for Aβ1–40, Aβ1–42 and Aβ1–x with a non-significant trend for Aβ reduction compared with vehicle-treated transgenic animals in the soluble fraction after 20 mg·kg−1·day−1 JNJ-40418677 treatment, a significant reduction after 60 mg·kg−1·day−1 JNJ-40418677 treatment and a further reduction after 120 mg·kg−1·day−1 JNJ-40418677 treatment (Figure 6A). The mean percentages of reduction of Aβ levels in the soluble fraction compared with vehicle-treated transgenic mice are summarized in Table 1. Aβ1–40, Aβ1–42 and Aβ1–x measured in the soluble fraction were reduced to a similar extent after 60 mg·kg−1·day−1 treatment (47%, 56% and 49% respectively). Soluble Aβ1–42 levels were reduced more than Aβ1–40 and Aβ1–x levels after 120 mg·kg−1·day−1 treatment (84% reduction for Aβ1–42 compared with 57% and 59% reduction for Aβ1–40 and Aβ1–x respectively). The reduction of Aβ levels in deposited fractions was larger than in soluble fractions after 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment for all Aβ species measured.
The effect of chronic JNJ-40418677 treatment of Tg2576 mice on APP expression and processing in brain was also evaluated. Western blot analysis showed that chronic JNJ-40418677 treatment with doses up to 120 mg·kg−1·day−1 did not affect the steady state levels of full-length APP, CTF-α and CTF-β when compared with vehicle-treated Tg2576 mice (Figure 6B).
Chronic JNJ-40418677 treatment inhibits plaque formation in Tg2576 mice
Plaque load in Tg2576 brain after a 7 month treatment with vehicle or JNJ-40418677 was analysed with immunohistochemistry utilizing antibodies 4G8 against the Aβ mid-domain, JRF/AbN/25 against full-length human Aβ (Aβ1–x), and carboxy-terminal antibodies J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26 against Aβ ending at amino acid 38, 40 and 42 respectively (Figure 7A). Immunohistochemistry with 4G8 and JRF/AbN/25 antibodies resulted in similar read-outs for measures of plaque load, suggesting that most of the Aβ peptides deposited in Tg2576 brain are full-length peptides, as described previously (Kawarabayashi et al., 2001).
Figure 7.
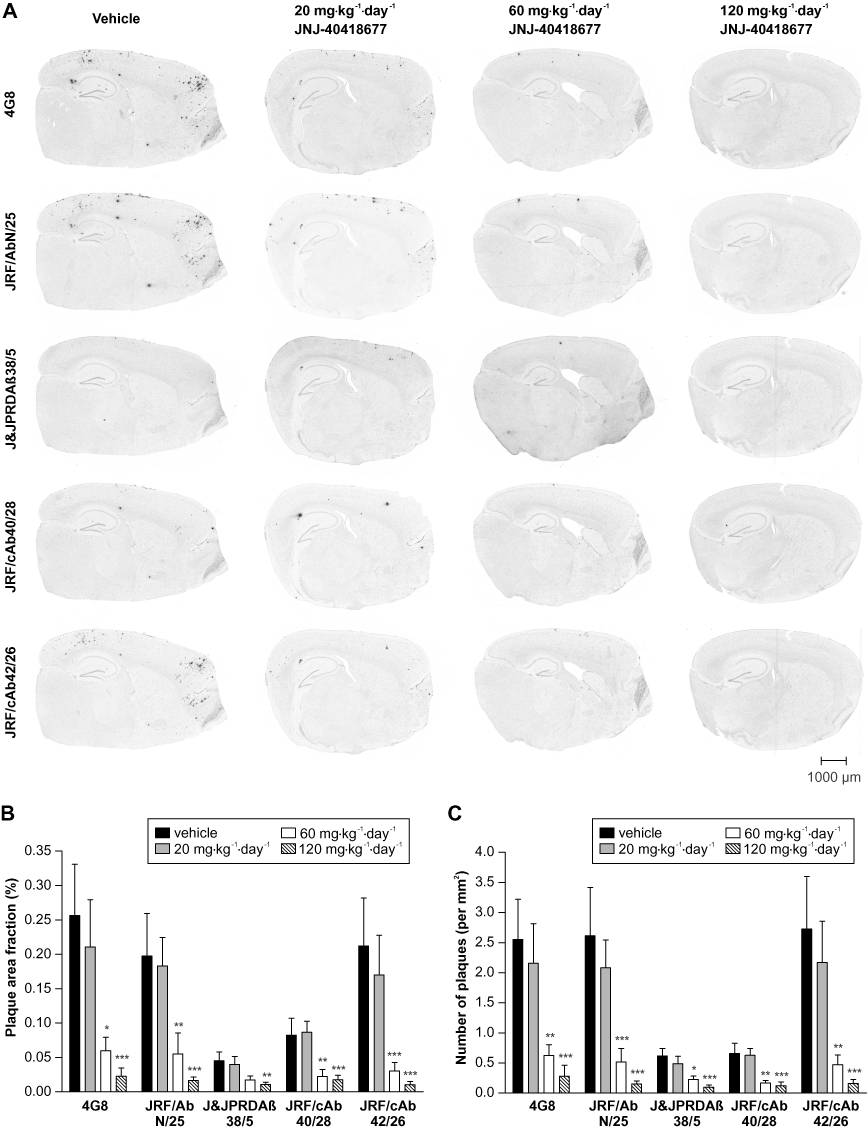
Plaque load was reduced in brains of Tg2576 mice after chronic treatment with JNJ-40418677 via a medicated diet. Female Tg2576 mice were treated from 6 to 13 months of age with 20 (n = 16), 60 (n = 13) or 120 mg·kg−1·day−1 (n = 16) JNJ-40418677 or vehicle (n = 15). (A) Representative images of immunohistochemical labelling of plaques in brains of Tg2576 mice treated with vehicle or 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677 as visualized with monoclonal antibodies 4G8 (against the Aβ mid-domain), JRF/AbN/25 (against full-length human Aβ) and J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26 against Aβ ending at amino acid 38, 40 and 42, respectively. Note the marked reduction of immunostaining in JNJ-40418677-treated animals for all antibodies utilized for visualization of plaques. Scale bar represents 1000 µm. Mean (+SEM) brain area fraction occupied by plaques (% of tissue area) (B) and number of plaques (mm2 of brain) (C) as visualized by immunostaining with 4G8, JRF/AbN/25, J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26 antibodies in brains of Tg2576 mice treated with vehicle or 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677. Chronic administration of JNJ-40418677 resulted in dose-dependent reductions of plaque load for all antibodies utilized for visualization of plaques. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle.
There were significant differences between treatment groups for the area occupied by plaques [F(3,56) = 18.63, P < 0.0001 for 4G8; F(3,56) = 17.95, P < 0.0001 for JRF/AbN/25; F(3,55) = 5.41, P = 0.0025 for J&JPRDAβ38/5; F(3,56) = 14.65, P < 0.0001 for JRF/cAb40/28; F(3,55) = 24.26, P < 0.0001 for JRF/cAb42/26] (Figure 7B). Similarly, the number of plaques per mm2 brain tissue was significantly different between treatment groups [F(3,56) = 22.52, P < 0.0001 for 4G8; F(3,56) = 30.66, P < 0.0001 for JRF/AbN/25; F(3,55) = 14.16, P < 0.0001 for J&JPRDAβ38/5; F(3,56) = 22.87, P < 0.0001 for JRF/cAb40/28; F(3,55) = 25.66, P < 0.0001 for JRF/cAb42/26] (Figure 7C). The effect was dose-dependent: compared with vehicle-treated transgenic mice, 20 mg·kg−1·day−1 JNJ-40418677 treatment caused a non-significant trend for a reduced plaque area fraction and number of plaques, while 60 mg·kg−1·day−1 JNJ-40418677 treatment resulted in a significant reduction in plaque area fraction and number of plaques and this reduction was even more pronounced after 120 mg·kg−1·day−1 JNJ-40418677 treatment. The mean percentages of reduction of plaque area fraction and number of plaques compared with vehicle-treated transgenic mice are summarized in Table 2. In general, the biochemical measure of deposited Aβ levels seemed to be an accurate reflection of the plaque load: the observed reductions versus vehicle-treated animals as observed with biochemical analysis of the deposited Aβ fraction and immunohistochemical measures of plaque load were very similar after 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment. Plaque area fraction and number of plaques were reduced to a similar extent when measured with 4G8, JRF/AbN/25 and carboxy-terminal antibodies in 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment groups (55–84% and 82–96% respectively).
Table 2.
Effect of chronic administration of JNJ-40418677 on plaque area fraction and number of plaques in Tg2576 brain
| 20 mg·kg−1·day−1 JNJ-40418677 | 60 mg·kg−1·day−1 JNJ-40418677 | 120 mg·kg−1·day−1 JNJ-40418677 | |||||
|---|---|---|---|---|---|---|---|
| Antibody | Measure of plaque burden | Mean reduction (%) | 95% CI reduction (%) | Mean reduction (%) | 95% CI reduction (%) | Mean reduction (%) | 95% CI reduction (%) |
| 4G8 | % area | 22 | −143, 75 | 78 | 26, 93 | 96 | 86, 99 |
| # plaques | 16 | −131, 69 | 76 | 32, 92 | 95 | 86, 98 | |
| JRF/AbN/25 | % area | −10 | −208, 60 | 77 | 32, 92 | 92 | 78, 97 |
| # plaques | 4 | −131, 60 | 82 | 54, 93 | 95 | 87, 98 | |
| J&JPRDAß38/5 | % area | 30 | −105, 76 | 55 | −41, 86 | 82 | 47, 94 |
| # plaques | 35 | −46, 71 | 65 | 18, 85 | 88 | 72, 95 | |
| JRF/cAb40/28 | % area | −21 | −229, 56 | 74 | 25, 91 | 88 | 68, 96 |
| # plaques | 0.5 | −131, 57 | 74 | 38, 89 | 91 | 79, 96 | |
| JRF/cAb42/26 | % area | 6 | −177, 68 | 84 | 51, 95 | 96 | 89, 99 |
| # plaques | 10 | −141, 66 | 80 | 44, 93 | 96 | 88, 98 | |
Female Tg2576 mice were treated with 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677 via a medicated diet from 6 to 13 months of age. Plaques were visualized with monoclonal antibodies 4G8 (against the Aβ mid-domain), JRF/AbN/25 (against full-length human Aβ) and J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26 against Aβ ending at amino acid 38, 40 and 42 respectively. Data are expressed as Dunnett adjusted geometric mean reduction (%) compared with vehicle-treated transgenic mice together with 95% CI (lower limit, upper limit). % area, % of total brain area occupied by plaques; # plaques, number of plaques mm2 brain.
Plasma and brain levels of JNJ-40418677 after chronic treatment of Tg2576 mice
Mean plasma and brain levels of JNJ-40418677 after a 7 month treatment of Tg2576 mice via medicated diet are summarized in Table 3. Mean plasma and brain concentrations seemed to increase more than dose proportional between 20 and 60 mg·kg−1·day−1 and between 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment groups. As was observed after treatment of non-transgenic mice with a single oral dose of JNJ-40418677, brain to plasma ratio was close to 1 in all JNJ-40418677-treated groups.
Table 3.
Mean plasma and brain concentrations of JNJ-40418677 after administration of JNJ-40418677 via medicated diet to Tg2576 mice from 6 to 13 months of age
| Dosing schedule (mg·kg−1·day−1 JNJ-40418677) | Plasma concentration (µM) | Brain concentration (µM) |
|---|---|---|
| 20 | 0.38 ± 0.02 | 0.42 ± 0.02 |
| 60 | 2.7 ± 0.4 | 2.4 ± 0.3 |
| 120 | 13 ± 1 | 12 ± 1 |
Data are expressed as mean ± SEM (µM).
Discussion and conclusions
In this study we describe the in vitro and in vivo characterization of a novel GSM, JNJ-40418677, with an improved potency towards Aβ42 inhibition and excellent brain penetration after oral administration in mice. Similar to other GSMs, JNJ-40418677 displays unique pharmacological properties compared with GSIs. First, JNJ-40418677 shifted the preferred γ-cleavage sites leading to increased Aβ38 and decreased Aβ42 levels and the compound also did not interfere with the formation of APP-CTF and AICD (Weggen et al., 2001; 2003a; Gasparini et al., 2004). Moreover, JNJ-40418677 differentially affected the proteolytic activities on APP and Notch in vitro (Weggen et al., 2001; Gasparini et al., 2004), indicating that a therapeutic window between the effects of JNJ-40418677 on Aβ42 formation and Notch processing seems possible. These pharmacological characteristics suggest a mechanism of action different from GSIs, and selective targeting of the toxic Aβ42 species without an effect on the formation of other APP cleavage products or Notch processing contributes to a potentially improved safety profile of GSM versus GSI treatment.
JNJ-40418677 treatment caused a selective reduction of Aβ42 secretion in supernatants of APP WT human neuroblastoma cells and primary rat neurones, with a wide separation between the IC50 for reduction of Aβ42 (200 nM) and the IC50 for total Aβ reduction (>10 µM). Compared with its parent compound R-flurbiprofen that shows an IC50 of 150–200 µM in APP-Swe human neuroglioma cells (Eriksen et al., 2003), JNJ-40418677 exhibits a markedly improved potency for reducing Aβ42 secretion of ∼1000-fold. Modulatory effects of JNJ-40418677 with a selective reduction of Aβ42 and a concomitant increase in Aβ38 were also observed in a cell-free γ-secretase assay and after acute treatment in brains of non-transgenic mice. Aβ1–38 was shown to be less toxic to mouse hippocampal and cortical neurones than Aβ1–42 (Singer and Dewji, 2006), suggesting that increased Aβ38 levels caused by GSM treatment should not lead to neuronal cell damage. Moreover, Aβ38 exhibited a low tendency to form β-sheets and had an inhibitory effect on Aβ42 aggregation (Watanabe et al., 2006), suggesting that enhanced Aβ38 production might even inhibit plaque formation that is thought to be initiated by Aβ42 (McGowan et al., 2005).
The ability of certain NSAIDs with GSM activity to lower brain amyloid pathology in transgenic mouse models of AD has been reported after long-term treatment; these include ibuprofen (Lim et al., 2000; Yan et al., 2003) and indomethacin (Sung et al., 2004). A major concern with chronic use of NSAIDs is their inhibition of COX-1 activity leading to severe gastrointestinal toxicity. One solution to avoid these side effects is to use the R-enantiomer of some NSAIDs (e.g. R-flurbiprofen) that displays lower anti-COX activity than the S-enantiomer (Jerussi et al., 1998) while maintaining Aβ42 inhibitory action (Eriksen et al., 2003). In this context, it was established that JNJ-40418677 does not inhibit COX-1/2 enzymes, even at concentrations much higher than those required to reduce Aβ42, thus supporting its suitability for chronic use.
The use of R-flurbiprofen and other NSAIDS also has some limitations due to their rather low potency and low brain penetration (Eriksen et al., 2003; Stock et al., 2006), characteristics that might have contributed to the disappointing results of a recently completed Phase III clinical trial with R-flurbiprofen for the treatment of mild AD (Green et al., 2009). As discussed above, JNJ-40418677 shows an improved potency for Aβ42 reduction and this study also demonstrates that JNJ-40418677 has excellent brain penetration both after acute oral treatment of non-transgenic mice and after chronic treatment of Tg2576 mice. After chronic treatment with 60 mg·kg−1·day−1 JNJ-40418677, compound concentrations reached in the brain of Tg2576 mice (2.4 ± 0.3 µM) were high enough to efficiently inhibit Aβ42 secretion, based on an IC50 of 200 nM as measured in cell-based assays.
To assess the effects of JNJ-40418677 on plaque formation, Tg2576 mice were treated for 7 months with 20, 60 or 120 mg·kg−1·day−1 JNJ-40418677 via medicated diet starting at the age of 6 months, that is, before the appearance of plaques (Hsiao et al., 1996; Kawarabayashi et al., 2001; Terai et al., 2001).
JNJ-40418677 was well-tolerated when administered for 7 months to Tg2576 mice via the diet at doses up to 120 mg·kg−1·day−1. There was no compound-related mortality: the number of animals that died before the end of the treatment period was similar in vehicle- and JNJ-40418677-treated groups, did not increase with increasing doses of JNJ-40418677 and was in accordance with previous studies on premature mortality in this mouse model (Lewis et al., 2004). Mean body weight was similar in vehicle and JNJ-40418677 treatment groups over the treatment period. The lack of overt toxicity in the present study indicates good tolerance of JNJ-40418677 after prolonged treatment in mice, although further chronic toxicological studies are needed to confirm the absence of Notch-related side effects in the gastrointestinal and immune system that are commonly associated with prolonged exposure to GSIs (Hadland et al., 2001; Searfoss et al., 2003; Milano et al., 2004; Wong et al., 2004).
Chronic administration of JNJ-40418677 significantly reduced soluble Aβ1–40 and Aβ1–42 levels in Tg2576 mouse brains, but also reduced soluble Aβ1–x levels and did not increase soluble Aβ1–38 levels, in contrast to the effects observed after acute dosing in non-transgenic mice. Our analysis of APP and APP-CTF levels showed this was not due to altered transgene APP expression or complete inhibition of γ-secretase activity after chronic treatment. JNJ-40418677 treatment causes an acute rise of Aβ38 levels, as observed in non-transgenic mice. However, chronic JNJ-40418677 treatment also leads to decreased Aβ38 deposition. This reduction might be reflected in the soluble fraction if there is some contamination of this fraction with plaque-derived Aβ from the deposited fraction. In this case, the combined presence of an acute increase and a chronic decrease of Aβ38 levels after JNJ-40418677 treatment might cause the observed equivalence of soluble Aβ38 levels in JNJ-40418677- and vehicle-treated mice. Interestingly, a decrease in brain Aβ38 levels in the soluble fraction after chronic GSM treatment of Tg2576 mice was also observed in another recent study (Kounnas et al., 2010).
Chronic administration of JNJ-40418677 significantly inhibited the formation of plaques in Tg2576 brain, as was shown by a marked decrease in brain area occupied by plaques as well as a reduced number of plaques compared with vehicle-treated transgenic controls. Treatment with JNJ-40418677 at a dose of 120 mg·kg−1·day−1 almost completely prevented plaque deposition in Tg2576 brain. We showed that JNJ-40418677 treatment reduced plaque deposition in a dose-dependent manner as measured with various monoclonal antibodies against all Aβ species (4G8), full-length human Aβ (JRF/AbN/25) and Aβ38, Aβ40 and Aβ42 (J&JPRDAβ38/5, JRF/cAb40/28 and JRF/cAb42/26, respectively) to a similar extent. This was confirmed by our biochemical analysis of the deposited Aβ fraction, where the amount of human Aβ1–x, Aβ1–38, Aβ1–40 and Aβ1–42 was significantly reduced after 60 and 120 mg·kg−1·day−1 JNJ-40418677 treatment and this reduction was similar for all Aβ species measured within a particular treatment group. While selective Aβ42 reduction is a characteristic feature of γ-secretase modulation, Aβ42 is also the initial deposited species in AD brain (Iwatsubo et al., 1994) that acts as a seed for co-accumulation of other Aβ species (McGowan et al., 2005). Consequently, reduction of Aβ42 secretion and thus deposition is expected to lead to reduced accumulation of other Aβ species.
In agreement with our data, plaque load was recently shown to be reduced after chronic treatment of Tg2576 mice via the diet with two other GSM compounds (Imbimbo et al., 2007; 2010; Kounnas et al., 2010). JNJ-40418677 treatment at the same dose (60 mg·kg−1·day−1) and via the same administration route (mixed in the diet) reduced plaque area fraction in Tg2576 mice to a similar extent to treatment with the R-flurbiprofen analogue CHF5074 when mice were killed at a similar age (Imbimbo et al., 2007) and to a larger extent when treatment was started at the same age (Imbimbo et al., 2010).
In conclusion, this study describes a novel, orally active modulator of γ-secretase activity that selectively reduces Aβ42 in vitro and in vivo. Chronic treatment of Tg2576 mice with JNJ-40418677 via the diet appeared to be well-tolerated and led to a significant decrease in brain Aβ levels and plaque burden in a dose-dependent manner when treatment was started before the onset of plaque deposition. Moreover, JNJ-40418677 displayed excellent brain penetration after oral treatment in mice and did not affect the formation of APP-CTF and AICD, Notch processing or the activity of COX enzymes. Further studies should shed light on possible side effects of the compound and reveal whether the compound is able to restore cognitive and behavioural deficits in transgenic mice. If successful, these studies will add to the potential of JNJ-40418677 as a safe and effective approach for a disease-modifying treatment for AD that would slow down or halt the progression of this debilitating disease.
Acknowledgments
We thank Dr Mathews (Nathan S. Kline Institute, Orangeburg) for the generous gift of the monoclonal C1/6.1 antibody, Bart Hermans and Ana Barao for technical assistance in elisa analysis, Marc Vandermeeren for monoclonal antibody generation, Greet Meulders and Marianne Borgers for Western blot analysis, Roel Straetemans for assistance with statistical analysis, the Bioanalysis department of Janssen Pharmaceutica for pharmacokinetic analysis and HistoGeneX (Edegem, Belgium) for immunohistochemistry and image analysis. This study was sponsored by Cellzome and Johnson & Johnson Pharmaceutical Research and Development, Janssen Pharmaceutica.
Glossary
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer's disease
- AICD
APP intracellular domain
- APP
amyloid precursor protein
- APP-Swe
APP with Swedish double mutation K670N/M671L
- CHF5074
1-(3′,4′-dichloro-2-fluoro[1,1′-biphenyl]-4-yl)-cyclopropanecarboxylic acid
- CHO
Chinese hamster ovary
- CTF
carboxy-terminal fragment
- DAB
3,3′-diaminobenzidine
- DAPT
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester
- elisa
enzyme-linked immunosorbent assay
- FL
full-length
- GSI
γ-secretase inhibitor
- GSM
γ-secretase modulator
- GuHCl
guanidinehydrochloride
- HRPO
horseradish peroxidase
- JNJ-40418677
2-(S)-(3,5-bis(4-(trifluoromethyl)phenyl)phenyl)-4-methylpentanoic acid
- MS
mass spectrometry
- NICD
Notch intracellular domain
- NSAID
non-steroidal anti-inflammatory drug
- PBS
phosphate buffered saline
- PS
presenilin
- WT
wild-type
Conflicts of interest
Bianca Van Broeck, Miek Desmidt, Eric Karran and Marc Mercken are employees of Johnson & Johnson Pharmaceutical Research and Development, Janssen Pharmaceutica. Jinq-May Chen, Carsten Hopf, Nigel Ramsden and Adele Rowley are employees of Cellzome Ltd or Cellzome AG.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Czirr E, Leuchtenberger S, Dorner-Ciossek C, Schneider A, Jucker M, Koo EH, et al. Insensitivity to Abeta42-lowering nonsteroidal anti-inflammatory drugs and gamma-secretase inhibitors is common among aggressive presenilin-1 mutations. J Biol Chem. 2007;282:24504–24513. doi: 10.1074/jbc.M700618200. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, Andrieu P, Fang LY, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. Gamma-secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280:41987–41996. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Rusconi L, Xu H, del Soldato P, Ongini E. Modulation of beta-amyloid metabolism by non-steroidal anti-inflammatory drugs in neuronal cell cultures. J Neurochem. 2004;88:337–348. doi: 10.1111/j.1471-4159.2004.02154.x. [DOI] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadland BK, Manley NR, Su D, Longmore GD, Moore CL, Wolfe MS, et al. Gamma-secretase inhibitors repress thymocyte development. Proc Natl Acad Sci USA. 2001;98:7487–7491. doi: 10.1073/pnas.131202798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Ho CY. Preparation of biphenyl derivatives as γ-secretase modulators. 2009. WO2009052341A1. 2009 Apr 23.
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP, Del Giudice E, Colavito D, D'Arrigo A, Dalle Carbonare M, Villetti G, et al. 1-(3′,4′-Dichloro-2-fluoro[1,1′-biphenyl]-4-yl)-cyclopropanecarboxylic acid (CHF5074), a novel gamma-secretase modulator, reduces brain beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease without causing peripheral toxicity. J Pharmacol Exp Ther. 2007;323:822–830. doi: 10.1124/jpet.107.129007. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP, Giardino L, Sivilia S, Giuliani A, Gusciglio M, Pietrini V, et al. CHF5074, a novel gamma-secretase modulator, restores hippocampal neurogenesis potential and reverses contextual memory deficit in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2010;20:159–173. doi: 10.3233/JAD-2010-1366. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jerussi TP, Caubet JF, McCray JE, Handley DA. Clinical endoscopic evaluation of the gastroduodenal tolerance to (R)- ketoprofen, (R)- flurbiprofen, racemic ketoprofen, and paracetamol: a randomized, single-blind, placebo-controlled trial. J Clin Pharmacol. 1998;38(2) Suppl:S19–S24. doi: 10.1002/j.1552-4604.1998.tb04413.x. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, et al. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer's disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, et al. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Lewis HD, Beher D, Smith D, Hewson L, Cookson N, Reynolds DS, et al. Novel aspects of accumulation dynamics and A beta composition in transgenic models of AD. Neurobiol Aging. 2004;25:1175–1185. doi: 10.1016/j.neurobiolaging.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken M, Brepoels E, De Jongh M, Laenen W, Raeymakers P, Geerts H. Specific ELISA systems for the detection of endogenous human and rodent Aβ40 and Aβ42. Neurobiol Aging. 2000;21:S41. [Google Scholar]
- Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- Morihara T, Chu T, Ubeda O, Beech W, Cole GM. Selective inhibition of Abeta42 production by NSAID R-enantiomers. J Neurochem. 2002;83:1009–1012. doi: 10.1046/j.1471-4159.2002.01195.x. [DOI] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Perez-Revuelta BI, Fukumori A, Jacobsen H, et al. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- Peretto I, La Porta E. Gamma-secretase modulation and its promise for Alzheimer's disease: a medicinal chemistry perspective. Curr Top Med Chem. 2008;8:38–46. doi: 10.2174/156802608783334097. [DOI] [PubMed] [Google Scholar]
- Pype S, Moechars D, Dillen L, Mercken M. Characterization of amyloid beta peptides from brain extracts of transgenic mice overexpressing the London mutant of human amyloid precursor protein. J Neurochem. 2003;84:602–609. doi: 10.1046/j.1471-4159.2003.01556.x. [DOI] [PubMed] [Google Scholar]
- Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem. 2003;278:46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–638. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- Singer SJ, Dewji NN. Evidence that Perutz's double-beta-stranded subunit structure for beta-amyloids also applies to their channel-forming structures in membranes. Proc Natl Acad Sci USA. 2006;103:1546–1550. doi: 10.1073/pnas.0509892103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock N, Munoz B, Wrigley JD, Shearman MS, Beher D, Peachey J, et al. The geminal dimethyl analogue of Flurbiprofen as a novel Abeta42 inhibitor and potential Alzheimer's disease modifying agent. Bioorg Med Chem Lett. 2006;16:2219–2223. doi: 10.1016/j.bmcl.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, et al. Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004;165:2197–2206. doi: 10.1016/s0002-9440(10)63269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Iwai A, Kawabata S, Tasaki Y, Watanabe T, Miyata K, et al. Beta-amyloid deposits in transgenic mice expressing human beta-amyloid precursor protein have the same characteristics as those in Alzheimer's disease. Neuroscience. 2001;104:299–310. doi: 10.1016/s0306-4522(01)00095-1. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Bernier F, Miyagawa T. Therapeutic agent for Abeta related disorders. 2006. US20060241038A1. 2006 Oct 26.
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. Abeta42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem. 2003a;278:30748–30754. doi: 10.1074/jbc.M304824200. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, et al. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003b;278:31831–31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- Winkler E, Hobson S, Fukumori A, Dumpelfeld B, Luebbers T, Baumann K, et al. Purification, pharmacological modulation, and biochemical characterization of interactors of endogenous human gamma-secretase. Biochemistry. 2009;48:1183–1197. doi: 10.1021/bi801204g. [DOI] [PubMed] [Google Scholar]
- Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.