

Abstract
Objective:
Since Alzheimer disease (AD) neuropathology is thought to develop years before dementia, it may be possible to detect subtle AD-related atrophy in preclinical AD. Here we hypothesized that the “disease signature” of AD-related cortical thinning, previously identified in patients with mild AD dementia, would be useful as a biomarker to detect anatomic abnormalities consistent with AD in cognitively normal (CN) adults who develop AD dementia after longitudinal follow-up.
Methods:
We studied 2 independent samples of adults who were CN when scanned. In sample 1, 8 individuals developing AD dementia (CN-AD converters) after an average of 11.1 years were compared to 25 individuals who remained CN (CN-stable). In sample 2, 7 CN-AD converters (average follow-up 7.1 years) were compared to 25 CN-stable individuals.
Results:
AD-signature cortical thinning in CN-AD converters in both samples was remarkably similar, about 0.2 mm (p < 0.05). Despite this small absolute difference, Cohen d effect sizes for these differences were very large (>1). Of the 11 CN individuals with baseline low AD-signature thickness (≥1 SD below cohort mean), 55% developed AD dementia over nearly the next decade, while none of the 9 high AD-signature thickness individuals (≥1 SD above mean) developed dementia. This marker predicted time to diagnosis of dementia (hazard ratio = 3.4, p < 0.0005); 1 SD of thinning increased dementia risk by 3.4.
Conclusions:
By focusing on cortical regions known to be affected in AD dementia, subtle but reliable atrophy is identifiable in asymptomatic individuals nearly a decade before dementia, making this measure a potentially important imaging biomarker of early neurodegeneration.
The dementia of Alzheimer disease (AD) develops insidiously, initially emerging as mild cognitive impairment before eventually progressing to rob the individual of independent function. Ten or more years may elapse from first symptoms until clinical dementia.1 The evolution of AD neuropathology is longer, developing in cognitively normal (CN) adults and causing dysfunction and cell death in neuronal systems subserving cognition, eventually leading to the clinical syndrome.
Cognitive test measures may change 5–10 years before AD dementia,2–6 and may be useful for predicting dementia in CN cohorts.7 A few imaging studies have reported subtle brain abnormalities, particularly in medial temporal lobe and parietal regions, in groups of CN individuals who subsequently declined.8–17 To date, there has been very little investigation of neuroimaging measures in CN subjects to predict AD dementia.
We undertook this study hypothesizing that neuroanatomic abnormalities consistent with very early AD pathology could be detected in CN individuals followed longitudinally and eventually diagnosed with AD dementia. Working with 2 independent samples with nearly identical demographic and cognitive characteristics, we investigated whether these measures could be translated into an AD–dementia risk MRI biomarker applicable to CN individuals. We employed a hypothesis-driven approach using regions of interest (ROIs) collectively called the cortical signature of AD based on our earlier work18 comparing patients with mild AD dementia to CN controls. After examining the 2 samples separately using these a priori ROIs, we then pooled them to obtain estimates of this measure's utility in risk assessment, including survival models predicting time to AD dementia.
METHODS
The 2 samples' characteristics are described briefly; details are provided in appendix e-1 on the Neurology® Web site at www.neurology.org. Sample 1 (Massachusetts General Hospital [MGH]) included community volunteers participating in a longitudinal study. The participants in sample 2 (Rush) were recruited for a longitudinal imaging project19 from the community by the Rush AD Center, the Religious Order Study, or the Rush Memory and Aging Project. In both studies, subjects were excluded if they had significant active medical, neurologic, or psychiatric illness, or major vascular risk factors or disease (i.e., atrial fibrillation, insulin-dependent diabetes mellitus, cerebral infarcts, cardiac bypass graft surgery). At baseline, both samples underwent comprehensive clinical evaluations, neuropsychological testing, and MRI scans. Composite episodic memory Z scores were computed from multiple neuropsychological memory measures. All subjects underwent annual clinical evaluations and were determined to be CN, to be mildly impaired, or to have dementia. For the present analyses, we included all subjects in the original cohorts with a baseline CN status and at least 4 annual follow-up visits (1 Rush subject had 3 follow-up visits) after MRI. Because our hypotheses were focused on the presence of subtle neurodegenerative change in preclinical AD, we restricted the sample to individuals who remained CN at most recent follow-up (CN-stable) or those diagnosed with probable AD dementia (CN-AD converter), excluding those diagnosed with mild cognitive impairment (MCI, since their longer-term outcome is not yet known) or non-AD dementia. This resulted in sample sizes of 33 (MGH: 25 CN-stable, 8 CN-AD converters) and 32 (Rush: 25 CN-stable, 7 CN-AD converters). Nearly all CN-AD converters in both samples were evaluated at an intermediate time when their clinical status was MCI; 2 individuals in the Rush sample changed from CN to AD dementia without having been seen during that transitional stage.
MRI data were acquired very similarly for both samples using a General Electric (Milwaukee, WI) 1.5-T scanner and a 3-dimensional T1-weighted spoiled gradient recalled echo pulse sequence. The MRI parameters and analysis methods are described in previous publications and appendix e-1. We used an a priori set of 9 ROIs derived from a previous analysis18 (figure e-1) and a primary visual cortex ROI as a control region. ROIs were mapped to each individual and thickness values were obtained; a single measure was derived from the average thickness of all 9 ROIs, the AD-signature summary measure.
Group statistical comparisons were performed using analysis of variance with post hoc pairwise comparisons for continuous measures or χ2 for proportions (SPSS 16.0, Chicago, IL). Potential confounding factors, including age, gender, and education, were used as covariates. Separate univariate Cox regression models were constructed to investigate the relationship of covariates, memory Z scores, and AD-signature cortical thickness summary measures to the likelihood of progression from CN to AD dementia. A multivariate Cox regression model was then constructed including independent variables that reached a trend-level effect (p < 0.1) in the univariate analyses (p value-to-enter <0.05).
Standard protocol approvals, registrations, and patient consents.
Each participant gave written informed consent in accordance with institutional Human Subjects Research Committee guidelines.
RESULTS
Clinical characteristics.
The 2 samples were remarkably similar (table 1). Baseline Mini-Mental State Examination (MMSE) was within 1 to 2 points of ceiling, and baseline episodic memory scores were above or only slightly below the mean of large normative groups. Over nearly a decade of follow-up, CN-stable subjects remained clinically stable, an observation supported by unchanged MMSE and memory scores. In contrast, CN-AD converters declined to AD dementia, illustrated by substantial decrements in MMSE and memory scores.
Table 1.
Demographic and clinical characteristics of samplesa

Abbreviations: AD = Alzheimer disease; CN = cognitively normal; MGH = Massachusetts General Hospital; MMSE = Mini-Mental State Examination.
Values are mean (SD) unless otherwise noted.
p < 0.01.
p < 0.1 (trend-level effect).
p < 0.05.
p < 0.001.
For CN-stable, these are the last available values; for CN-AD converter, these are the values at the visit associated with time of diagnosis of AD dementia.
The trajectories of progressive decline are illustrated in figure 1, which shows (top) memory performance data, demonstrating the stability of CN-stable performance compared to substantial declines in the CN-AD converters, and (bottom) the trajectories of clinical measures for each individual CN-AD converter.
Figure 1. Baseline and longitudinal clinical characteristics of the 2 samples.

(A, B) Baseline and follow-up episodic memory Z scores from the 2 samples, illustrating the stability (or even slight improvement in sample 2) of performance in the cognitively normal (CN)-stable groups over nearly a decade of follow-up. In contrast, the CN-Alzheimer disease (AD) converters performed normally at baseline but declined substantially over nearly a decade of follow-up by the time of diagnosis of AD dementia. (C, D) Detailed illustration of cognitive decline in each of the CN-AD converter individuals. (C) Annual Clinical Dementia Rating (CDR)–sum of boxes scores increased from normal (0) at baseline to mild dementia in each of the 8 individuals in the Massachusetts General Hospital sample. (D) Annual episodic memory Z scores declined from normal to substantially impaired in each of the 7 individuals in the Rush sample. Breaks in lines indicate years for which data were not available.
Regional cortical thinning in relation to future AD dementia.
In both samples, the CN-AD converters showed thinner medial temporal lobe (MTL), temporal pole, and superior frontal gyrus (p < 0.05) than CN-stables. The MGH sample also demonstrated thinning in the inferior parietal lobule (angular and supramarginal gyri) and middle frontal gyrus (p < 0.05). Although not reaching statistical significance in the Rush sample, these and other AD-signature ROIs showed consistent effects in the expected direction (table 2). In contrast, the primary visual cortex did not show such a pattern (p > 0.1).
Table 2.
Mean (SD) region of interest measures by subject group (in mm)
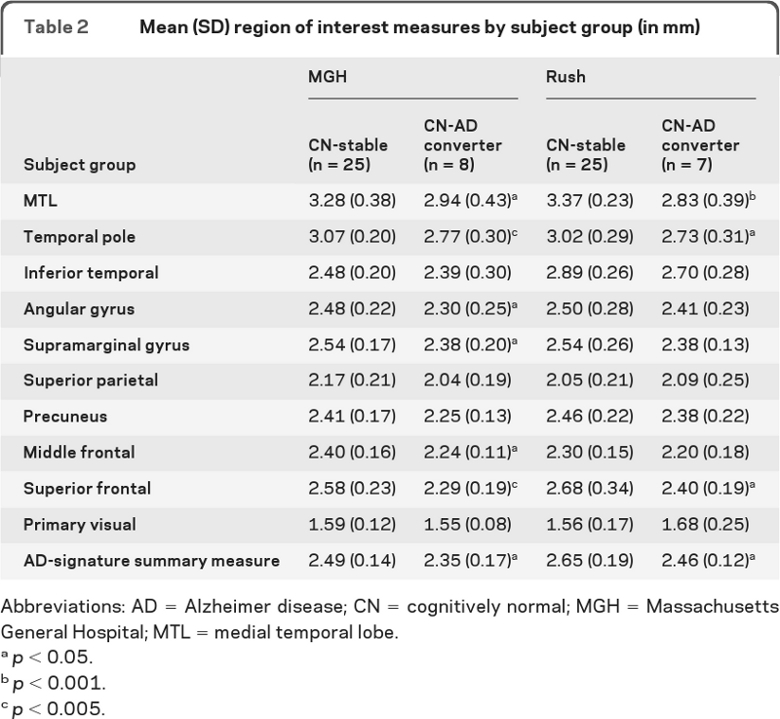
Abbreviations: AD = Alzheimer disease; CN = cognitively normal; MGH = Massachusetts General Hospital; MTL = medial temporal lobe.
p < 0.05.
p < 0.001.
p < 0.005.
Finally, the AD signature thickness summary measure demonstrated strikingly similar effects, with the CN-AD converters being 0.20 mm (MGH) or 0.19 mm (Rush) thinner than CN-Stable (figure 2, top). Cohen's d effect sizes were 1.3 and 1.2, indicating very large effects even though absolute magnitudes of group differences were less than ¼ millimeter. For comparison, the pooled-sample between-group effect size for baseline Episodic Memory Z scores was 0.73. Table e-1 contains effect sizes for each ROI.
Figure 2. Thinner cortex in regions associated with Alzheimer disease (AD) in asymptomatic adults who eventually develop dementia.

(A) For each of the 2 samples, the average cortical thickness of the AD-signature regions was slightly thinner in the cognitively normal (CN)-AD converter group than the CN-stable group at the time of the baseline MRI scan. The absolute difference in the means of the groups for each sample is less than ¼ mm, but because variance is small the Cohen d effect size between the groups is very large for each sample (>1.2). Inset illustrates AD-signature regions of interest. (B) Illustration of the risk of AD dementia as a function of AD-signature thickness from baseline MRI scans in the pooled group of all participants in this study. For the group of 9 individuals with high AD-signature thickness, none developed AD dementia in the follow-up period, while for the group of 45 individuals with average AD-signature thickness, 20% developed AD dementia in the follow-up period. A striking 55% of the 11 individuals with low AD-signature thickness developed AD dementia in the follow-up period.
Since the 2 samples' characteristics were so similar, they were pooled for subsequent analyses. To obtain a preliminary sense of whether the thickness measure might be a useful predictor, we divided the sample into 3 subgroups based on AD-signature thickness: 1) ≥1 SD below the entire cohort mean (low thickness subgroup), 2) within 1 SD of the mean (average), and 3) ≥1 SD above the mean (high). Of the 11 CN individuals with baseline low AD-signature thickness, 55% developed AD dementia over nearly the next decade. In contrast, 20% of the 45 average AD-signature thickness individuals developed AD dementia, while none of the 9 high AD-signature thickness individuals developed dementia, a highly significant difference (χ2 = 8.3, p < 0.005; figure 2, bottom). Figure e-1 presents a scatterplot illustrating these data.
In the 2 samples analyzed separately, low/average/high AD-signature thickness was associated with conversion-to-dementia rates of 67%/17%/0% (MGH) and 40%/24%/0% (Rush). This replication shows that low/average/high AD-signature thickness indicates high/middle/low risk for AD dementia nearly a decade in the future, with the pooled analysis above providing the best estimates of the actual likelihood for each level of risk.
Prediction of time to dementia in asymptomatic older adults.
Univariate Cox regression models of covariates indicated that gender was a predictor of time to dementia (greater risk for men, p < 0.005) but age, education, APOE status, and MMSE were not (p > 0.1), at least in this small sample. Univariate and multivariate (with gender) Cox regression models indicated that the AD-signature thickness measure was a strong predictor of time to dementia (univariate model: hazard ratio [HR] = 3.5 for 1 SD decrease in thickness; 95% confidence interval [CI] 2.0–6.4; p < 0.00005; multivariate model: HR = 3.4 for 1 SD thickness decrease; 95% CI 1.7–6.9; p < 0.0005). Figure 3 presents a survival plot illustrating these results. The mean time to a diagnosis of AD dementia in the pooled sample was 8 years (SD = 1.9).
Figure 3. Alzheimer disease (AD)-signature MRI biomarker predicts time to dementia in people who were cognitively normal when scanned.

Univariate survival plot of predicted time to AD dementia for hypothetical average study participants with AD-signature cortical thickness in the lowest (smallest) tertile, middle tertile, and highest tertile. The displayed survival curves are therefore model predictions and do not directly represent subject results. AD-signature thickness was predictive of progression from normal cognition to AD dementia (hazard ratio 3.5 for 1 SD decrease in thickness; 95% confidence interval 2.0–6.4; p < 0.00005) in the model. The mean thickness of the lowest tertile was 1.1 standard deviations thinner than the mean of the entire group of all participants, the mean of the middle tertile was at approximately the mean of the entire group, and the mean of the highest tertile was 1.1 standard deviations thicker than the mean.
In comparison, episodic memory Z score was also predictive of conversion but at a slightly weaker level than AD-signature thickness (univariate model: HR = 3.1 for 1 SD decrease in memory performance; 95% CI 1.6–6.1; p < 0.001; multivariate model: HR = 3.2 for 1 SD decrease in performance; 95% CI 1.5–6.6; p < 0.001).
DISCUSSION
Two decades of investigation have led to a mature understanding of the brain regions that are consistently atrophic in patients with AD dementia. Here we looked further back in the trajectory of AD and demonstrated that subtle neuroanatomic abnormalities are detectable in asymptomatic individuals nearly a decade before they are diagnosed with AD dementia, and are useful not only for assessing risk of AD dementia but also for predicting time to onset. Despite the small sample size, the striking consistency of these findings in 2 independent samples supports their generalizability.
Although sophisticated tools are now available for investigating changes in brain structure, function, molecular profiles, and behavior, the challenges of longitudinal research have made it difficult to study the full course of AD from preclinical to prodromal to dementia. Partly because they are time-, labor-, and cost-intensive, very few longitudinal studies have been completed of CN individuals who are followed until a clinical diagnosis of AD dementia. In one of the earliest studies of the decline of elderly individuals without dementia to AD dementia, smaller baseline hippocampal volume was present,10 as also observed in a more recent epidemiologic study.14 Another investigation of CN individuals demonstrated that ventricular volume was predictive of cognitive decline (11 converted to MCI, 2 to AD), but hippocampal, entorhinal, or whole brain volume were not.20
A voxel-based morphometry study found reduced MTL and parietal gray matter in 23 CN individuals who later developed MCI,13 and another analysis of the same sample using manual ROIs demonstrated hippocampal/entorhinal atrophy.15 Two other investigations found subtle shape abnormalities in hippocampal subregions in CN individuals who progressed to MCI.16,17
Very recently, a group of 9 CN individuals harboring brain amyloid who progressed to very mild AD dementia were shown to have lower baseline parahippocampal volumes compared to a stable CN group.8 We previously demonstrated that CN adults with brain amyloid have subtle AD-signature cortical thinning compared to CN adults without brain amyloid, but these individuals had not yet been followed longitudinally.18
Finally, a few studies have identified MTL and whole brain atrophy in the presymptomatic stage of genetically determined early-onset familial AD, again supporting the concept that atrophy, while subtle, may be present for years before AD symptoms.9,11,12
From the perspective of quantitative imaging biomarkers, the present study adds to prior literature in several ways. First, most prior work has employed manual tracings of one or a few ROIs. While neuropathologic studies21–23 made MTL structures a logical place to focus when investigating brain regions in which the earliest atrophy occurs in AD, recent imaging research has highlighted ventrolateral temporal, lateral parietal, and the posterior cingulate/precuneus as also involved early in the disease course.11,13,24,25
Here a relatively new technique for quantitative neuroanatomic measurement (cortical thickness analysis)—employed using a novel a priori approach focusing on brain regions known to be consistently affected early in AD—enabled the detection of statistically robust (large effect sizes due to small measurement error), replicable group differences of very subtle absolute magnitude (∼¼ mm) in MRI scans now considered previous generation technology. Such observations highlight the value of returning to older MRI datasets when new measurement technology is developed.
More importantly, these measures can be efficiently, reliably obtained from single individuals. A determination of whether AD-signature thickness is low, average, or high relative to similarly aged individuals appears valuable in assessing risk of future AD dementia, a finding replicated across both samples. Furthermore, the Cox regression model results are powerful in that they augment the risk assessment with an estimate of the potential time to AD dementia. Whether such an approach will prove useful in a generalizable manner is an important research topic.
A critical element of studies aiming to identify biomarkers of asymptomatic AD is the cognitive assessment methodology. Contemporary study designs include the use of questionnaires or careful structured interviews to ascertain the presence and severity of symptoms of declining memory and cognition in daily life. Questions are asked of both the individual and usually a knowledgeable informant with regard to whether the individual has experienced a decline in his or her ability to remember the details of recent events or conversations, items to purchase while shopping, or how to navigate to familiar places. Changes of this sort, particularly when corroborated by an informant, are in some cases associated with atrophy in neural circuits subserving memory26 and may potentially be predictors of future decline and dementia,27 although reports are inconsistent.28
Some individuals who do not endorse symptoms of memory impairment in daily life may exhibit signs of early impairment on detailed neuropsychological performance-based testing. Verbal list learning, paired associates, and similar challenging memory tests may provide objective evidence of mild impairments in individuals who otherwise appear normal when tested but who ultimately develop cognitive decline and AD dementia after follow-up.7 With longitudinal cognitive testing, evidence of decline (or in some cases absence of a practice effect) can be seen for 5–10 years or more prior to AD dementia.1–6 Given the challenges in these types of assessments, the clinical designation of “cognitively normal” should not be used lightly and the methods for making this determination deserve further investigation and discussion.
Three points regarding the cognitive test performance characteristics of the present participants are worth discussion. First, coloring the interpretation of all of these tests, the participants were relatively well-educated with all but 3 individuals having at least a high-school education and more than half having at least a college degree. Second, no individual performed below 28 on the MMSE at baseline. Third, although the preclinical AD dementia groups performed slightly lower on baseline memory testing than those remaining stable, performance would have been considered within the normal range for nearly all individuals. Only 3 individuals performed more than 1 SD below the mean (scores were 1.0–1.5 SD below the mean), and one remained clinically stable as CN nearly a decade later. Nevertheless, episodic memory measures were useful in prediction of AD dementia, with similar predictive power in the Cox model as the MRI measure. We interpret this to mean that the CN individuals with preclinical AD may perform within the low-normal range on memory testing (presumably lower than they would have performed previously) because of the early neurodegenerative change being detected by this MRI technique. Longitudinal studies support the idea that a causal link can be detected between subtle cortical atrophy at baseline in CN individuals and subsequent decline in memory performance.29
Studies going back to the 1960s have highlighted the presence of AD pathology in individuals who die within a relatively short time after having been assessed as CN,30–34 and extensive investigations have demonstrated the dissociations that can be present between the quantitative burden of AD pathology and the degree of cognitive impairment.23,35 These and other observations illustrate the distinction between the neuropathology of AD as a biological disease as opposed to the clinical syndrome of AD dementia, which, although strongly coupled, are by no means synonymous. Factors such as cognitive reserve have been invoked to explain why some individuals may remain CN despite the presence of substantial AD neuropathology.36–38 Nevertheless, it is becoming clear that the presence of AD pathology, as ascertained via amyloid imaging or CSF analysis, in CN individuals appears to elevate the probability of subsequent cognitive decline and dementia.8,39 Efforts are increasing to elucidate relationships between the presence and severity of these surrogate molecular markers of AD neuropathology and the timing of the development of symptoms. The present data demonstrate that in CN older adults a thinner cerebral cortex in regions typically affected by AD is predictive of time to onset of AD dementia, providing an additional tool to link the biological changes of the disease with the symptoms of the illness. We conceptualize this measure as a marker of early neurodegenerative change which appears to be of potential value among the candidate biomarkers being proposed as part of the new research criteria for the diagnosis of preclinical AD.40
The major limitation of this study is the relatively small sample size. Yet most prior studies that resemble the present one also include small samples. The field is now primed for larger, prospectively designed studies of this sort, although many challenges remain in making such cumbersome investigations as efficient as possible. Such studies will require as much as 10 years of follow-up and may need to employ new methodologic advances, including not only molecular imaging and CSF analysis, but also telephone-based cognitive screening and imaging-guided neuropathologic assessment, to make them at once cost-effective and informative. Extending these investigations to more representative samples would also be a valuable goal, since prior cohorts including the present ones include highly selected, generally healthy individuals who are motivated to participate in this type of research. Another limitation is that the computational analysis technology required to produce these measurements, while publicly available, requires nontrivial expertise and computer systems. Finally, although prior work has demonstrated that cortical thickness measures are highly reliable across different scanner platforms and field strengths,41 it is not clear whether the differences between the measures in the 2 samples reported here are attributable to true biological variability, instrument variability, or other factors. Further investigation of these topics will be required.
ACKNOWLEDGMENT
The authors thank the faculty and staff of the Massachusetts ADRC and the Rush ADC for their expertise in coordinating and evaluating participants; and the participants in this study and their families for their contributions.
Supplemental data at www.neurology.org
- AD
- Alzheimer disease
- CI
- confidence interval
- CN
- cognitively normal
- HR
- hazard ratio
- MCI
- mild cognitive impairment
- MGH
- Massachusetts General Hospital
- MMSE
- Mini-Mental State Examination
- MTL
- medial temporal lobe
- ROI
- region of interest
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Dickerson.
DISCLOSURE
Dr. Dickerson serves on the editorial board of Hippocampus and receives research support from the NIH and the Alzheimer's Association. Dr. Stoub reports no disclosures. Dr. Shah receives research support from Ceregene, Danone Research B.V., Eisai Inc., Elan Corporation, Merck Serono, Pamlab, L.L.C., Orasi, Inc., Pfizer Inc, the NIH, and the Illinois Department of Public Aid Alzheimer's Disease Assistance Center. Dr. Sperling served on the editorial board of Alzheimer's Disease and Associated Disorders; has served as a consultant for Elan Corporation, Wyeth, Bristol-Myers Squibb, Pfizer Inc, and Bayer Schering Pharma; and receives research support from Elan Corporation, Janssen, Bristol-Myers-Squibb, NIH/NIA, the Alzheimer's Association, the Anonymous Foundation, and the American Health Assistance Foundation. Dr. Killiany receives research support from the NIH. Dr. Albert serves as a consultant for Genentech Inc. and Eli Lilly and Company and receives research support from the NIH/NIA. Dr. Hyman serves on a scientific advisory board for NeuroPhage; serves as a consultant for FoldRx Pharmaceuticals, Pfizer Inc, EMD Serono, Inc., Janssen, Takeda Pharmaceutical Company Limited, Bristol-Myers Squibb, NeuroPhage, Campbell Alliance, Quanterix, VBI Belgium, and Schlesinger Associates & The Research House; receives research support from Fidelity Biosciences, the NIH and the Alzheimer's Association; and holds stock in Novartis. Dr. Blacker receives research support from the NIH and the Alzheimer's Association. Dr. deToledo-Morrell serves on the editorial board of Neurobiology of Aging and receives research support from the NIH/NIA.
REFERENCES
- 1. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol 2008;64:492–498 [DOI] [PubMed] [Google Scholar]
- 2. Rubin EH, Storandt M, Miller JP, et al. A prospective study of cognitive function and onset of dementia in cognitively healthy elders. Arch Neurol 1998;55:395–401 [DOI] [PubMed] [Google Scholar]
- 3. Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. J Int Neuropsychol Soc 2008;14:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hall CB, Lipton RB, Sliwinski M, Stewart WF. A change point model for estimating the onset of cognitive decline in preclinical Alzheimer's disease. Stat Med 2000;19:1555–1566 [DOI] [PubMed] [Google Scholar]
- 5. Howieson DB, Dame A, Camicioli R, Sexton G, Payami H, Kaye JA. Cognitive markers preceding Alzheimer's dementia in the healthy oldest old. J Am Geriatr Soc 1997;45:584–589 [DOI] [PubMed] [Google Scholar]
- 6. Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol 2009;66:1254–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blacker D, Lee H, Muzikansky A, et al. Neuropsychological measures in normal individuals that predict subsequent cognitive decline. Arch Neurol 2007;64:862–871 [DOI] [PubMed] [Google Scholar]
- 8. Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009;66:1469–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fox NC, Warrington EK, Freeborough PA, et al. Presymptomatic hippocampal atrophy in Alzheimer's disease: a longitudinal MRI study. Brain 1996;119:2001–2007 [DOI] [PubMed] [Google Scholar]
- 10. Kaye JA, Swihart T, Howieson D, et al. Volume loss of the hippocampus and temporal lobe in healthy elderly persons destined to develop dementia. Neurology 1997;48:1297–1304 [DOI] [PubMed] [Google Scholar]
- 11. Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN. Imaging of onset and progression of Alzheimer's disease with voxel-compression mapping of serial magnetic resonance images. Lancet 2001;358:201–205 [DOI] [PubMed] [Google Scholar]
- 12. Schott JM, Fox NC, Frost C, et al. Assessing the onset of structural change in familial Alzheimer's disease. Ann Neurol 2003;53:181–188 [DOI] [PubMed] [Google Scholar]
- 13. Smith CD, Chebrolu H, Wekstein DR, et al. Brain structural alterations before mild cognitive impairment. Neurology 2007;68:1268–1273 [DOI] [PubMed] [Google Scholar]
- 14. den Heijer T, Geerlings MI, Hoebeek FE, Hofman A, Koudstaal PJ, Breteler MM. Use of hippocampal and amygdalar volumes on magnetic resonance imaging to predict dementia in cognitively intact elderly people. Arch Gen Psychiatry 2006;63:57–62 [DOI] [PubMed] [Google Scholar]
- 15. Martin SB, Smith CD, Collins HR, Schmitt FA, Gold BT. Evidence that volume of anterior medial temporal lobe is reduced in seniors destined for mild cognitive impairment. Neurobiol Aging 2011;31:1099–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Apostolova LG, Mosconi L, Thompson PM, et al. Subregional hippocampal atrophy predicts Alzheimer's dementia in the cognitively normal. Neurobiol Aging 2010;31:1077–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Csernansky JG, Wang L, Swank J, et al. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005;25:783–792 [DOI] [PubMed] [Google Scholar]
- 18. Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009;19:497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeToledo-Morrell L, Stoub TR, Bulgakova M, et al. MRI-derived entorhinal volume is a good predictor of conversion from MCI to AD. Neurobiol Aging 2004;25:1197–1203 [DOI] [PubMed] [Google Scholar]
- 20. Jack CR, Jr, Shiung MM, Weigand SD, et al. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 2005;65:1227–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259 [DOI] [PubMed] [Google Scholar]
- 22. Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging 1991;12:295–312 [DOI] [PubMed] [Google Scholar]
- 23. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42:631–639 [DOI] [PubMed] [Google Scholar]
- 24. Thompson PM, Mega MS, Woods RP, et al. Cortical change in Alzheimer's disease detected with a disease-specific population-based brain atlas. Cereb Cortex 2001;11:1–16 [DOI] [PubMed] [Google Scholar]
- 25. Good CD, Scahill RI, Fox NC, et al. Automatic differentiation of anatomical patterns in the human brain: validation with studies of degenerative dementias. Neuroimage 2002;17:29–46 [DOI] [PubMed] [Google Scholar]
- 26. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology 2006;67:834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmand B, Jonker C, Hooijer C, Lindeboom J. Subjective memory complaints may announce dementia. Neurology 1996;46:121–125 [DOI] [PubMed] [Google Scholar]
- 28. Jorm AF, Masaki KH, Davis DG, et al. Memory complaints in nondemented men predict future pathologic diagnosis of Alzheimer disease. Neurology 2004;63:1960–1961 [DOI] [PubMed] [Google Scholar]
- 29. Murphy EA, Holland D, Donohue M, et al. Six-month atrophy in MTL structures is associated with subsequent memory decline in elderly controls. Neuroimage 2010;53:1310–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomlinson BE, Blessed G, Roth M. Observations on the brains of non-demented old people. J Neurol Sci 1968;7:331–356 [DOI] [PubMed] [Google Scholar]
- 31. Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 2009;30:1026–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 1988;38:1682–1687 [DOI] [PubMed] [Google Scholar]
- 33. Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988;23:138–144 [DOI] [PubMed] [Google Scholar]
- 34. Dickson DW, Crystal HA, Mattiace LA, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 1992;13:179–189 [DOI] [PubMed] [Google Scholar]
- 35. Hyman BT, Marzloff K, Arriagada PV. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol 1993;52:594–600 [DOI] [PubMed] [Google Scholar]
- 36. Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010;67:353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roe CM, Mintun MA, D'Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Arch Neurol 2008;65:1467–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20:S69–S74 [DOI] [PubMed] [Google Scholar]
- 39. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007;64:343–349 [DOI] [PubMed] [Google Scholar]
- 40. Sperling RA, Aisen PS, Beckett LA, et al. Towards defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging and the Alzheimer Association Workgroup. Alzheimers Dement (in press 2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dickerson BC, Fenstermacher E, Salat DH, et al. Detection of cortical thickness correlates of cognitive performance: Reliability across MRI scan sessions, scanners, and field strengths. Neuroimage 2008;39:10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]