

Abstract
BACKGROUND AND PURPOSE
The mechanisms underlying increased renal noradrenaline in renal failure are still unclear. In this study, the role of α2A-adrenoceptors in controlling sympathetic neurotransmission in chronic renal failure was evaluated in a subtotal nephrectomy model. Also, the influence of this receptor subtype on angiotensin II (Ang II)-mediated noradrenaline release was evaluated.
EXPERIMENTAL APPROACH
α2A-Adrenoceptor-knockout (KO) and wild-type (WT) mice underwent subtotal (5/6) nephrectomy (SNx) or SHAM-operation (SHAM). Kidneys of WT and KO mice were isolated and perfused. Renal nerves were stimulated with platinum electrodes and noradrenaline release was measured by HPLC.
KEY RESULTS
Noradrenaline release induced by renal nerve stimulation (RNS) was significantly increased in WT mice after SNx. RNS-induced noradrenaline release was significantly higher in SHAM-KO compared with SHAM-WT, but no further increase in noradrenaline release could be observed in SNx-KO. α-Adrenoceptor antagonists increased RNS-induced noradrenaline release in SHAM-WT but not in SHAM-KO. After SNx, the effect of α2-adrenoceptor blockade on renal noradrenaline release was attenuated in WT mice. The mRNA expression of α2A-adrenoceptors was not altered, but the inhibitory effect of α2-adrenoceptor agonists on cAMP formation was abolished after SNx. Ang II facilitated RNS-induced noradrenaline release in SHAM-WT but not in SHAM-KO and SNx-WT.
CONCLUSION AND IMPLICATIONS
In our model of renal failure autoregulation of renal sympathetic neurotransmission was impaired. Presynaptic inhibition of noradrenaline release was diminished and the facilitatory effect of presynaptic angiotensin AT1 receptors on noradrenaline release was markedly decreased in renal failure and depended on functioning α2A-adrenoceptors.
Keywords: chronic renal failure, sympathetic overactivity, subtotal nephrectomy, α2-adrenoceptors, α2A-adrenoceptor-knockout, mouse kidney, presynaptic receptors, angiotensin II, cAMP
Introduction
Chronic renal failure (CRF) and end stage renal disease (ESRD) are major health issues in modern populations. Patients suffering from CRF or ESRD are at higher risk for cardiovascular events leading to increased morbidity and mortality (London, 2000; Guérin et al., 2008). It is well known that CRF is associated with an activation of the sympathetic nervous system which contributes to the progression of CRF and cardiovascular complications, such as hypertension (Converse et al., 1992; Rump et al., 2000; Hausberg et al., 2002; Vonend et al., 2004; 2008; Bakris et al., 2006; Schlaich et al., 2009b). Moreover, elevated catecholamine plasma levels correlate with cardiovascular events (Zoccali et al., 2002). However, the mechanisms that lead to the impaired regulation of sympathetic neurotransmitter release in CRF are poorly understood. Insights into the underlying mechanism(s) might help to improve treatment of hypertension and CRF.
Only recently has it been recognized that even a minor injury to the kidney (Ye et al., 2006) triggers afferent nerve traffic to the hypothalamus and thereby increases efferent sympathetic nerve activity and blood pressure (Rump et al., 2003). It is not entirely clear which signals in the damaged kidney trigger afferent signals [e.g. hypoxia, adenosine, angiotensin II (Ang II) or others], but sympathetic overactivity has been observed in a variety of experimental models of renal damage including subtotal nephrectomy. Campese has shown that selective renal de-afferentiation (dorsal rhizotomy) prevents the development of hypertension in rats after 5/6-nephrectomy (Campese, 2000). In patients with resistant hypertension, renal denervation reduced sympathetic nerve activity and blood pressure (Krum et al., 2009; Schlaich et al., 2009a).
α2-Adrenoceptors play a major role in regulation of sympathetic neurotransmitter release (Starke et al., 1989). Three subtypes (α2A, α2B, α2C) have been described (Bylund et al., 1994; Hein, 2006; receptor nomenclature follows Alexander et al., 2009). The α2A-adrenoceptor subtype represents the major autoinhibitory receptor modulating neurotransmitter release from sympathetic nerve endings (Altman et al., 1999; Vonend et al., 2007). Besides affecting noradrenaline release, the α2A-receptor acts as a heteroreceptor and also mediates release of ATP (Vonend et al., 2007).
In addition, the sympathetic nervous system interacts with vasoactive hormone systems such as nitric oxid (Sartoria et al., 2005) or the renin-angiotensin system (DiBona, 2001). Local Ang II facilitates presynaptic noradrenaline release in the human or murine heart and kidney. There is strong evidence that this interaction between the renin-angiotensin system and the sympathetic nervous system plays a role in the genesis of hypertension in CRF (Rump et al., 1995; Stegbauer et al., 2005).
The aim of the present study was to investigate local mechanisms that regulate sympathetic neurotransmitter release in the kidney in a model of CRF. In this respect, the roles of presynaptic α2-adrenoceptors and angiotensin AT1 receptors were evaluated using a mouse model with CRF.
Methods
All animal care and experimental investigations were in accordance with the current European Communities regulations (Office Journal of the European Communities L358/1 18 December 1986) and approved by a University-independent governmental Ethics Committee. α2A-Adrenoceptor-knockout (KO) and wild-type (WT) mice, both on FVB-background, were obtained from Experimental Animal Centre of the Ruhr University of Bochum. Primarily, α2A-adrenoceptor-KO mice were generated on C57Bl/6-background by Lutz Hein, Department of Pharmacology, University of Freiburg, Germany (Hein et al., 1999) and later backcrossed on FVB-background for seven generations. Backcrossing the C57Bl/6-α2A-adrenoceptor-KO mice on FVB-background was necessary because mice on C57Bl/6-background are not susceptible to glomerulosclerosis after subtotal nephrectomy (Ma and Fogo, 2003). Adult (60–75 days) male mice with a body weight of 21–26 g were used for the experiments. All animals were housed under standard conditions and had free access to water and food.
Isolated perfused kidneys
Mice were anaesthetized by intraperitoneal injection of sodium pentobarbital (0.270 mg·g−1 body weight). Kidneys were isolated microscopically (Olympus CO11) and perfused with Krebs–Henseleit solution [composition (mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 0.45, NaHCO3 25, KH2PO4 1.03, d-(+)-glucose 11.1, Na2EDTA 0.067 and ascorbic acid 0.07; all from Fluka, Switzerland] at a constant rate (7.25 mL·min−1·g−1 kidney) as described previously (Vonend et al., 2005). The perfusion medium was gassed continuously with a mixture of 95% O2 and 5% CO2 and passed through a 0.45 µm filter before reaching the kidney. The kidneys were transferred into a jacketed glass chamber maintained at a temperature of 37°C. Bipolar platinum electrodes were placed around the renal arteries to stimulate the renal sympathetic nerves. The perfusate was collected from the renal vein and ureter. Five minutes before nerve stimulation, drugs were infused into the perfusion line by a perfusion apparatus (Harvard Pump 11 Plus Dual Syringe) at a constant flow rate of 0.158 mL·min−1. To test the viability of the preparation, the renal nerve was stimulated (RNS) at a frequency of 5 Hz (3 s, 1 ms pulse width, 40 mA) followed by the administration of 60 mM KCl 15 min later.
Effect of renal nerve stimulation on renal noradrenaline release
After a stabilization period of 30 min, cocaine (10 µM) and corticosterone (20 µM) were added to the perfusion solution in order to prevent neuronal and extraneuronal uptake of released noradrenaline respectively. After another 20 min, 3 min fractions of the perfusate were collected by a fraction collector (LKB, Bromma, Sweden) into vials containing 125 µL HCl (1 M), 13.3 µL of EDTA (0.067 M) and 2.5 µL Na2SO3 (1 M). Kidneys were stimulated at frequencies of 5, 7.5 and 10 Hz (3 s, 1 ms pulse width, 40 mA) as described above.
Effect of α-adrenoceptor blockade on renal noradrenaline release
The experiment started after a stabilization period of 30 min. Then, six RNS (S0–S5) at a frequency of 5 Hz (3 s, 1 ms pulse width, 40 mA) were applied. Using a perfusion apparatus (Harvard Pump 11 Plus Dual Syringe) the non-selective α-adrenoceptor antagonist phentolamine was applied in a cumulative manner at 0.01, 0.03, 0.1, 0.3 and 1 µM, the selective α2-adrenoceptor antagonist rauwolscine at 0.01, 0.1 and 1 µM and the selective α1-adrenoceptor antagonist prazosin at 0.01, 0.1, 1 and 10 µM at a constant rate of 0.158 mL·min−1 accompanying the stimulations. The interval of perfusion started 5 min before RNS and lasted until 1 min after. The perfusate of the kidneys was collected in two fractions of 3 min after each RNS. Noradrenaline release was measured by HPLC.
Effect of angiotensin II on renal noradrenaline release
Five RNS (S0–S4), each at 5 Hz (3 s, 1 ms pulse width, 40 mA), were applied at 6, 22, 38, 54 and 72 min after the start of fraction collection. Ang II was added directly to the perfusion system via perfusion apparatus (Harvard Pump 11 Plus Dual Syringe) in a cumulative manner (0.01, 0.1, 1 and 10 nM), starting 5 min before and stopping 1 min after S1, S2, S3 and S4. In the set of experiments evaluating the effect of α2-adrenoceptor blockade on Ang II-mediated facilitation of noradrenaline release the selective α2-adrenoceptor antagonist rauwolscine was added 10 min before the first stimulation period (S0) at a concentration of 1 µM to the perfusion solution for the total time of experiment.
Quantitative determination of release of endogenous noradrenaline
Noradrenaline in the collected samples was extracted (adsorption onto alumina, elution with HClO4). The quantity of noradrenaline in each sample was determined by reversed-phase HPLC detection (Stegbauer et al., 2005) and corrected for recovery using an internal standard (3,4-dihydroxybenzylamine 12 pg·mL−1, Chromsystems, Germany). RNS-induced outflow of noradrenaline was determined as the difference between the content of noradrenaline present in two 3-min samples collected immediately after onset of stimulation and spontaneous noradrenaline content present in the 3-min sample collected immediately before RNS (Stegbauer et al., 2005).
Calculation of data
In some experiments (Figures 1, 2 and 3A',B') the absolute amounts of noradrenaline release were calculated and expressed in pg·g−1 kidney wet weight. S0 served as a reference stimulation, and the RNS-induced noradrenaline outflow in S1–S3 in experiments with rauwolscine and S1–S4 in those with application of Ang II and prazosin was expressed as a percentage of the outflow in S0 (Sn as % of S0). For further evaluations of the effects of rauwolscine, prazosin and Ang II, the Sn/S0 ratios were calculated as a percentage of values, which were determined in the corresponding control experiments presented in Figure 4 (Sn/S0 as % of control).
Figure 1.
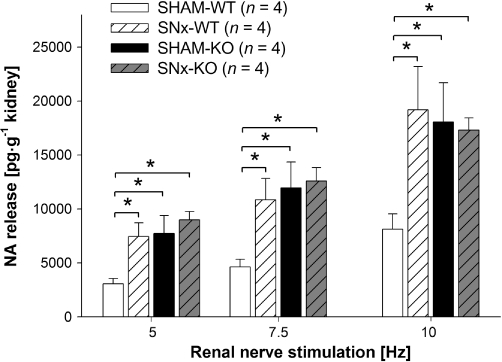
Renal nerve stimulation (RNS)-induced endogenous noradrenaline (NA) release in isolated kidneys of SHAM-operated (SHAM) and subtotal nephrectomized (SNx) wild-type (WT) and knockout (KO) mice at frequencies of 5, 7.5 and 10 Hz (each for 3 s, with 1 ms pulse width at 40 mA). RNS-induced NA release was measured and expressed in pg·g−1 kidney wet weight (data given are mean and vertical lines show SEM). Noradrenaline release was significantly increased in WT mice after SNx. In SHAM-KO mice, NA release was significantly greater than in SHAM-WT mice. There was no increase in NA release in KO mice after SNx. Experiments in SNx-animals were performed 60 days postoperatively. *P < 0.05 indicates significant differences.
Figure 2.
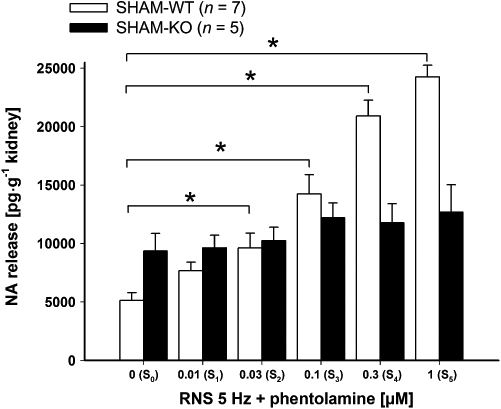
Effect of the non-selective α-adrenoceptor antagonist phentolamine on renal nerve stimulation (RNS)-induced endogenous noradrenaline (NA) release in isolated kidneys. There were six stimulation periods (S0–S5, each at 5 Hz for 3 s, 1 ms pulse width, 40 mA). Phentolamine was applied in increasing concentrations (0.01, 0.03, 0.1, 0.3 and 1 µM) and was added to the perfusion solution to S1–S5. RNS-induced NA release was measured and expressed in pg·g−1 kidney wet weight (data given are mean and vertical lines show SEM). A significant increase in RNS-induced NA release after α-adrenoceptor blockade by phentolamine was found in wild-type (WT) but not in knockout (KO) mice. *P < 0.05 indicates a significant difference in NA release by increasing concentrations phentolamine in WT mice.
Figure 3.
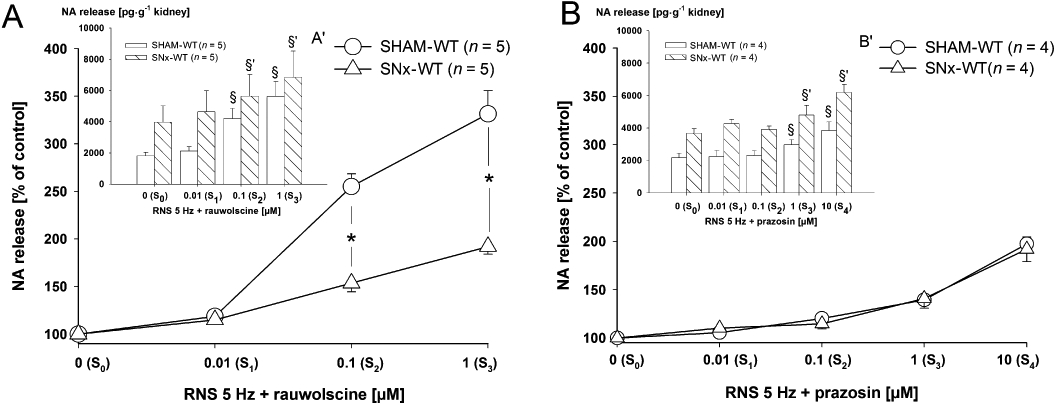
(A,B) Effects of the selective α2-adrenoceptor antagonist rauwolscine (A) and the selective α1-adrenoceptor antagonist prazosin (B) on renal nerve stimulation (RNS)-induced endogenous noradrenaline (NA) release in isolated kidneys of SHAM-operated (SHAM) and subtotal nephrectomized (SNx) wild-type (WT) mice. There were four stimulation periods in experiments with rauwolscine (S0–S3) and five stimulation periods in experiments with prazosin (S0–S4). Each stimulation period was performed at 5 Hz for 3 s, 1 ms width, 40 mA. Rauwolscine was applied in increasing concentrations of 0.01, 0.1 and 1 µM to the perfusion solution to S1–S3 and prazosin in increasing concentrations of 0.01, 0.1, 1 and 10 µM to S1–S4. Noradrenaline release was measured and expressed as a percentage of control S0 (Sn as % of S0) (A, B). In (A') and (B') the same data are given but expressed in pg·g−1 kidney wet weight for SHAM and SNx mice. Data given are mean and vertical lines show SEM. The increase in NA release after α2-adrenoceptor blockade by rauwolscine was significantly reduced in SNx-WT, compared with SHAM-WT mice (A; *P < 0.05 indicates a significant difference). The increase in NA release after α1-adrenoceptor blockade was similar in kidneys of SHAM and SNx-WT mice (B). Graphs indicating absolute NA release demonstrate the higher level of stimulation-induced transmitter release in SNx compared with SHAM animals (A',B'). Rauwolscine and prazosin lead to a concentration-dependent increase in NA release in SHAM and SNx animals (A',B') (§P < 0.05 indicates significant differences compared with S0, §P within SHAM group and §'P within SNx group).
Figure 4.
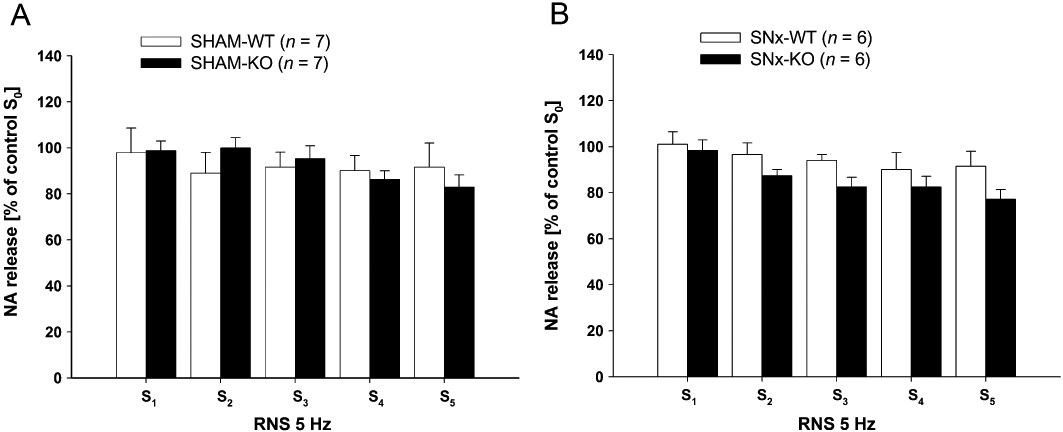
(A,B) Renal nerve stimulation (RNS)-induced endogenous noradrenaline (NA) release in isolated kidneys of SHAM-operated (SHAM) (A) and subtotal nephrectomized (SNx) (B) wild-type (WT) and knockout (KO) mice, six times consecutively (S0–S5) at a frequency of 5 Hz (each for 3 s, with 1 ms pulse width at 40 mA) without application of any drugs. The stimulation-induced NA release was measured and expressed in percentage of S0 (S1/S0, to S5/S0) (data given are mean and vertical lines show SEM). Noradrenaline release after RNS was stable over time in all four groups. Experiments in SNx animals were performed 30 days postoperatively.
Subtotal (5/6) nephrectomy
The animals were placed on a heating pad to maintain a constant body temperature (37°C). Under anaesthesia with sodium pentobarbital (0.270 mg·g−1 body weight), right flank incisions were made, and the right kidney was decapsulated and removed. To achieve a subtotal (5/6) nephrectomy (SNx), upper and lower pole of the left kidney were removed. To control for the effect of the surgery, WT and KO mice were treated with a SHAM-operation (SHAM), in which the right and left flank incisions were made in anaesthetized mice and sutured without any tissue removal. Thirty days after the operation, the serum urea (mean ± SEM; in SNx-WT mice, 1781 ± 758 mg·L−1; in SHAM-WT mice, 553 ± 106 mg·L−1) and the albumin to creatinine ratio in urine (mean ± SEM; in SNx-WT mice, 16715 ± 1237 mg·g−1; in SHAM-WT mice, 73 ± 31 mg·g−1) were significantly increased, indicating CRF.
Quantification of mRNA expression
The α2A-, α2B- and α2C-adrenoceptor mRNA expression was estimated by real-time-quantitative PCR analyses. RNA was isolated from tissue of α2A-adrenoceptor-KO and -WT mice using RNeasy Plus Mini Kit (Qiagen). The extracted RNA was transcribed into cDNA by QuantiTect Reverse Transcription Kit (Qiagen). Quantitative mRNA analysis was performed by 7300 real-time PCR system (Applied Biosystems) using TaqMan Gene Expression assays (Applied Biosystems). Changes in expression level were calculated using Rest 2008 Software (Corbett Research, USA) according to Pfaffl et al. (Pfaffl et al., 2002).
Quantification of cAMP accumulation
The SHAM-operated and SNx-WT mice were anaesthetized by intraperitoneal injection of sodium pentobarbital (0.270 mg·g−1 body weight) and perfusion was performed through the abdominal aorta with Dulbecco's modified Eagle's medium (DMEM) in order to remove blood from the kidneys. The kidneys were removed and cut by a sharp blade (Apollo, Germany) into thin tissue slices. The tissue slices were transferred into 2 mL of prewarmed (37°C) DMEM, gassed with 5% CO2 and 95% O2 and incubated for 90 min. After the incubation period, 3-isobutyl-1-methylxanthine (IBMX) was added to the medium culture. After 20 min IBMX treatment, forskolin was added in increasing concentration (0, 1 and 5 µM) in the presence or absence of the selective α2-adrenoceptor agonist UK14.304 (25 µM). Tissue slices were collected after 30 min of incubation with forskolin and cAMP accumulation was measured by cAMP EIA kit (Cayman Chemical, USA). The effect of forskolin and the combination of forskolin and UK14.304 on cAMP formation was expressed as a percentage of basal cAMP formation in the absence of other drugs except IBMX.
Statistics
All data were expressed as mean ± SEM. Differences were analysed by one-way anova for repeated measurement combined with a post hoc test (Bonferroni). Probability levels of P < 0.05 were considered statistically significant. The number of experiments indicates the number of individual kidneys.
Materials
The following drugs were used: corticosterone, phentolamine HCL, rauwolscine, prazosin, Ang II, IBMX, forskolin, UK14.304 (Sigma-Aldrich, Germany); cocaine HCl (Merck, Germany). Drugs were dissolved in distilled water before being diluted with the Krebs–Henseleit solution, except corticosterone (absolute ethanol).
Results
Effect of subtotal nephrectomy on renal noradrenaline release
The WT and KO mice were treated with SNx or a SHAM. RNS was performed to induce noradrenaline release from renal sympathetic nerve endings. RNS-induced noradrenaline release at all frequencies (5, 7.5 and 10 Hz) was significantly higher in SNx-WT than in kidneys of SHAM-WT mice (Figure 1). In SHAM-KO mice, RNS-induced noradrenaline release was already at same level as in WT mice after SNx and significantly higher at all frequencies compared with SHAM-WT mice (Figure 1). Experimental CRF had no effect on renal noradrenaline release in KO mice. There was no significant difference in noradrenaline release at all frequencies in KO mice after SNx- compared with SHAM-KO mice (Figure 1).
Renal nerve stimulation-induced noradrenaline release in the presence of α-adrenoceptor blockade
The non-selective α-adrenoceptor antagonist phentolamine increased RNS (5 Hz)-induced noradrenaline release in a concentration-dependent manner (0.01, 0.03, 0.1, 0.3 and 1 µM) in kidneys of SHAM-WT mice (Figure 2). In WT mice the increase in noradrenaline release by phentolamine was significant at concentrations of 0.03, 0.1, 0.3 and 1 µM. Only a marginal increase in noradrenaline release by phentolamine was observed in SHAM-KO mice. In the absence of α-adrenoceptor antagonists, noradrenaline release, induced by RNS (S0–S5 at 5 Hz), was similar in kidneys of WT and KO mice (Figure 4A).
Quantitative expression of α2A-, α2B- and α2C-adrenoceptors
The expression of mRNA for α2A-, α2B- and α2C-adrenoceptors was determined by quantitative PCR analysis in renal tissue of SHAM and SNx α2A-adrenoceptor-KO and -WT mice. The expression of α2B- and α2C-adrenoceptors revealed no significant difference between SHAM-KO and SHAM-WT mice (Figure 5A). SNx did not change the mRNA expression level of α2A-adrenoceptors (Figure 5B).There was also no difference between expression of mRNA for α2B- and α2C-adrenoceptors in SNx-KO and SHAM-KO mice as well as SNx-KO and SNx-WT mice (data not shown).
Figure 5.
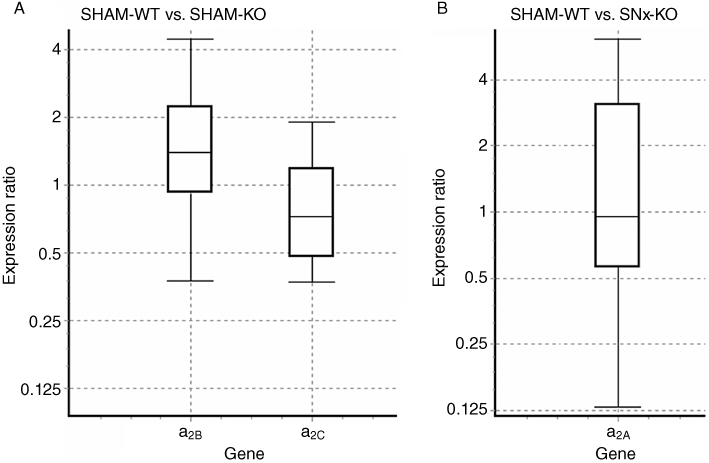
(A,B) Effect of knockout (KO) compared with WT (A) and of subtotal nephrectomized (SNx)-WT compared with SHAM-operated (SHAM)-WT (B) on mRNA expression of α2A-, α2B- and α2C-adrenoceptor subtypes in the kidneys. The mRNA expression was quantified by real-time-quantitative PCR. Results indicated no significant differences in mRNA expression (data are shown as medians (black lines in boxes), with box (interquartile range) and whiskers (maximum and minimum); n = 4 for each group).
Renal nerve stimulation-induced noradrenaline release in the presence of selective α1- and α2-adrenoceptor blockade
The selective α2-adrenoceptor antagonist rauwolscine facilitated RNS (5 Hz)-induced noradrenaline release in a concentration-dependent manner (0.01, 0.1 and 1 µM) in kidneys of SHAM-WT and SNx-WT mice (Figure 3A/A′). Application of rauwolscine induced an increase in noradrenaline release in SHAM-WT up to 331.5%, and in SNx-WT up to 192%. This increase was significantly lower in SNx-WT compared with SHAM-WT mice (Figure 3A). The selective α1-adrenoceptor antagonist prazosin facilitated RNS (5 Hz)-induced noradrenaline release at 1 and 10 µM in kidneys of SHAM-WT and SNx-WT mice (Figure 3B/B′). Application of prazosin induced an increase in noradrenaline release in SHAM-WT up to 197.3% similar to that seen in SNx-WT (Figure 3B).
Inhibition of forskolin-induced cAMP accumulation
Kidney tissue slices of SHAM- and SNx-WT mice were stimulated by the adenylyl cyclase activator forskolin (0, 1 and 5 µM) in the presence and in the absence of the α2-adrenoceptor agonist UK14.304 (25 µM). Forskolin 1 µM and 5 µM increased cAMP accumulation in SHAM-WT to 457% and 773% respectively (Figure 6). Application of UK14.304 inhibited forskolin-induced cAMP accumulation significantly in SHAM- WT mice. In contrast, the inhibiting effect of UK14.304 on forskolin-induced cAMP accumulation was abolished in SNx-WT mice (Figure 6).
Figure 6.
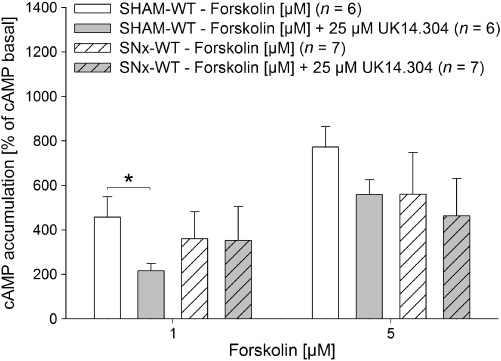
Effect of UK14.304 (25 µM) on forskolin-induced (0, 1 and 5 µM) cAMP accumulation in kidney slices of SHAM-operated (SHAM) and subtotal nephrectomized (SNx) mice. At 1 µM forskolin, a significant inhibition of cAMP accumulation by UK14.304 was observed in SHAM-wild-type (WT) but not in SNx-WT mice. Experiments in SNx animals were performed 30 days after subtotal nephrectomy. cAMP accumulation is presented as percentage of cAMP basal level (data given are mean and vertical lines show SEM.). *P < 0.05 indicates a significant difference.
Effect of angiotensin II on renal noradrenaline release
Ang II application facilitated noradrenaline release, induced by RNS (5 Hz), in a concentration-dependent manner (0.01, 0.1, 1 and 10 nM) in kidneys of SHAM-WT mice up to 197% (Figure 7). The increase in noradrenaline release by 1 nM Ang II was significantly attenuated in SNx-WT mice. The facilitatory effect of 1 nM Ang II on RNS-induced noradrenaline release was significantly smaller in SHAM-KO compared with SHAM-WT mice. In addition, no difference in the facilitatory effect of noradrenaline release could be observed between SHAM-KO and SNx-KO (Figure 7). As demonstrated in α2A-adrenoceptor KO mice, the pharmacological blockade of α2A-adrenoceptors using rauwolscine attenuated the facilitatory effect of Ang II in SHAM-WT in a similar manner. Noradrenaline release induced by consecutive RNS at 5 Hz (S0–S4) decreased slightly over time in SHAM and SNx mice.
Figure 7.
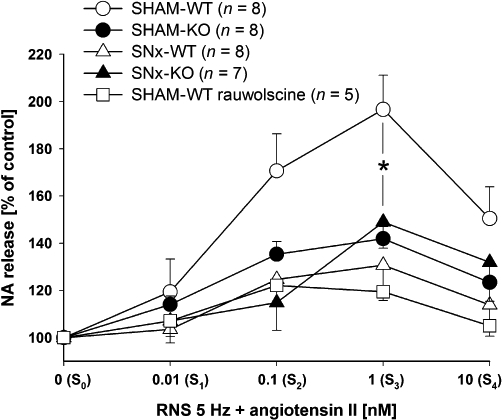
Effect of angiotensin II (Ang II) on renal nerve stimulation (RNS)-induced endogenous noradrenaline (NA) release in isolated kidneys of SHAM-operated (SHAM) and subtotal nephrectomized (SNx) knockout (KO) and wild-type (WT) mice. There were five stimulations periods (S0–S4, each at 5 Hz for 3 s, 1 ms pulse width, 40 mA). Ang II was applied in increasing concentrations (0.01, 0.1, 1 and 10 nM) to the perfusion solution to S1–S4. The RNS-induced NA release was measured and calculated as a percentage of control S0 (Sn as % of S0) (data given are mean and vertical lines show SEM.). Experiments in SNx mice were performed 30 days postoperatively. The enhancement of NA release by Ang II was reduced in mice lacking the α2A-adrenoceptor (KO) and in mice after SNx, compared with SHAM-WT mice. The antagonism of α2A-adrenoceptors by rauwolscine (1 µM) reduced the enhancing effect in a similar manner. *P < 0.05 indicates significant differences in the facilitatory effect of Ang II on NA release between SHAM-WT and all other groups.
Discussion
Impaired function of presynaptic α2A-adrenoceptors in chronic renal failure
In the present study, we showed that noradrenaline release induced by RNS was significantly increased in SNx mice, our model of CRF. There is evidence for an impaired autofeedback regulation in uraemia in the CNS (Klein et al., 2005). In the present set of experiments, we tested whether the function of presynaptic α2-adrenoceptors was impaired in CRF. Autoinhibition by presynaptic α2-adrenoceptors is one of the most potent mechanisms regulating sympathetic neurotransmitter release. Indeed, the increase in noradrenaline release after SNx, as observed in WT mice, could not be seen in α2A-adrenoceptor SNx-KO mice. However, SHAM-KO mice were characterized by a higher RNS-induced renal noradrenaline release compared with SHAM-WT mice.
In order to prove that in FVB-WT mice, similar to previous observations in C57Bl/6 mice (Vonend et al., 2007), the presynaptic autoinhibition of noradrenaline release was mediated by the α2A-adrenoceptor subtype, a non-specific α-adrenoceptor antagonist phentolamine was applied. In WT mice, phentolamine uncoupled RNS-induced noradrenaline release in a concentration-dependent manner. In contrast, the application of phentolamine led only to a marginal increase in noradrenaline release in KO mice. It is possible that other α2-adrenoceptor subtypes (α2B-and α2C-adrenoceptors) could participate in renal sympathetic neurotransmission (Hein, 2001; Starke, 2001). However, in our experimental setup, a compensatory overexpression of α2B- and α2C-adrenoceptors on mRNA level was not observed in mice lacking the α2A-adrenoceptor.
The marginal increase in renal noradrenaline release in KO mice by phentolamine could be due to inhibition of α1-adrenoceptors. Previously, facilitatory effects of α1-adrenoceptor blocking drugs on noradrenaline release have been described in rats (Rump and Majewski, 1987; Bohmann et al., 1993a,b;). It was proposed that prazosin increased noradrenaline release induced by RNS, by preventing a transjunctional inhibitiory feedback loop which is mediated through release of prostaglandins and adenosine, following stimulation of postjunctional α1-adrenoceptors. Furthermore, these authors discussed a blockade of α2-adrenoceptors at higher prazosin concentrations. Indeed, in the present study, prazosin facilitated RNS-induced noradrenaline release in SHAM and SNx mice. It remains unclear which of the above mentioned mechanisms may be responsible for the observed effect of phentolamine in KO mice. In any case, the facilitatory effect of prazosin on noradrenaline release was unaffected in mice with CRF.
The α2-adrenoceptor antagonist rauwolscine increased the RNS-induced noradrenaline release to a significantly greater extent in SHAM than in SNx mice, indicating impaired α2-adrenoceptor function in CRF. These observations are in contrast to previous results in rat kidneys in which an α2-adrenoceptor antagonist had a greater facilitatory effect on RNS-induced noradrenaline release after SNx, compared with control animals (Amann et al., 2000). We do not have a definite explanation, but species differences may account for the contrasting findings. The present data suggest that the inhibitory α2A-adrenoceptor function was impaired in kidneys of SNx mice and this might be one reason for the observed increase in presynaptic release of noradrenaline.
To further elucidate the underlying mechanism for this impairment we analysed α2A-adrenoceptor mRNA expression level. The SNx per se did not alter the mRNA expression of α2A-adrenoceptors, compared to SHAM-WT mice. We carried out additional experiments to test α2-adrenoceptor signalling. As α2A-adrenoceptors are Gi coupled receptors, their activation leads to a reduction in cAMP accumulation (Bylund et al., 1994). Incubation of kidney slices with the selective α2-adrenoceptor agonist UK14.304 inhibited cAMP accumulation in SHAM-WT mice. This effect was attenuated in kidneys of SNx-WT mice indicating α2-adrenoceptor desensitization in CRF. The reduced activity of α2A-adrenoceptors might be caused by desensitization after receptor phosphorylation or internalization by the binding of arrestins to phosphorylated receptors as suggested previously (Olli-Lahdesmaki et al., 2004). However, as most tissues, including the kidney, contain both pre- and postsynaptic α2-adrenoceptors, it is difficult to distinguish between these two sites by binding or signalling studies. A transgenic mouse model expressing the dopamine β-hydroxylase promoter combined with general deletion of the endogenous α2A-adrenoceptor gene could be used to unequivocally distinguish between pre- and postsynaptic α2-adrenoceptors (Gilsbach et al., 2009; 2010;).
Angiotensin AT1 receptor-mediated noradrenaline release depends on presynaptic α2-adrenoceptors
Apart from α2A-adrenoceptor function, other presynaptic mechanisms might be affected in this model of CRF. And it is well known that the renin-angiotensin system is activated in CRF. In this respect, Ang II has been described as a potent modulator of sympathetic neurotransmitter release (Stegbauer et al., 2005) and it was possible that the facilitatory effect of Ang II on noradrenaline release was also altered in CRF. There is evidence that angiotensin AT1 receptors interact with α2-adrenoceptors. Cox and colleagues showed that a marked presynaptic facilitatory effect of Ang II in atria isolated from mice required an ongoing presynaptic α2-autoinhibition (Cox et al., 2000). Trendelenburg et al. confirmed these findings and pointed out that the α2C-adrenoceptor subtype in particular was responsible for this receptor interaction (Trendelenburg et al., 2003).
Indeed, exogenously applied Ang II caused a significant facilitation of renal noradrenaline release in SHAM-WT mice. According to Stegbauer et al., the effect is mediated by presynaptic angiotensin AT1 receptors (Stegbauer et al., 2003). This facilitatory effect of Ang II was significantly diminished in WT mice with CRF. As described above, the α2A-adrenoceptor-mediated autoinhibition was impaired in our model of CRF and, in case of a functional cross talk with presynaptic angiotensin AT1 receptors, might disrupt the facilitatory effect of Ang II on noradrenaline release. To test this hypothesis, we repeated the experiments using α2A-adrenoceptor-KO mice. As expected, the facilitatory effect of Ang II on noradrenaline release was significantly diminished in SHAM-KO mice. In addition, pharmacological α2-adrenoceptor blockade by rauwolscine attenuated the facilitatory effect of Ang II on noradrenaline release in a similar way.
In this set of experiments, it was shown for the first time that α2A-adrenoceptors are involved in the receptor interaction between α-adrenoceptors and angiotensin AT1 receptors. This interaction probably represents a necessary mechanism of fine regulation of sympathetic neurotransmission under physiological conditions and may also have important pathophysiological implications.
In summary, we conclude that the α2A-adrenoceptor plays a key role in regulating sympathetic activity in the kidney and its function is impaired in CRF. Moreover, there is an interaction between α2A-adrenoceptors and angiotensin AT1 receptors that represents an interface between the sympathetic nervous system and the renin-angiotensin system.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (RU 401/9-1). The expert technical assistances of Blanka Duvnjak and Christina Schwandt are greatly acknowledged.
Glossary
Abbreviations
- Ang II
angiotensin II
- CRF
chronic renal failure
- HPLC
high performance liquid chromatography
- IBMX
3-isobutyl-1-methylxanthine
- KO
knockout
- RNS
renal nerve stimulations
- SHAM
SHAM-operation
- SNx
subtotal (5/6) nephrectomy
- WT
wild-type
Conflict of interest
None.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th ednition. Br J Pharmacol. 2009;158(Suppl 1):S1–254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman JD, Trendelenburg AU, MacMillan L, Bernstein D, Limbird L, Starke K, et al. Abnormal regulation of the sympathetic nervous system in alpha2A-adrenergic receptor knockout mice. Mol Pharmacol. 1999;56:154–161. doi: 10.1124/mol.56.1.154. [DOI] [PubMed] [Google Scholar]
- Amann K, Rump LC, Simonaviciene A, Oberhauser V, Wessels S, Orth SR, et al. Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J Am Soc Nephrol. 2000;11:1469–1478. doi: 10.1681/ASN.V1181469. [DOI] [PubMed] [Google Scholar]
- Bakris GL, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006;70:1905–1913. doi: 10.1038/sj.ki.5001835. [DOI] [PubMed] [Google Scholar]
- Bohmann C, Schollmeyer P, Rump LC. Alpha 2-autoreceptor subclassification in rat isolated kidney by use of short trains of electrical stimulation. Br J Pharmacol. 1993a;108:262–268. doi: 10.1111/j.1476-5381.1993.tb13472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmann C, Schollmeyer P, Rump LC. Methoxamine inhibits noradrenaline release through activation of alpha 1- and alpha 2-adrenoceptors in rat isolated kidney: involvement of purines and prostaglandins. Naunyn Schmiedebergs Arch Pharmacol. 1993b;347:273–279. doi: 10.1007/BF00167445. [DOI] [PubMed] [Google Scholar]
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, et al. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;26:121–136. [PubMed] [Google Scholar]
- Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int. 2000;57:2–6. [PubMed] [Google Scholar]
- Converse RL, Jacobsen TN, Toto RD, Jost CM, Cosentino F, Fouad-Tarazi F, et al. Sympathetic overactivity in patients with chronic renal failure. N Engl J Med. 1992;327:1912–1918. doi: 10.1056/NEJM199212313272704. [DOI] [PubMed] [Google Scholar]
- Cox SL, Schelb V, Trendelenburg AU, Starke K. Enhancement of noradrenaline release by angiotensin II and bradykinin in mouse atria: evidence for cross-talk between G(q/11) protein- and G(i/o) protein-coupled receptors. Br J Pharmacol. 2000;129:1095–1102. doi: 10.1038/sj.bjp.0703167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona GF. Peripheral and central interactions between the renin-angiotensin system and the renal sympathetic nerves in control of renal function. Ann N Y Acad Sci. 2001;940:395–406. doi: 10.1111/j.1749-6632.2001.tb03693.x. [DOI] [PubMed] [Google Scholar]
- Gilsbach R, Roser C, Beetz N, Brede M, Hadamek K, Haubold M, et al. Genetic dissection of alpha2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Mol Pharmacol. 2009;75:1160–1170. doi: 10.1124/mol.109.054544. [DOI] [PubMed] [Google Scholar]
- Gilsbach R, Schneider J, Lother A, Schickinger S, Leemhuis J, Hein L. Sympathetic alpha(2)-adrenoceptors prevent cardiac hypertrophy and fibrosis in mice at baseline but not after chronic pressure overload. Cardiovasc Res. 2010;86:432–442. doi: 10.1093/cvr/cvq014. [DOI] [PubMed] [Google Scholar]
- Guérin AP, Pannier B, Marchais SJ, London GM. Arterial structure and function in end-stage renal disease. Cur Hypertens Rep. 2008;2:107–111. doi: 10.1007/s11906-008-0021-2. [DOI] [PubMed] [Google Scholar]
- Hausberg M, Kosch M, Harmelink P, Barenbrock M, Hohage H, Kisters K, et al. Sympathetic nerve activity in end-stage renal disease. Circulation. 2002;106:1974–1979. doi: 10.1161/01.cir.0000034043.16664.96. [DOI] [PubMed] [Google Scholar]
- Hein L. Transgenic models of alpha 2-adrenergic receptor subtype function. Rev Physiol Biochem Pharmacol. 2001;142:161–185. doi: 10.1007/BFb0117493. [DOI] [PubMed] [Google Scholar]
- Hein L. Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 2006;326:541–551. doi: 10.1007/s00441-006-0285-2. [DOI] [PubMed] [Google Scholar]
- Hein L, Altman JD, Kobilka BK. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999;402:181–184. doi: 10.1038/46040. [DOI] [PubMed] [Google Scholar]
- Klein K, Daschner M, Vogel M, Oh J, Feuerstein TJ, Schaefer F. Impaired autofeedback regulation of hypothalamic norepinephrine release in experimental uremia. J Am Soc Nephrol. 2005;16:2081–2087. doi: 10.1681/ASN.2004100830. [DOI] [PubMed] [Google Scholar]
- Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet. 2009;373:1275–1281. doi: 10.1016/S0140-6736(09)60566-3. [DOI] [PubMed] [Google Scholar]
- London GM. Alterations of arterial function in end-stange renal disease. Nephron. 2000;2:111–118. doi: 10.1159/000045557. [DOI] [PubMed] [Google Scholar]
- Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int. 2003;64:350–355. doi: 10.1046/j.1523-1755.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- Olli-Lahdesmaki T, Tiger M, Vainio M, Scheinin M, Kallio J. Ligand-induced alpha2-adrenoceptor endocytosis: relationship to Gi protein activation. Biochem Biophys Res Commun. 2004;321:226–233. doi: 10.1016/j.bbrc.2004.06.131. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rump LC, Majewski H. Modulation of norepinephrine release through alpha 1- and alpha 2-adrenoceptors in rat isolated kidney. J Cardiovasc Pharmacol. 1987;9:500–507. doi: 10.1097/00005344-198704000-00016. [DOI] [PubMed] [Google Scholar]
- Rump LC, Bohmann C, Schaible U, Schultze-Seemann W, Schollmeyer PJ. Beta-adrenergic, angiotensin II, and bradykinin receptors enhance neurotransmission in human kidney. Hypertension. 1995;26:445–451. doi: 10.1161/01.hyp.26.3.445. [DOI] [PubMed] [Google Scholar]
- Rump LC, Amann K, Orth S, Ritz E. Sympathetic overactivity in renal disease: a window to understand progression and cardiovascular complications of uraemia? Nephrol Dial Transplant. 2000;15:1735–1738. doi: 10.1093/ndt/15.11.1735. [DOI] [PubMed] [Google Scholar]
- Rump LC, Amann K, Ritz E. Sympathetic innervation of the kidney in health and disease. In: Bolis C, editor. Handbook of the Autonomic Nervous System in Health and Disease. New York: Dekker; 2003. pp. 561–587. [Google Scholar]
- Sartoria C, Leporib M, Scherrera U. Interaction between nitric oxide and the cholinergic and sympathetic nervous system in cardiovascular control in humans. Pharmacol Ther. 2005;106:209–220. doi: 10.1016/j.pharmthera.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med. 2009a;361:932–934. doi: 10.1056/NEJMc0904179. [DOI] [PubMed] [Google Scholar]
- Schlaich PM, Socratous F, Hennebry S, Eikles N, Lambert EA, Straznicky N, et al. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009b;20:933–939. doi: 10.1681/ASN.2008040402. [DOI] [PubMed] [Google Scholar]
- Starke K. Presynaptic autoreceptors in the third decade: focus on alpha2-adrenoceptors. J Neurochem. 2001;78:685–693. doi: 10.1046/j.1471-4159.2001.00484.x. [DOI] [PubMed] [Google Scholar]
- Starke K, Göthert M, Kilbinger H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- Stegbauer J, Vonend O, Oberhauser V, Rump LC. Effects of angiotensin-(1–7) and other bioactive components of the renin–angiotensin system on vascular resistance and noradrenaline release in rat kidney. J Hypertens. 2003;21:1391–1399. doi: 10.1097/00004872-200307000-00030. [DOI] [PubMed] [Google Scholar]
- Stegbauer J, Vonend O, Habbel S, Quack I, Sellin L, Gross V, et al. Angiotensin II modulates renal sympathetic neurotransmission through nitric oxide in AT2 receptor knockout mice. J Hypertens. 2005;23:1691–1698. doi: 10.1097/01.hjh.0000179763.02583.8e. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Klebroff W, Guimarães S, Starke K. Crosstalk between presynaptic angiotensin receptors, bradykinin receptors and alpha2-autoreceptors in sympathetic neurons: a study in alpha2-adrenoceptor-deficient mice. Br J Pharmacol. 2003;138:1389–1402. doi: 10.1038/sj.bjp.0705223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonend O, Apel T, Amann K, Sellin L, Stegbauer J, Ritz E, et al. Modulation of gene expression by moxonidine in rats with chronic renal failure. Nephrol Dial Transplant. 2004;19:2217–2222. doi: 10.1093/ndt/gfh374. [DOI] [PubMed] [Google Scholar]
- Vonend O, Stegbauer J, Sojka J, Habbel S, Quack I, Robaye B, et al. Noradrenaline and extracellular nucleotide cotransmission involves activation of vasoconstrictive P2X1,3- and P2Y6-like receptors in mouse perfused kidney. Br J Pharmacol. 2005;145:66–74. doi: 10.1038/sj.bjp.0706151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonend O, Habbel S, Stegbauer J, Roth J, Hein L, Rump LC. Alpha(2A)-adrenoceptors regulate sympathetic transmitter release in mice kidneys. Br J Pharmacl. 2007;150:121–127. doi: 10.1038/sj.bjp.0706961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonend O, Rump LC, Ritz E. Sympathetic overactivity – the Cinderella of cardiovascular risk factors in dialysis patients. Semin Dial. 2008;21:326–330. doi: 10.1111/j.1525-139X.2008.00456.x. [DOI] [PubMed] [Google Scholar]
- Ye S, Zhong H, Yanamadala S, Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309–315. doi: 10.1161/01.HYP.0000231307.69761.2e. [DOI] [PubMed] [Google Scholar]
- Zoccali C, Mallamaci F, Parlongo S, Cutrupi S, Benedetto FA, Tripepi G, et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation. 2002;105:1354–1359. doi: 10.1161/hc1102.105261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.