

Abstract
BACKGROUND AND PURPOSE
The effects of metformin, an antidiabetic agent that improves insulin sensitivity, on endothelial function have not been fully elucidated. This study was designed to assess the effect of metformin on impaired endothelial function, oxidative stress, inflammation and advanced glycation end products formation in type 2 diabetes mellitus.
EXPERIMENTAL APPROACH
Goto-Kakizaki (GK) rats, an animal model of nonobese type 2 diabetes, fed with normal and high-fat diet during 4 months were treated with metformin for 4 weeks before evaluation. Systemic oxidative stress, endothelial function, insulin resistance, nitric oxide (NO) bioavailability, glycation and vascular oxidative stress were determined in the aortic rings of the different groups. A pro-inflammatory biomarker the chemokine CCL2 (monocyte chemoattractant protein-1) was also evaluated.
KEY RESULTS
High-fat fed GK rats with hyperlipidaemia showed increased vascular and systemic oxidative stress and impaired endothelial-dependent vasodilatation. Metformin treatment significantly improved glycation, oxidative stress, CCL2 levels, NO bioavailability and insulin resistance and normalized endothelial function in aorta.
CONCLUSION AND IMPLICATIONS
Metformin restores endothelial function and significantly improves NO bioavailability, glycation and oxidative stress in normal and high-fat fed GK rats. This supports the concept of the central role of metformin as a first-line therapeutic to treat diabetic patients in order to protect against endothelial dysfunction associated with type 2 diabetes mellitus.
Keywords: endothelial dysfunction, metformin, type 2 diabetes, oxidative stress, glycation
Introduction
The worldwide prevalence of diabetes mellitus, particularly type 2 diabetes, has increased significantly in recent years. Type 2 diabetes alters the vascular responsiveness to several vasoconstrictors and vasodilators and is a major factor underlying development of cardiovascular disease (De Vriese et al., 2000a). Many of the complications in diabetes are related to increased serum glucose and increased generation of reactive oxygen species, which lead to endothelial dysfunction. Impaired endothelium-dependent relaxation to vasodilators such as acetylcholine is a common feature in both conduit and resistance arteries from experimental models of type 1 and type 2 diabetes (Cosentino and Luscher, 1999; De Vriese et al., 2000b; Ding et al., 2005; Pannirselvam et al., 2005; Sena et al., 2008).
Metformin is a biguanide derivative that exerts an anti-hyperglycaemic effect with minimal risk of hypoglycaemia. It has been widely used in the management of type 2 diabetes, with a 31% reduction in incidence (Knowler et al., 2002). Metformin lowers blood glucose concentration and improves insulin sensitivity by reducing hepatic gluconeogenesis and enhancing insulin-stimulated peripheral glucose uptake (Yoshida et al., 2009). It also inhibits adipose tissue lipolysis, thereby reducing circulating levels of free fatty acids (FFA) (Kirpichnikov et al., 2002). In addition, metformin improves lipid profile and lowers blood pressure in both patients and animals with impaired glucose tolerance and type 2 diabetes (Verma et al., 1994; Kirpichnikov et al., 2002). It has also been described that metformin not only improves the antioxidant defence but is also a powerful activator of AMP-kinase, which is a major regulator of glucose and lipid metabolism (Leverve et al., 2003). In overweight type 2 diabetic patients, metformin use is associated with decreases in macrovascular morbidity and mortality, effects that appear to be independent of the improvement in glycaemic control (UKPDS, 1998). Moreover, metformin seems to improve endothelial function. Indeed, metformin restores the microvascular reactivity to histamine, bradykinin or acetylcholine, of arterioles and venules from neonatal streptozotocin-diabetic rats (Sartoretto et al., 2005). More recently, metformin has been shown that it improves the flow-mediated dilatation of normoinsulinemic subjects (Romualdi et al., 2008).
On the basis of these observations, we hypothesized that metformin, already used clinically for diabetes, would be beneficial at the macrovascular level through a mechanism that involves glycation, oxidative stress, nitric oxide (NO) bioavailability and inflammation in aortic vessels of diabetic rats. As model of type 2 diabetes, we used GK rats, some of which were fed with high-fat diet for 4 months (GK + AD) to induce hyperlipidaemia and more pronounced macrovascular complications. GK rats exhibit a spontaneous polygenic mild type 2 diabetes. They are characterized by an early increase in serum insulin, normolipidaemia and also by mild hyperglycaemia and insulin resistance (Nunes et al., 2007). There are several reports of abnormalities of vascular function in this diabetic model (Cheng et al., 2001; Rösen and Wiernsperger, 2006; Sena et al., 2008).
The purpose of the present study was to evaluate the effect of metformin on endothelial dysfunction and analyse the influence of the drug on glycation, oxidative stress and early inflammation in a model of type 2 diabetes, using the aorta from Goto-Kakizaki diabetic rats fed with normal or high-fat diets.
Methods
Animals
All animal care and experimental protocols were in accordance with the Portuguese Law on Experimentation with Laboratory Animals (last amendment, 2004), which is based on the principles of laboratory animal care as adopted by the EC Directive 86/609/EEC for animal experiments. Adult male Wistar and spontaneously diabetic GK rats were obtained from the local breeding colony in Coimbra (Portugal). GK rats are characterized by an early increase in serum insulin and also by mild hyperglycaemia, insulin resistance and mild type 2 diabetes (Nunes et al., 2007). There are several reports of abnormalities of vascular function in this diabetic model (Cheng et al., 2001; Rösen and Wiernsperger, 2006; Sena et al., 2008). Control animals were fed, ad libitum, with a standard commercial pellet chow (Diet AO4-Panlab). As a model of type 2 diabetes, we used GK rats, some of which were fed with high-fat diet for 4 months (GK + AD). The GK rats were divided into four groups: (i) GK diabetic control rats (n = 16, GK) maintained with standard diet until 6 months of age; (ii) GK diabetic rats treated with metformin (60 mg·kg−1·day−1, in drinking water) during the last 4 weeks (n = 6, GK + M) and maintained with standard diet; (iii) high-fat diet fed GK diabetic rats for 4 months (n = 16, GK + AD, containing: 70% AO4 standard Chow, 7.5% cocoa butter and 1.25% cholesterol) and (iv) high-fat diet fed GK diabetic rats treated with metformin (60 mg·kg−1·day−1, in drinking water) during the last 4 weeks (n = 16, GK + AD + M). All animals were allowed free access to water; they were kept in rooms with 12 h periods of light and darkness. Urine and blood were collected at the end of treatment. The aorta was excised and used for vascular function studies and histological examination.
Determination of metabolic and oxidative stress parameters
After a 15 h fast, animals were anaesthetized with ketamine/chlorpromazine [ketamine chloride (75 mg·kg−1, i.m., Parke-Davis, Ann Arbor, MI, USA) and chlorpromazine chloride (2.65 mg·kg−1, i.m., Lab. Vitória, Portugal)] and killed by decapitation. Blood was taken by heart puncture for determination of lipids, carbonyl compounds, free fatty acid levels and insulin. For glucose tolerance tests, rats were fasted overnight and were given an intraperitoneal injection of glucose (1.75 g·kg−1 body weight) in phosphate-buffered saline (PBS). Blood glucose was determined by sampling from the tail vein at 0, 60 and 120 min after injection by a glucose-oxidase method using a glucometer (Glucometer-Elite-Bayer, Portugal S.A.) and compatible reactive test strips. Fasting plasma lipids [total and high density lipoprotein (HDL) cholesterol, triglycerides and phospholipids] and plasma insulin levels were quantified using commercially available kits and by an in-house enzyme-linked immunosorbent assay (Seiça et al., 2004) respectively. To assess insulin resistance in the fasted state, the homeostasis model assessment of insulin resistance (HOMA) and quantitative insulin-sensitivity check index (QUICKI) were calculated, as previously described (Sena et al., 2007). HOMA was calculated as [(G0) × (I0) / 22.5] where G0 is the fasting glucose level (mmol·L−1), and I0 is the fasting insulin level (µU·mL−1). QUICKI was calculated as 1 / [log(G0) + log(I0)], where G0 is fasting glycaemia (mg·dL−1), and I0 is fasting insulin level (µU·mL−1).
Plasma FFA levels were evaluated using enzymatic assay kits (Roche Applied Science, Amadora, Portugal). Rats were placed in metabolic cages for 24 h and urine collected. Urinary 8-hydroxydeoxyguanosine (8-OHdG), albumin and plasma carbonyl protein concentrations were measured using ELISA kits (OXIS health Products, Portland, OR, USA; Nephrat II, Exocell, Philadelphia, PA, USA and Cayman Chemical Company, Ann Arbor, MI, USA, respectively).
Isometric tension studies
Aorta were rapidly excised and freed of connective tissue. The aorta was divided into two segments (4 mm width). Ring segments were mounted between stainless steel triangles into individual organ chambers filled with oxygenated (95% O2, 5% CO2) modified Krebs-Henseleit buffer (37°C, pH 7.4) (composition in mM: NaCl 119; KCl 4.7; CaCl2 1.6; MgSO4 1.2; NaHCO3 25; KH2PO4 1.2; glucose 11.0). Indomethacin (10 µM) was present in the experiments to inhibit prostaglandin synthesis. Aortic rings were subject to a resting tension of 14.7 mN. After equilibration for 60 min, all vessels were preconstricted with 0.3 µM phenylephrine. Ligand-stimulated receptor-mediated NO bioavailability was assessed by a concentration-dependent relaxation to acetylcholine (ACh, 10−9 to 10−2 M), whereas sodium nitroprusside (SNP, 10−9 to 10−2 M) was used as an endothelium-independent agonist. Relaxation responses to ACh and SNP were expressed as percentage of relaxation from a submaximal phenylephrine-induced constriction and concentration–response curves were obtained as previously described (Sena et al., 2008, 2009).
Detection of superoxide anion
Unfixed frozen, 30 µm-thick sections of proximal aorta were incubated with dihydroethidium (DHE) (2 × 10−6 M) in PBS for 30 min at 37°C in a humidified chamber protected from light. DHE is oxidized on reaction with O2·- to ethidium bromide (EtBr), which binds to DNA in the nucleus and fluoresces red (Miller et al., 1998). Polyethylene glycol-superoxide dismutase (PEG-SOD, 500 U·mL−1) abolished EtBr fluorescence, confirming specificity of the fluorescent signal for O2·- (data not shown). For EtBr detection, images were obtained with a fluorescence microscope (Leica DMIRE200, Wetzlar, Germany). Fluorescence was detected with a 568 nm filter. Normal and diabetic tissues were processed and imaged in parallel with identical settings. Microscope and camera settings were kept constant for all preparations. Fluorescence was quantified using ImageJ (1.40 g, NIH).
Assessment of aortic immunofluorescence
Sections (6 µm) of abdominal aorta were washed with PBS and fixed in ice-cold acetone for 10 min. Sections were then permeabilized for 10 min in 1% Triton X-100 in PBS, pH 7.4, and blocked with 10% goat serum for 30 min. Primary antibodies were diluted in PBS containing 0.02% BSA (PBS/BSA). The primary antibodies were added, and the sections were incubated overnight at 4°C. After incubation, the sections were extensively washed with PBS/BSA solution. After, sections were incubated with the secondary antibodies and diluted in PBS/BSA for 1 h. The coverslips were washed before mounting with Glycergel Dako mounting medium (Dako, Carpinteria, CA, USA). Immunostained aortic sections were visualized with a Leica DMIRE200 fluorescence microscope. Immunostained aortic sections were counterstained with 4′,6-diamidino-2-phenylindole and examined, photographed and quantified as described above for DHE fluorescence.
Aortic nitrite levels
Nitrite levels were determined as an index of NO generation in aortic homogenates by the Griess reaction after conversion of nitrate to nitrite by nitrate dehydrogenase (Green et al., 1982) as previously described (Majithiya et al., 2005). An aliquot of the supernatant was mixed with an equal volume of Griess reagent (sulfanilamide 1% w/v; naphtylethylenediamine dihydrochloride, 0.1% w/v; and orthophosphoric acid, 25% v/v) and incubated at room temperature for 10 min. The absorbance of the samples at 540 nm was determined and compared with those of known concentrations of sodium nitrite. The amount of nitrite formed was normalized to the protein content of the respective aorta.
Determination of advanced glycation end products (AGEs)
After incubation for the required time, AGE production was assessed by fluorescence and Western blotting. The fluorescence was assessed with excitation at 370 nm and emission at 440 using a microplate reader (Synergy HT, Biotek Instruments, Alcobendas, Madrid, Spain). The amounts of carboxymethyl-lysine (CML) were measured by Western blotting using anti-CML antibody, as described previously (Röcken et al., 2003).
Western blot analysis
Segments of endothelium-intact thoracic aortas were washed with cold PBS and chilled in buffer containing in mM the following: Tris–HCl 50, NaCl 150, EDTA 1, EGTA 0.1, as well as NP-40, 0.1%, SDS 0.1% and deoxycholate 0.5%. Phenylmethylsulfonyl fluoride (1 mM), aprotonin (10 µg·mL−1), leupeptin (10 µg·mL−1) and pepstatin (10 µg·mL−1) all from Sigma Chemicals (St. Louis, MO, USA) were added as the protease inhibitors. Tissues were homogenized in a standard fashion followed by centrifugation at 14 000×g for 20 min at 4°C. The supernatants were collected, and total protein concentration was determined. Samples containing 40 µg of protein were loaded on to a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel, run and electroblotted onto polyvinylidene difluoride membrane. Prestained molecular weight marker proteins were used as standards for the SDS-PAGE. Ponceau staining was performed to confirm the quality of the transfer and to ensure equal protein loading. Blots were blocked in 5% skimmed nonfat milk in PBS for 1 h, treated overnight with antibody against CML, VASP, pVASP or CCL2 and then incubated with alkaline phosphatase secondary antibodies for 1 h. Anti-VASP phosphoserine 239 antibody was used for the analysis of the phosphorylation state of VASP at Ser239 (pVASP), which is a reliable biochemical marker of vascular cGMP-dependent protein kinase-1 activity. Commercial VASP (20 ng) was used as an internal standard for Western blotting. Activation of VASP was indicated by the intensity ratio pVASP/VASP. Immunoblots were developed with an ECF Western blotting detection system (Amersham Biosciences, Carnaxide, Portugal).
Protein content was determined using a Bio-Rad protein assay kit.
Statistical analysis
All data were analysed by standard computer programs (GraphPad Prism PC Software version 3.0, anova) and are expressed as mean ± SE (n = 6–16 individual animals per group). Significant differences were evaluated using either the t-test or anova. P < 0.05 was considered significant. Dose–response curves were fitted by nonlinear regression with simplex algorithm. Relaxation responses were given as the percentage of phenylephrine preconstriction. Comparisons of dose–response curves were evaluated by two-way anova for repeated measures.
Materials
1,1-Dimethylbiguanide hydrochloride (metformin), phenylephrine, acetylcholine, N-nitro-L-arginine- methyl ester (L-NAME) and polyethylene glycol-superoxide dismutase were obtained from SIGMA (St. Louis, MO, USA). Anti-CML, VASP, pVASP and nitrotyrosine were obtained from Trans Genic, Inc. (Tokyo, Japan), Cell Signaling Technology (Danvers, MA, USA) and Upstate Biotechnology (Lake Placid, NY, USA) respectively. Anti-CCL2 and β-actin were obtained from Chemicon International, Inc. (Temecula, CA, USA), whereas DHE was obtained from Invitrogen (Barcelona, Spain). All other chemicals and reagents used in the study were of high grade.
Results
Animal characteristics
The rats used in our experiments exhibited similar fasting plasma glucose levels at the beginning of the study. Food consumption and water intake did not significantly change over the experimental period between the different groups studied (data not shown). Body weight was significantly lower in GK rats compared with age-matched Wistar rats. GK + AD significantly increased body weight, while metformin treatment (W + M; GK + M; GK + AD + M) did not change this parameter (Table 1). Glycaemia, fasting and 2 h after glucose load and FFA during the post-absorptive state were elevated in GK and GK + AD rats when compared with their corresponding control Wistar values (Figure 1A,B and Table 1). Treatment with metformin for 4 weeks effectively reduced fasting glucose and circulating concentrations of FFA in diabetic rats (Figure 1A and Table 1).
Table 1.
Body weight, and plasma lipid levels in 6 months old nondiabetic Wistar and diabetic Goto-Kakizaki (GK) rats and in GK rats fed with atherogenic diet (AD) with (W + M; GK + M; GK + M + AD) or without metformin treatment (W; GK; GK + AD)
| Wistar | W + M | GK control | GK + M | GK + AD | GK + AD + M | |
|---|---|---|---|---|---|---|
| Body weight (g) | 403.9 ± 6.9 | 439.6 ± 7.4 | 312.7 ± 5.8a | 351 ± 8.9 | 387.3 ± 4.3b | 383.6 ± 10.6b |
| Total cholesterol (mM) | 1.82 ± 0.04 | 2.46 ± 0.17c | 2.3 ± 0.05c | 2.6 ± 0.03a | 2.6 ± 0.14a | 2.79 ± 0.08a,b |
| Non-HDL cholesterol (mM) | 0.7 ± 0.04 | 1.1 ± 0.05 | 0.71 ± 0.03 | 0.99 ± 0.02 | 1.29 ± 0.12a,b | 1.5 ± 0.07a,b |
| Triglycerides (mM) | 1.23 ± 0.08 | 1.24 ± 0.1 | 1.69 ± 0.13 | 0.6 ± 0.02d | 2.52 ± 0.23a,d | 2.99 ± 0.13a,b |
| FFA (mM) | 0.71 ± 0.08 | 0.69 ± 0.05 | 0.79 ± 0.04 | 0.53 ± 0.02 | 1.13 ± 0.04a,b | 0.77 ± 0.05e |
Data are expressed as mean ± SE (n = 6–16 animals in each group).
P < 0.001 versus Wistar rats.
P < 0.001 versus GK control group.
P < 0.01 versus Wistar rats.
P < 0.01 versus GK control group.
P < 0.001 versus GK + AD group.
Figure 1.
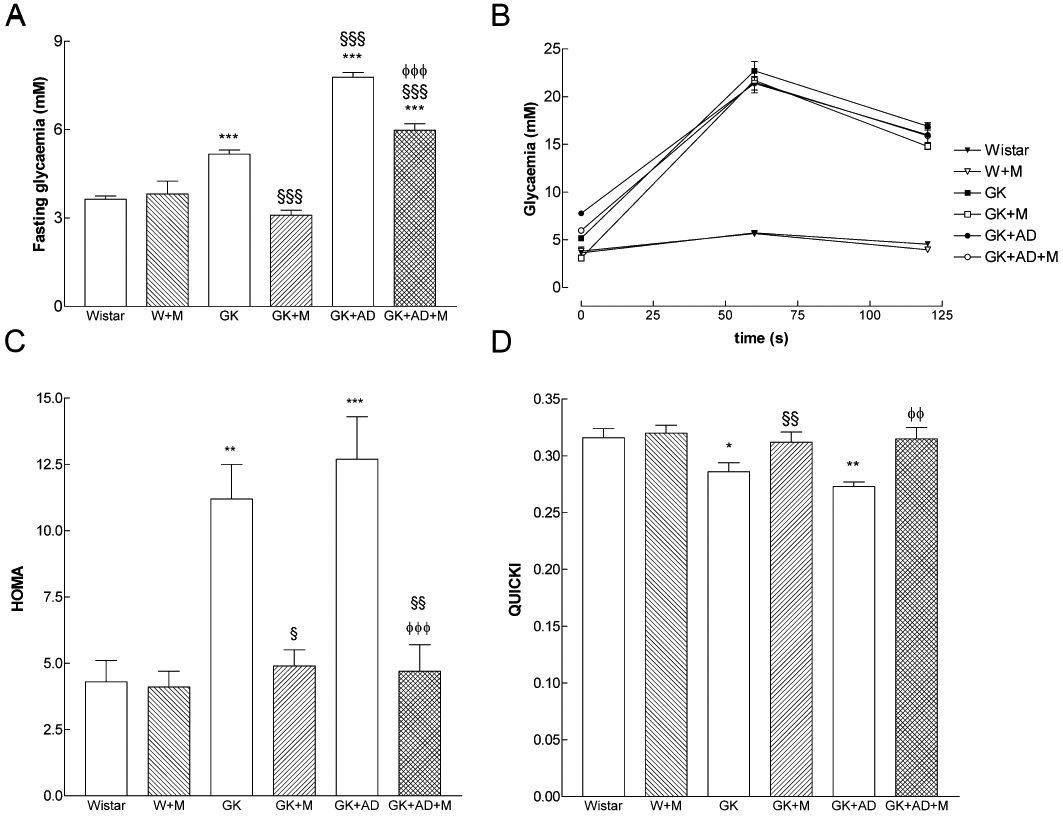
Influence of metformin on fasting glycaemia (A), blood glucose levels during an intraperitoneal glucose tolerance test (B), homeostasis model assessment of insulin resistance (HOMA, C) and quantitative insulin-sensitivity check index (QUICKI, D) in normal (GK) and high-fat fed GK rats (GK + AD) as compared with nondiabetic Wistar rats. Blood glucose levels were determined at the end of treatment with metformin. The blood glucose was determined using a blood glucose monitoring system at 0, 60 and 120 min after the glucose injection. Results are mean ± SE (n = 6–16 animals in each group). *P < 0.05, **P < 0.01, ***P < 0.001 versus Wistar group; §P < 0.05, §§P < 0.01, §§§P < 0.001 versus GK control group; φφP < 0.01, φφφP < 0.001 versus GK + AD group.
HOMA was significantly higher in GK and GK + AD rats when compared with Wistar rats and significantly reduced to nondiabetic levels after treatment with metformin (Figure 1C). In addition, QUICKI sensitivity index was significantly lower in GK and GK + AD groups than in Wistar rats and significantly increased in the diabetic groups treated with metformin (Figure 1D).
Although GK rats exhibited normal non-HDL-cholesterol and triglycerides, GK rats fed with the high-fat diet had elevated levels of these variables, compared with GK rats fed the control diet (control diabetics). The plasma levels of total cholesterol remained high in GK + AD rats and did not change in the diabetic groups treated with metformin, while triglycerides were significantly reduced in the GK + M group (Table 1).
NO-dependent vascular relaxation in rat aorta
In 6 month old GK rats, endothelium-mediated vascular relaxation of phenylephrine-precontracted aorta arterial rings in response to ACh was impaired compared with age-matched Wistar rats, but the endothelium-independent relaxations to SNP were similar in both strains (Figure 2A, B). Preincubation of the arterial rings with the NOS inhibitor L-NAME and the cyclooxygenase inhibitor indomethacin almost completely abolished relaxation by ACh in GK rats and Wistar rats (Figure S1 online supplement). The residual component due to other vasodilators is around 15% in our experimental conditions. High-fat diet further impaired vascular relaxation in response to ACh in GK rats. Indeed, maximal endothelium-mediated relaxation of phenylephrine-precontracted rings in response to ACh declined by 42% (Figure 2A). No differences on maximal relaxation were observed in the concentration–effect curves for SNP between any of the groups of rats. Vascular sensitivity to SNP was decreased in GK + AD rats (Figure 2B, Table 2). The metformin treatment normalized endothelium-dependent and independent vascular relaxation (Figure 2A, B). Detailed data on maximal relaxations and EC50 values are summarized in Table 2. These results indicated that treatment with metformin normalized this index of endothelial function in normal and high-fat fed diabetic GK rats. Note that treating normal Wistar rats with metformin did not change endothelium-mediated relaxation in response to ACh (Figure 2A).
Figure 2.
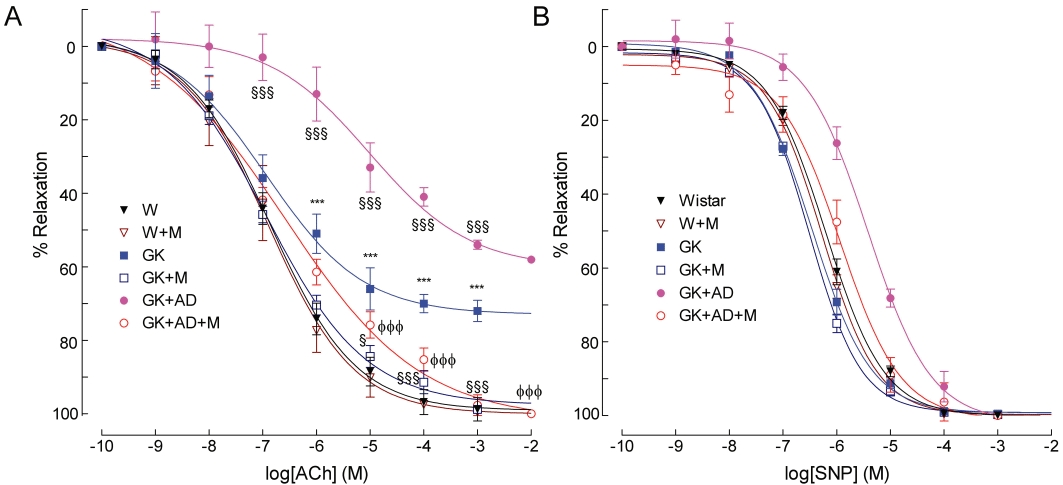
Effects of high-fat diet and metformin treatment on relaxant responses to acetylcholine (A) and sodium nitroprusside (B) in aortic segments from GK rats, compared with those from nondiabetic Wistar rats, after phenylephrine preconstriction. Relaxation of aortic rings was measured using an isometric force displacement transducer. Data are expressed as mean ± SE (n = 6–16). ***P < 0.001 versus Wistar group; §§§P < 0.001 versus GK control group; φφφP < 0.001 versus GK + AD group.
Table 2.
Maximal relaxation responses (%) and pEC50 (-logEC50) in isolated aorta arteries of 6 month old variously treated spontaneously diabetic Goto-Kakizaki (GK) rats and age-matched nondiabetic Wistar (W) rats
| Wistar | W + M | GK control | GK + M | GK + AD | GK + AD + M | |
|---|---|---|---|---|---|---|
| Ach | ||||||
| pEC50 | 6.87 ± 0.04 | 6.88 ± 0.02 | 6.94 ± 0.09 | 6.94 ± 0.05 | 5.02 ± 0.14a,b | 6.6 ± 0.17c |
| Maximal relaxation (%) | 98.9 ± 4.8 | 99.9 ± 3.8 | 72 ± 2.1a | 98.8 ± 3.9b | 58 ± 1.8a,b | 99.7 ± 4.2b,c |
| SNP | ||||||
| pEC50 | 6.20 ± 0.04 | 6.27 ± 0.3 | 6.43 ± 0.04 | 6.51 ± 0.03 | 5.42 ± 0.03a,b | 5.93 ± 0.11b,c |
| Maximal relaxation (%) | 99.95 ± 0.11 | 99.75 ± 0.2 | 99.5 ± 0.12 | 99.3 ± 0.3 | 99.41 ± 0.4 | 99.9 ± 0.12 |
Data are expressed as mean ± SE (n = 6–16 animals in each group). pEC50 values are presented as the negative logarithm (–logEC50) of concentration of the agonist.
P < 0.001 versus Wistar rats.
P < 0.001 versus GK control group.
P < 0.001 versus GK + AD group.
P < 0.05 versus GK control group.
Systemic oxidative stress biomarkers
Urinary levels of 8-OHdG were significantly higher in GK rats, when compared to age-matched Wistar rats, while plasma levels of protein carbonyl compounds were not statistically different between the two groups (Table S1, online supplement). Feeding AD diet for 4 months to GK rats significantly increased protein carbonyl compounds levels by 62% and did not change the levels of 8-OHdG (Table S1, online supplement). Supplementation with metformin significantly reduced 8-OHdG and protein carbonyl compounds levels (Table S1, online supplement).
Oxidative stress in the vascular wall
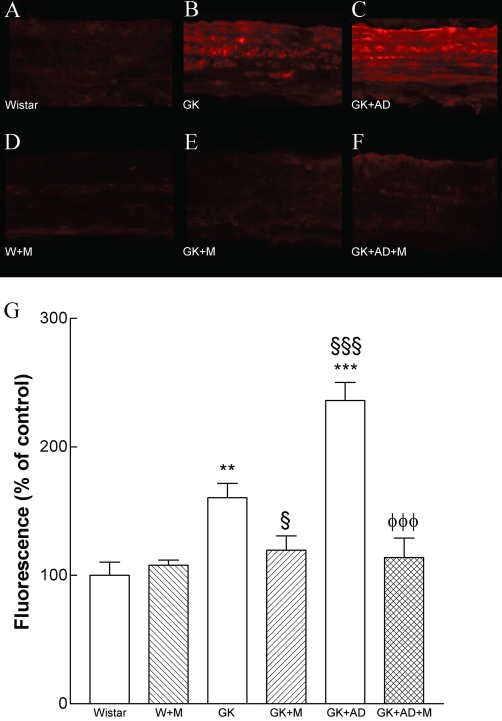
We determined the potential impact of metformin on the generation of superoxide anion in the diabetic vasculature assessed by DHE. Interestingly, diabetes induced a 2.8-fold increase in superoxide production in diabetic aorta (P < 0.001; Figure 3B, G). The density of DHE was 1.5-fold higher in the aorta of diabetic rats fed with high-fat diet compared with GK controls (P < 0.001; Figure 3C, G). Metformin significantly reduced superoxide production in normal and high-fat fed diabetic GK rats (P < 0.01; P < 0.001, Figure 3E, F, G). L-NAME (100 µM) significantly reduced superoxide production in diabetic GK rats fed with high-fat diet (data not shown). Accordingly, we sought to investigate whether the enhanced O2•− production in aortic tissue of GK rats was associated with peroxynitrite formation and the nitration of tyrosine residues. Diabetic GK rats also had increased immunoreactive nitrotyrosine levels in their aortas, suggesting increased peroxynitrite-mediated protein oxidation (Figure 4B). High-fat diet significantly increased nitrotyrosine staining in the GK + AD rats (Figure 4C).
Figure 3.
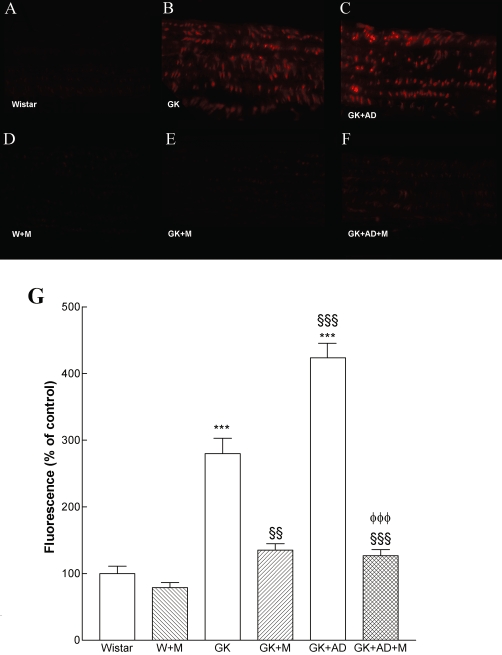
In situ detection of superoxide anion in rat aorta. Representative DHE-stained aorta artery sections reflect O2·- production with the different treatments. The endothelium is facing up in all layers. At identical settings, fluorescence in diabetic GK and GK + AD aorta (B and C respectively) was markedly increased, compared with vessels form normal rats (Wistar, A). Note the increased fluorescence reflecting O2·- levels in the endothelium, intima and media of GK aorta. DHE fluorescence decreased to basal (Wistar) levels in the GK + M (E) and GK + DA + M treated groups (F). Panel G shows quantification of the fluorescence ethidium signal in the different groups of arteries. Data are mean ± SE. ***P < 0.001 versus Wistar group; §§P < 0.01, §§§P < 0.001 versus GK control group; φφφP < 0.001 versus GK + AD group.
Figure 4.
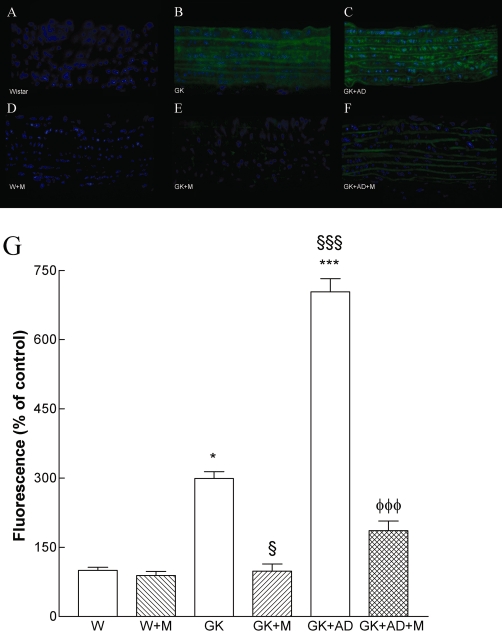
Nitrotyrosine expression is enhanced in aortas from diabetic GK and in GK + AD rats, indicative of increased peroxynitrite formation. Representative aortic sections showing nitrotyrosine staining in control Wistar (A), diabetic GK (B), diabetic GK fed with AD diet (C), Wistar treated with metformin (W + M; D) and diabetic GK rats with normal GK + M (E), and high-fat fed diet GK + AD + M (F) rats treated with metformin. The endothelium is facing up in all layers. Panel G shows quantification of the green fluorescence in the different groups of arteries. Data are mean ± SE. *P < 0.05, ***P < 0.001 versus Wistar group; §P < 0.01, §§§P < 0.001 versus GK control group; φφφP < 0.001 versus GK + AD group.
NO metabolites were assessed in aortic homogenates using the Griess reaction. Acetylcholine-stimulated NO synthesis was significantly decreased in GK rats when compared with the Wistar counterpart (Table S1, online supplement) and further diminished with high-fat diet (GK + AD). As with other parameters, we observed a reversal with metformin treatment (Figure 4E, F, G, Table S1, online supplement).
pVASP and total VASP in the aorta
The phosphorylation of VASP at Ser239 has been shown to be an indicator of bioactive NO and the activity of the NO–cGMP–protein kinase G signalling pathway (Oelze et al., 2000). To establish NO bioavailability, we measured pVASP and total VASP in the aorta. The ratio of pVASP/VASP in normal and high-fat fed GK rats was significantly decreased compared with that in Wistar rats (Figure 5A, B). Metformin treatment significantly increased pVASP/VASP ratio in diabetic GK rats (GK + M, GK + AD + M; Figure 5A, B). No changes were observed in the Wistar treated group.
Figure 5.
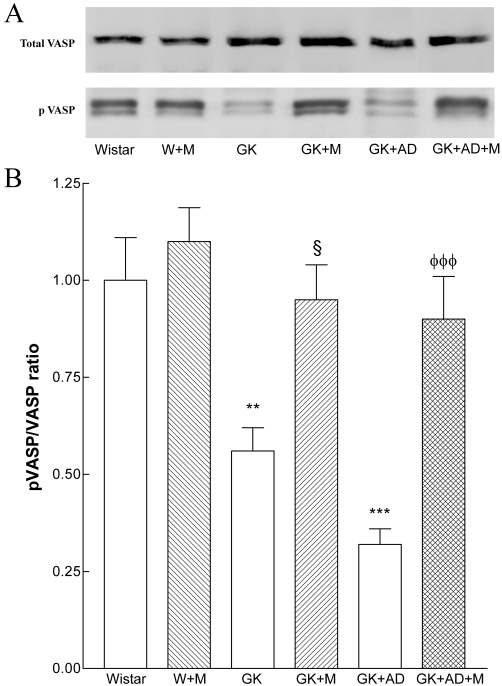
Effects of metformin on aortic pVASP expression. To examine NO–cGMP signal activation, total vasodilator-stimulated phosphoprotein (VASP) and phosphorylated (Ser239) VASP (pVASP) expression were assessed. Aortic lysates were analysed by SDS-PAGE. (A) Representative Western blot analyses of total VASP and pVASP expression in aortas of the different groups of arteries. (B) Averaged densitometric data for pVASP/total VASP ratio. Densitometry data are presented as mean ± SE. **P < 0.01, ***P < 0.001 versus Wistar group; §P < 0.05 versus GK control group; φφφP < 0.001 versus GK + AD group.
Diabetes-associated accumulation of AGEs
The accumulation of AGEs and CML, in settings such as diabetes is considered to play a role in progressive vascular injury. As expected, diabetic GK had a significant elevation in vascular AGE deposition when compared with the Wistar counterpart, as detected by immunostaining for the well-described AGE, CML (Figure 6A, B, G). CML immunostaining was further enhanced in high-fat fed GK rats (Figure 6C, G) and reduced in the aortas of metformin treated to levels not significantly different from control animals (Figure 6E, F, G). We also measured plasma AGEs concentrations (Figure 7A). Interestingly, plasma AGEs levels were higher in diabetic GK rats and GK + AD compared with nondiabetic W rats. A similar pattern was seen by Western blot evaluation of aortic AGE levels (Figure 7B–C) with increased expression AGEs in GK and GK + AD aortas, compared with control Wistar rats, and with normalization in metformin-treated GK rats (Figure 7).
Figure 6.
Effects of metformin on aortic carboxymethyl-lysine (CML) accumulation in Wistar (W + M) and diabetic GK rats (GK + M; GK + AD + M). Representative aortic sections demonstrating increased CML staining in the diabetic and GK + AD aortas. Panel presents aorta from control Wistar (A), diabetic GK (B), diabetic GK fed with AD diet (C), Wistar treated with metformin (D), GK treated with metformin (E) and diabetic GK + AD treated with metformin (F) rats. (G) Graph presenting percentage of CML-positive aortic area in each group. Data are mean ± SE. **P < 0.01, ***P < 0.001 versus Wistar group; §P < 0.05, §§§P < 0.001 versus GK control group; φφφP < 0.001 versus GK + AD group.
Figure 7.
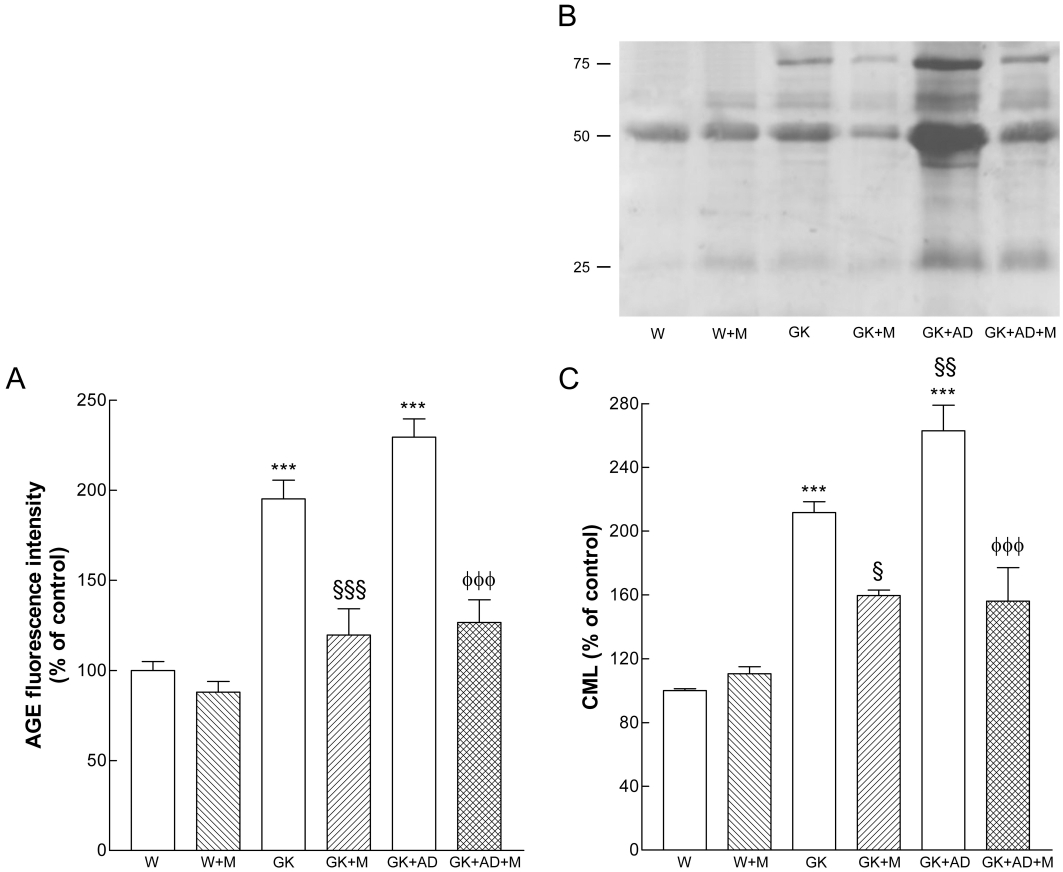
Effects of metformin on AGE levels in Wistar and diabetic GK rats fed with normal and AD diet. (A) Plasma fluorescence AGE intensity as a percentage of elevation over the control value (set to 100%). (B) Representative Western blot analyses of carboxymethyl-lysine (CML) expression in aortas of the different groups of arteries. (C) Averaged densitometric data for W + M and diabetic groups expressed as a percentage of elevation over the control value established as 100%. Data are mean ± SE. ***P < 0.001 versus Wistar group; §P < 0.05, §§§P < 0.001 versus GK control group; φφφP < 0.001 versus GK + AD group.
Marker of vascular inflammation
We next focused on a central mediator of inflammation and retrieved abdominal aortas of the different groups. Levels of the chemokine CCL2 (or MCP-1), one of the earliest molecular markers of vascular inflammation in atherogenesis (Piga et al., 2007), were significantly increased in the aortas of GK and GK + AD rats compared with nondiabetic Wistar rats (Figure 8A, B). By immunofluorescence, CCL2 expression was increased by two- and threefold in diabetic GK and GK + AD rats, respectively (Figure 9B, C).
Figure 8.
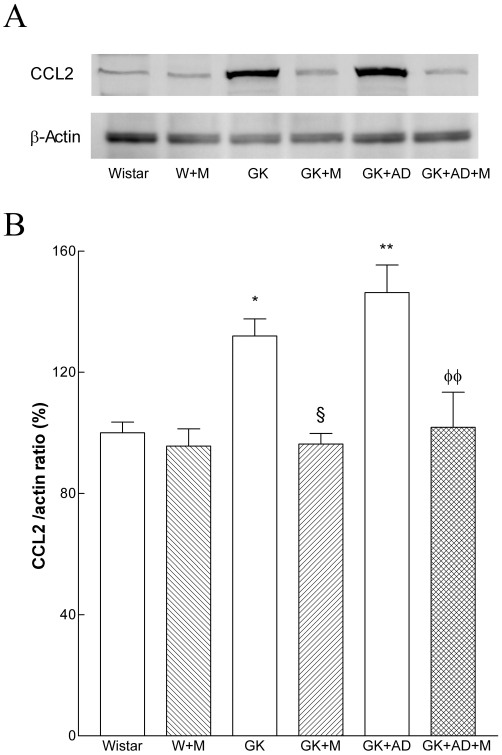
Effects of metformin on aortic contents of the chemokine CCL2 in Wistar and diabetic GK rats fed with normal and AD diet. (A) Representative Western blot analyses of CCL2 expressed in aortas of the different groups of rats. (B) Averaged densitometric data for W + M and diabetic groups expressed as a percentage of elevation over the control value (set to 100%), after first normalizing against actin. Data are expressed as mean ± SE. *P < 0.05, **P < 0.01 versus Wistar group; §P < 0.05 versus GK control group; φφP < 0.01, versus GK + AD group.
Figure 9.
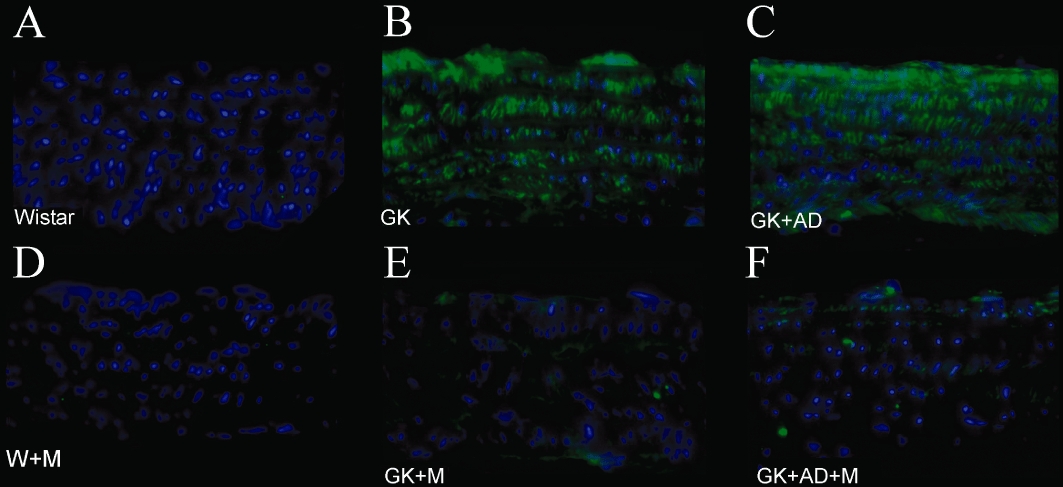
Effects of metformin on aortic CCL2 accumulation in Wistar and diabetic GK rats fed with normal and AD diet. Representative aortic sections demonstrating increased CCL2 staining in the diabetic and GK + AD aortas. Panel presents aorta from control Wistar (A), diabetic GK (B), diabetic GK fed with AD diet (C), Wistar treated with metformin (D), diabetic GK treated with metformin (E) and diabetic GK + AD treated with metformin (F) rats.
To assess the effects of metformin on expression of this inflammatory marker, we examined levels of expression of CCL2 and found that they were significantly attenuated in the aortae treated with metformin in GK and GK + AD rats (Figures 8A, B and 9E, F).
Discussion
Metformin is one of the most frequently used drugs to treat patients with type 2 diabetes. In addition to reducing hyperglycaemia and improving insulin sensitivity, it also independently promotes vasculoprotection (Katakam et al., 2000; Wiernsperger, 2000; Mather et al., 2001; Beisswenger and Ruggiero-Lopez, 2003; Iida et al., 2003). It has recently been suggested that metformin improves endothelial function in humans and animal models (Romualdi et al., 2008; Matsumoto et al., 2008). The precise mechanisms by which this drug prevents the development of atherogenesis have not yet been fully elucidated. Thus, we thought to investigate this oral antidiabetic drug on endothelial function, oxidative stress, inflammation and AGE formation in aorta of normal and high-fat fed GK diabetic rats.
Type 2 diabetes is associated with elevated levels of oxidative stress, glycation and endothelial dysfunction and these are further aggravated by a high-fat diet. Furthermore, metformin treatment was able to reduce oxidative stress as well as glycation and early inflammation observed in this animal model, and these actions were associated with a recovery in NO bioavailability and endothelial function in aorta.
In our GK rats fed with high-fat diet, plasma insulin, glucose, triglyceride, cholesterol, FFA levels and insulin resistance were increased compared with the corresponding values in the Wistar rats. When we administered metformin for 4 weeks, it did not improve the triglyceride or cholesterol levels, but it did significantly lower FFA levels and blood glucose. Nevertheless, these plasma glucose levels were still significantly higher than those of Wistar controls. However, the vasculoprotective actions of metformin were evident under these conditions, suggesting that the complete restoration of normoglycaemia was not a prerequisite for vasculoprotection. This is in accordance with findings of previous studies (Mather et al., 2001; Kirpichnikov et al., 2002; Matsumoto et al., 2008). It is also in agreement with the UK Prospective Diabetes Study clinical trial where metformin use was associated with decreases in macrovascular morbidity and mortality, effects that appear to be independent of the improvement in glycaemic control (UKPDS, 1998). Moreover, metformin significantly decreased the insulin resistance index, HOMA, and concomitantly increased insulin sensitivity index, QUICKI, which can partially explain the beneficial effects on vascular function. These results are consistent with other studies (Yoshida et al., 2009) that have recently reported that the major mechanism of metformin action is to stimulate cellular glucose utilization in GK rats.
Mechanisms that explain the beneficial effects of metformin on endothelial dysfunction are still undefined. Increased NO production/efficacy might be one logical mechanism, and some data exist, which point towards an involvement of this mediator. Metformin treatment results in increased production of NO by increasing AMP-activated protein kinase-dependent activation of endothelial nitric oxide synthase (eNOS) (Davis et al., 2006). In addition to enhancing NO production, metformin decreases circulating endothelin-1 levels in insulin-resistant women (Orio et al., 2005). Sartoretto and co-workers (2005) has reported that metformin increases NO activity, but not expression, and that it improves microvascular reactivity in type 2 diabetic animal models. It has also been suggested that metformin can favour NO efficacy indirectly (Davis et al., 2006), reducing plasma levels of asymmetric dimethylarginine (Asagami et al., 2002), a metabolite of arginine that is considered a good predictor of cardiovascular events in diabetes. Here we have shown a marked reduction in aortic NO metabolites and pVASP expression, an index of NO bioavailability (Oelze et al., 2000), during diabetes and diabetes with hyperlipidaemia. Metformin treatment was able to increase NO bioavailability and normalize endothelium-dependent relaxation in both normal and high-fat fed GK rats. Moreover, metformin had no effect when used in normal Wistar rats. The observation of a decrease in vascular NO bioavailability led us to measure O2•− anion, a well-known factor in the degradation of NO and its conversion to peroxynitrite (Pacher et al., 2007). In agreement with our previous studies in aged GK rats (Sena et al., 2008), we observed a significant reduction in the acetylcholine-induced relaxation of aortic rings in GK rats fed with high-fat diet and an increment in vascular O2•− anion and nitrotyrosine staining in aortic rings. Metformin significantly reduced O2•− anion and nitrotyrosine accumulation in aortic arteries along with a reduction in oxidative stress. These effects are observed in normal and high-fat fed GK diabetic rats.
Oxidative stress is now considered to play a key role in metabolic and vascular derangements in diabetes with an imbalance arising from exaggerated production and reduced elimination of radicals. In this study, we present evidence that metformin exerts an antioxidative effect in vivo: the amounts of protein carbonyl compounds in serum, urinary 8-OHdG and of tissue O2•− anion and nitrotyrosine accumulation were diminished by metformin in diabetic high-fat fed GK rats. This antioxidative effect of metformin is one important mechanism underlying the normalization in endothelial dysfunction observed. Several studies have reported the inhibitory effect of metformin on oxidative stress under various conditions: in vitro in either hyperglycaemic or hyperlipidaemic environments as well as in vivo (Gargiulo et al., 2002; Bellin et al., 2006; Faure et al., 1999; Matsumoto et al., 2008; Scarpello and Howlett, 2008). It inhibits oxidative stress production predominantly in mitochondria (Batandier et al., 2006). Recently, it was reported that metformin may also diminish oxidative stress–related DNA damage (Kanigür-Sultuybek et al., 2007). Also, metformin treatment (100 mg·kg−1·day−1 during 6 weeks) diminished the amounts of oxidized proteins in serum and of tissue lipid peroxides in 12 week old diabetic GK rats (Rösen and Wiernsperger, 2006). The reduction of oxidative stress by metformin may partly be due to inhibition of glycation – a process that directly causes free-radical production. A recent study suggests that the intracellular oxidant properties of metformin may result in the inhibition of both the receptor for AGEs and the lectin-like oxidized receptor 1 (Ouslimani et al., 2007).
In diabetic animals, oxidative stress is elevated in proportion to the accumulation of AGEs (Forbes et al., 2002). AGEs impair endothelial-dependent vascular relaxation mainly by reducing the bioavailability and activity of NO. In the present study, we show that chronic administration of high-fat diet to GK rats significantly increased serum and aortic oxidative stress parameters, enhanced CML accumulation in aorta and caused early inflammation in the arterial wall, leading to a significant decrease in endothelial dependent vasorelaxation and NO bioavailability. Metformin treatment normalized endothelial dysfunction in normal and high-fat fed GK rats increasing NO bioavailability and decreasing oxidative stress and AGEs formation. Metformin did not enhance endothelium-dependent relaxation in normal Wistar rats treated with this antidiabetic drug. Thus, it is not a general endothelium-dependent relaxation effect of metformin, but specifically, it restored endothelial function in diabetic GK rats.
Long-term benefit from metformin could be due to its anti-glycating properties. Glycation of proteins inhibits their function and leads to accumulation of extracellular matrix material. Due to its chemical structure, metformin can directly bind some potent glycating sugars such as methylglyoxal. It forms an adduct that has been identified (Ruggiero-Lopez et al., 1999) and detected in blood from metformin-treated diabetic patients (Beisswenger and Ruggiero-Lopez, 2003). Since non-enzymatic glycation is considered as a key pathological process in the development of diabetic vascular complication, this unique action of metformin is of major significance.
The idea of a ‘metabolic memory’ suggests that agents reducing cellular reactive oxygen species and glycation might be ideal to minimize long-term vascular complications of diabetes and emphasizes the central place metformin has in this medical setting (Ihnat et al., 2007). Most importantly, almost all the effects, whether on macro- or microcirculatory events, have also been demonstrated in non-hyperglycaemic conditions, i.e. in insulin-resistant or in ischaemic situations (Scarpello and Howlett, 2008). These properties are intrinsic to metformin and therefore add to the benefit obtained by its improvement of metabolic control.
In vitro studies have shown that metformin is able to significantly inhibit AGE formation (Ruggiero-Lopez et al., 1999; Kiho et al., 2005). Chronic treatment with metformin can also reduce AGE levels in lens, kidney and nerves in diabetic animals (Tanaka et al., 1999). Several mechanisms by which metformin inhibit glycation processes have been proposed. It is suggested that metformin traps reactive carbonyl species such as methylglyoxal and glyoxal (Ruggiero-Lopez et al., 1999; 2000;). It has also been proposed that metformin reduces methylglyoxal levels enhancing methylglyoxal detoxification through the glyoxalase pathway (Beisswenger et al., 1999). Metformin may also react with post-Amadori products (Rahbar et al., 2000).
In addition, metformin not only improves the antioxidant defence but is also a powerful activator of AMP-kinase (Zou et al., 2004), which is a major regulator of glucose and lipid metabolism and of glucose-6-phosphate dehydrogenase (Leverve et al., 2003). The unique efficacy in the treatment of hyperglycaemia and insulin resistance–dependent complications is presumably not only an expression of one mechanism alone but of the diverse and manifold properties of this compound (Kirpichnikov et al., 2002; Seufert et al., 2004]. Previous studies also show early evidence of metformin actions beyond its effects on glucose metabolism, including reduction of plasminogen activator inhibitor-1, von Willebrand factor and smooth muscle cell contractility via either agonist-induced increase in intracellular [Ca2+] or a secondary increase in nitric oxide (Nagi and Yudkin, 1993; Bhalla et al., 1996; Dominguez et al., 1996).
Clinical studies further suggest that metformin may alter inflammation as determined by decreased inflammatory markers in plasma, including soluble intercellular adhesion molecule, vascular cell adhesion molecule-1, nuclear factor κ-B and C-reactive protein in some cases of polycystic ovary syndrome, indicating modulation of inflammation (Morin-Papunen et al., 2003; Caballero et al., 2004; Isoda et al., 2006). It has also been shown that metformin causes a reduction in the plasma concentrations of macrophage migration inhibitory factor in obese subjects (Dandona et al., 2004). In this work, we present evidence that levels of the chemokine CCL2 were increased in diabetic rats (GK and GK + AD) and were significantly attenuated after metformin treatment, indicating an inhibition of early inflammation in aorta of type 2 diabetic rats.
The unique efficacy of metformin in the normalization (with a reversal of 40%) of aortic endothelial dysfunction is presumably not only an expression of one mechanism alone but of the diverse and manifold properties of this antidiabetic compound.
We conclude that, in GK rats fed with a high-fat diet, endothelial dysfunction is very clearly expressed in the aorta. Metformin normalized endothelial dysfunction enhancing NO bioavailability and reducing oxidative stress, nitrotyrosine formation and protein glycation and inflammation in the aorta, in control and high-fat fed diabetic GK rats. These findings support the concept of the central role of metformin as a first line treatment for diabetic patients in order to protect against endothelial dysfunction associated with type 2 diabetes mellitus. This has important implications for the investigation and treatment of vascular disease in patients with type 2 diabetes mellitus.
Acknowledgments
We wish to thank Prof João Patrício and coworkers (Laboratory of Animals Research Center, University Hospitals, Coimbra) for maintaining the animals. We also wish to thank Ilda da Conceição (Department of Pathological Anatomy, University Hospitals of Coimbra) for their technical assistance in histological preparations. The present work was supported by the Interdisciplinary Research Institute, Faculty of Medicine, University of Coimbra and University Hospitals of Coimbra, Portugal.
Glossary
Abbreviations
- 8- OHdG
8-hydroxy-2′-deoxyguanosine
- AD
atherogenic diet
- AGEs
advanced glycation end products
- CCL2 (MCP-1)
monocyte chemoattractant protein-1
- CML
carboxymethyl-lysine
- DHE
dihydroethidium
- eNOS
endothelial nitric oxide synthase
- FFA
free fatty acids
- GK rats
Goto-Kakizaki rats
- L-NAME
N-nitro-L-arginine- methyl ester
- pVASP
phosphorylated vasodilator-stimulated phosphoprotein
- SNP
sodium nitroprusside
- VASP
vasodilator-stimulated phosphoprotein
Conflict of interest
The authors report no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of metformin treatment on vasodilatory responses to acetylcholine in the presence of Nω-nitro-L-arginine methyl ester (L-NAME) 100 µM during 30 min after phenylephrine preconstriction of aortic segments. Vasorelaxation was measured using an isometric force displacement transducer. Data are expressed as mean ± SE (n = 16, 32 vascular ring preparations in 16 animals per group).
Table S1 Effects of metformin on systemic oxidative stress parameters and nitrite/nitrate levels in aortic tissue
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Asagami T, Abbasi F, Stuelinger M, Lamendola C, McLaughlin T, Cooke JP, et al. Metformin treatment lowers asymmetric dimethylarginine concentrations in patients with type 2 diabetes. Metabolism. 2002;51:843–846. doi: 10.1053/meta.2002.33349. [DOI] [PubMed] [Google Scholar]
- Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr. 2006;38:33–42. doi: 10.1007/s10863-006-9003-8. [DOI] [PubMed] [Google Scholar]
- Beisswenger P, Ruggiero-Lopez D. Metformin inhibition of glycation processes. Diabetes Metab. 2003;29:6S95–6103. doi: 10.1016/s1262-3636(03)72793-1. [DOI] [PubMed] [Google Scholar]
- Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999;48:198–202. doi: 10.2337/diabetes.48.1.198. [DOI] [PubMed] [Google Scholar]
- Bellin C, de Wiza BH, Wiernsperger NF, Rösen P. Generation of reactive oxygen species by endothelial and smooth muscle cells: influence of hyperglycemia and metformin. Horm Metab Res. 2006;38:732–739. doi: 10.1055/s-2006-955084. [DOI] [PubMed] [Google Scholar]
- Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. Vascular effects of metformin. Possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens. 1996;9:570–576. doi: 10.1016/0895-7061(95)00356-8. [DOI] [PubMed] [Google Scholar]
- Caballero AE, Delgado A, Aguilar-Salinas CA, Herrera AN, Castillo JL, Cabrera T, et al. The differential effects of Metformin on markers of endothelial activation and inflammation in subjects with impaired glucose tolerance: a placebo-controlled, randomized clinical trial. J Clin Endocrinol Metab. 2004;89:3943–3948. doi: 10.1210/jc.2004-0019. [DOI] [PubMed] [Google Scholar]
- Cheng ZJ, Vaskonen T, Tikkanen I, Nurminen K, Ruskoaho H, Vapaatalo H, et al. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats. Hypertension. 2001;37:433–439. doi: 10.1161/01.hyp.37.2.433. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial nitric oxide synthase activity. Cardiovasc Res. 1999;43:274–278. doi: 10.1016/s0008-6363(99)00134-0. [DOI] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Ghanim H, Mohanty P, Tripathy C, Hofmeyer D, et al. Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab. 2004;89:5043–5047. doi: 10.1210/jc.2004-0436. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000a;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vriese AS, Van De Voorde J, Blom HJ, Vanhoutte PM, Verbeke M, Lameire NH. The impaired renal vasodilator response attributed to endothelium-derived hyperpolarizing factor in streptozotocin-induced diabetic rats is restored by 5-methyltetrahydrofolate. Diabetologia. 2000b;43:1116–1125. doi: 10.1007/s001250051502. [DOI] [PubMed] [Google Scholar]
- Ding H, Hashem M, Wiehler WB, Lau W, Martin J, Reid J, et al. Endothelial dysfunction in the streptozotocin-induced diabetic apoE deficient mouse. Br J Pharmacol. 2005;146:1110–1118. doi: 10.1038/sj.bjp.0706417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez LJ, Davidoff AJ, Srinivas PR, Standley PR, Walsh MF, Sowers JR. Effects of metformin on tyrosine kinase activity, glucose transport, and intracellular calcium in rat vascular smooth muscle. Endocrinology. 1996;137:113–121. doi: 10.1210/endo.137.1.8536601. [DOI] [PubMed] [Google Scholar]
- Faure P, Rossini E, Wiernsperger N, Richard MJ, Favier A, Halimi S. An insulin sensitizer improves the free radical defense system potential and insulin sensitivity in high fructose-fed rats. Diabetes. 1999;48:353–357. doi: 10.2337/diabetes.48.2.353. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME, Thallas V, Burns WC, Thomas MC, Brammar GC, et al. Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes. 2002;51:3274–3282. doi: 10.2337/diabetes.51.11.3274. [DOI] [PubMed] [Google Scholar]
- Gargiulo P, Caccese D, Pignatelli P, Brufani C, De Vito F, Marino R, et al. Metformin decreases platelet superoxide anion production in diabetic patients. Diabetes Metab Res Rev. 2002;18:156–159. doi: 10.1002/dmrr.282. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite and 15[N] nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Ihnat MA, Thorpe JE, Ceriello A. Hypothesis: the ‘metabolic memory’, the new challenge of diabetes. Diabet Med. 2007;24:582–586. doi: 10.1111/j.1464-5491.2007.02138.x. [DOI] [PubMed] [Google Scholar]
- Iida KT, Kawakami Y, Suzuki M, Shimano H, Toyoshima H, Sone H, et al. Effect of thiazolidinediones and metformin on LDL oxidation and aortic endothelium relaxation in diabetic GK rats. Am J Physiol. 2003;284:E1125–E1130. doi: 10.1152/ajpendo.00430.2002. [DOI] [PubMed] [Google Scholar]
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin inhibits proinflammatory responses and nuclear Factor-κB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26:611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- Kanigür-Sultuybek G, Ozdas SB, Curgunlu A, Tezcan V, Onaran I. Does metformin prevent short-term oxidant-induced DNA damage? In vitro study on lymphocytes from aged subjects. J Basic Clin Physiol Pharmacol. 2007;18:129–140. doi: 10.1515/jbcpp.2007.18.2.129. [DOI] [PubMed] [Google Scholar]
- Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000;35:108–112. doi: 10.1161/01.hyp.35.1.108. [DOI] [PubMed] [Google Scholar]
- Kiho T, Kato M, Usui S, Hirano K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta. 2005;358:139–145. doi: 10.1016/j.cccn.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137:25–33. doi: 10.7326/0003-4819-137-1-200207020-00009. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;46:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverve XM, Guigas B, Detaille D, Batandier C, Koceir EA, Chauvin C, et al. Mitochondrial metabolism and type-2 diabetes: a specific target of metformin. Diabetes Metab. 2003;29:6S88–6S94. doi: 10.1016/s1262-3636(03)72792-x. [DOI] [PubMed] [Google Scholar]
- Majithiya JB, Parmar AN, Balaraman R. Pioglitazone, a PPAR γ agonist, restores endothelial function in aorta of streptozotocin-induced diabetic rats. Cardiovasc Res. 2005;66:150–161. doi: 10.1016/j.cardiores.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Mather KJ, Verna S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37:1344–1350. doi: 10.1016/s0735-1097(01)01129-9. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada N, Kamata K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am J Physiol. 2008;295:H1165–H1176. doi: 10.1152/ajpheart.00486.2008. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- Morin-Papunen L, Rautio K, Ruokonen A, Hedberg P, Puukka M, Tapanainen JS. Metformin reduces serum C-reactive protein levels in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:4649–4654. doi: 10.1210/jc.2002-021688. [DOI] [PubMed] [Google Scholar]
- Nagi DK, Yudkin JS. Effects of metformin on insulin resistance, risk factors for cardiovascular disease, and plasminogen activator inhibitor in NIDDM subjects. A study of two ethnic groups. Diabetes Care. 1993;16:621–629. doi: 10.2337/diacare.16.4.621. [DOI] [PubMed] [Google Scholar]
- Nunes E, Peixoto F, Louro T, Sena CM, Santos MS, Matafome P, et al. Soybean oil treatment impairs glucose-stimulated insulin secretion and changes fatty acids composition of normal and diabetic islets. Acta Diabetol. 2007;44:121–130. doi: 10.1007/s00592-007-0252-8. [DOI] [PubMed] [Google Scholar]
- Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, et al. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000;87:999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- Orio JF, Palomba S, Cascella T, De Simone B, Manguso F, Savastano S, et al. Improvement in endothelial structure and function after metformin treatment in young normal-weight women with polycystic ovary syndrome: results of a 6-month study. J Clin Endocrinol Metab. 2005;90:6072–6076. doi: 10.1210/jc.2005-0965. [DOI] [PubMed] [Google Scholar]
- Ouslimani N, Mahrouf M, Peynet J, Bonnefont-Rousselot D, Cosson C, Legrand A, et al. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin-like oxidized receptor 1. Metabolism. 2007;56:308–313. doi: 10.1016/j.metabol.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannirselvam M, Wiehler WB, Anderson T, Triggle CR. Enhanced vascular reactivity of small mesenteric arteries from diabetic mice is associated with enhanced oxidative stress and cyclooxygenase products. Br J Pharmacol. 2005;144:953–960. doi: 10.1038/sj.bjp.0706121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis. 2007;193:328–334. doi: 10.1016/j.atherosclerosis.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Rahbar S, Natarajan R, Yerneni K, Scott S, Gonzales N, Nadler JL. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin Chim Acta. 2000;301:65–77. doi: 10.1016/s0009-8981(00)00327-2. [DOI] [PubMed] [Google Scholar]
- Röcken C, Kientsch-Engel R, Mansfeld S, Stix B, Stubenrauch K, Weigle B, et al. Advanced glycation end products and receptor for advanced glycation end products in AA amyloidosis. Am J Pathol. 2003;162:1213–1220. doi: 10.1016/S0002-9440(10)63917-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi D, Costantini B, Selvaggi L, Giuliani M, Cristello F, Macri F, et al. Metformin improves endothelial function in normoinsulinemic PCOS patients: a new prospective. Hum Reprod. 2008;23:2127–2133. doi: 10.1093/humrep/den230. [DOI] [PubMed] [Google Scholar]
- Rösen P, Wiernsperger NF. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto-Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes Metab Res Rev. 2006;22:323–330. doi: 10.1002/dmrr.623. [DOI] [PubMed] [Google Scholar]
- Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol. 1999;58:1765–1773. doi: 10.1016/s0006-2952(99)00263-4. [DOI] [PubMed] [Google Scholar]
- Ruggiero-Lopez D, Howell SK, Szwergold BS, Wiernsperger N, Beisswenger PJ. Metformin reduces methylglyoxal levels by formation of a stable condensation product (Triazepinone) Diabetes. 2000;49:A124. [Google Scholar]
- Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C, et al. Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci. 2005;77:2676–2689. doi: 10.1016/j.lfs.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Scarpello JH, Howlett HCS. Metformin therapy and clinical uses. Diab Vasc Dis Res. 2008;5:157–167. doi: 10.3132/dvdr.2008.027. [DOI] [PubMed] [Google Scholar]
- Seiça R, Suzuki K, Santos RM, Rosário LM. Deficiência primária da secreção de insulina de ilhéus isolados de ratos Goto-Kakizaki, um modelo animal de diabetes tipo 2 não obesa. Acta Med Port. 2004;17:42–48. [PubMed] [Google Scholar]
- Sena C, Barosa C, Nunes E, Seiça R, Jones JG. Sources of endogenous glucose production in the Goto-Kakizaki diabetic rat. Diabetes Metab. 2007;33:296–302. doi: 10.1016/j.diabet.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Sena CM, Nunes E, Louro T, Proença T, Fernandes R, Boarder MR, et al. Effects of α-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br J Pharmacol. 2008;153:894–906. doi: 10.1038/sj.bjp.0707474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena CM, Louro T, Matafome P, Nunes E, Monteiro P, Seiça RM. Antioxidant and vascular effects of gliclazide in type 2 diabetic rats fed high-fat diet. Physiol Res. 2009;58:203–209. doi: 10.33549/physiolres.931480. [DOI] [PubMed] [Google Scholar]
- Seufert J, Lubben G, Dietrich K, Bates PC. A comparison of the effects of thiazolidinediones and metformin on metabolic control in patients with type 2 diabetes mellitus. Clin Ther. 2004;26:805–818. doi: 10.1016/s0149-2918(04)90125-7. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Uchino H, Shimizu T, Yoshii H, Niwa M, Ohmura C, et al. Effect of metformin on advanced glycation endproduct formation and peripheral nerve function in streptozotocin-induced diabetic rats. Eur J Pharmacol. 1999;376:17–22. doi: 10.1016/s0014-2999(99)00342-8. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- Verma S, Bhanot S, McNeill JH. Antihypertensive effects of Metformin in fructose-fed hyperinsulinemic, hypertensive rats. J Pharmacol Exp Ther. 1994;271:1334–1337. [PubMed] [Google Scholar]
- Wiernsperger NF. Metformin: intrinsic vasculoprotective properties. Diabetes Technol Ther. 2000;2:259–272. doi: 10.1089/15209150050025230. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Okuno A, Tanaka J, Takahashi K, Nakashima R, Kanda S, et al. Metformin primarily decreases plasma glucose not by gluconeogenesis suppression but by activating glucose utilization in a non-obese type 2 diabetes Goto-Kakizaki rats. Eur J Pharmacol. 2009;623:141–147. doi: 10.1016/j.ejphar.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WG, 4th, Schlattner U, et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.