

Summary
The vitamin D receptor (VDR) functions as an obligate heterodimer with the retinoid X receptor (RXR). These nuclear receptors (NRs) are multidomain proteins and it is unclear how various domains interact with one another within the NR heterodimer. Here we show that binding of intact heterodimer to DNA alters the receptor dynamics in regions remote from the DNA binding domains (DBDs), including the coactivator binding surfaces of both coreceptors, and the sequence of the DNA response element can specify the dynamics. Furthermore, agonist binding to the heterodimer results in changes in the stability of the VDR DBD, indicating that ligand itself may play a role in DNA recognition. These data suggest a mechanism by which NRs can display promoter-specific activity and impart differential effects on various target genes, which provides mechanistic insight for the function of selective NR modulators.
Keywords: DNA, allosteric communication, co-activator binding surfaces
Introduction
VDR plays a critical role in mineral homeostasis and has been implicated in a range of human disorders and diseases such as osteoporosis1, obesity2, autoimmune disease3, and cancer4-5. VDR is a ligand-dependent transcription factor and member of the NR superfamily and as such is composed of four major functional domains. The highly variable amino-terminal A/B domain is known to be important for NR transactivation but its structural elements are poorly defined. Adjacent to the A/B domain is the highly conserved zinc finger containing DBD (C domain). The hinge domain (D domain) provides the link between the DBD and LBD (E domain). The LBDs of the NRs are multifunctional and have secondary domain structure that is characteristic of all NRs. The LBD facilitates ligand binding, nuclear localization, dimerization, and interaction with coactivator and corepressor proteins. NRs can function as monomers, homodimers, or heterodimers with RXR. VDR functions as an obligate heterodimer with RXR and recognizes specific DNA elements known as vitamin D response elements (VDREs). The activated RXR-VDR heterodimer recruits coregulator complexes in proximity to DNA to remodel chromatin and alter gene transcription in a ligand-dependent manner6.
Our functional understanding of NRs has relied greatly on structural studies, involving either the LBD or DBD fragment alone. However, there is little structural information about full-length nuclear receptors, and to date, there has been only a single crystal structure of a nearly intact NR (PPARγ-RXRα) co-interacting with DNA solved7. Thus, there is a paucity of structural information regarding how the domains of NRs communicate with one another both intra-molecularly and inter-molecularly within a functional dimer. To address these issues, we employed hydrogen/deuterium exchange (HDX) to probe the influence of ligand, DNA, and coactivator on receptor dynamics in an effort to understand the molecular mechanism of activation of the VDR-RXR complex. HDX, particularly when coupled with mass spectrometry (MS), has emerged as a rapid and sensitive approach for characterization of protein dynamics and protein-ligand interactions8-10. Our laboratory and others have successfully applied HDX to the mechanistic analysis of nuclear receptor activation9,11-17. We have described a novel mechanism of ligand-activation of PPARγ (peroxisome proliferator-activated receptor gamma) where different binding modes were detected between full and partial agonists and determined that partial agonists activate the receptor in a helix 12 (H12) independent fashion relying on stabilization of other regions of the ligand binding pocket (LBP)11. This work has led to new insights into an alternative mechanism correlating action of anti-diabetic drugs with modulation of genes dysregulated in obesity18. We have also applied HDX to classify various selective ERα modulators (SERMs) based on their HDX signatures and it was determined that these signatures were correlative to the pharmacological profiles of these ligands in both pre-clinical and clinical settings16. While these studies were performed with isolated LBDs of the specific NR, there are several examples of a strong correlation between the HDX profiles of the LBD and full length forms of the same NRs when bound to specific ligands. For instance, HDX analysis of intact PPARγ-RXRα heterodimer was published along with the crystal structure of the intact complex7. The HDX profile of PPARγ in the intact complex is in good agreement with that previously published for the isolated LBD. Moreover, we have compared HDX profiles obtained with ligands in complex with both the human VDR LBD and the full-length RXRα-VDR heterodimer and found that the perturbation in receptor conformation induced by ligand binding to the isolated LBD is well maintained in the full length heterodimer with deviation observed only within the RXRα-VDR dimer interface19.
Here we investigated the dynamics of the human RXRα-VDR heterodimer upon interaction with VDR agonist (1,25D3) and RXRα agonist (9-cis-RA), along with DNA, and the receptor interacting domain (RID) of human steroid receptor coactivator 1 (SRC1). These studies provide for the first time a detailed map of local and global cooperative influence of both ligands and DNA on the coactivator binding surface of an NR complex and convey the essential nature of long-range allosteric interactions between domains within an NR heterodimer. Combined the studies presented here confirm the orientation of the heterodimer on DNA and provide insight into the permissive nature of the RXRα-VDR heterodimer.
Results
Differential HDX was employed to probe the conformational dynamics of the VDR-RXRα heterodimer upon binding cognate ligands, DNA, and coactivator. A schematic of all experiments is shown in Supplementary Table 1a. Independent measures of affinities were done to confirm that complexes used under HDX conditions were expected to be well formed. In the absence of a crystal structure of the RXRα-VDR heterodimer, we produced a model of the RXRα-VDR LBD and RXRα-VDR DBD heterodimer separately by docking the VDR LBD into PPARγ-RXRα crystal structure7 by superimposition with Coot20 and minimization with Chimera21; and remodeled the structure of the DBDs on VDRE DR3 using the structure of RXR-VDR DBDs on VDRE DR322.
The RXRα-VDR heterodimer interface
We investigated changes in conformational dynamics or stabilization that occur within VDR and RXRα upon heterodimerization. First we compared the HDX profile of VDR and RXRα alone to the HDX profile of the RXRα-VDR heterodimer (Fig. 1). Although the apo VDR was relatively unstable, addition of RXRα produced a stable heterodimer as expected19. Due to the poor stability of full-length VDR alone, we performed HDX on the isolated LBD of VDR in the absence and presence of RXRα to facilitate analysis of the dimer interface from the VDR perspective. The addition of RXRα to the VDR LBD induced a statistically significant increase in protection to solvent exchange in the region 317-325 (H7) of the VDR subunit versus the VDR LBD alone (Fig. 1a and Supplementary Table 1b(i–1)) indicating increased stabilization (less conformationally dynamic) in this region. Moreover, the addition of VDR to RXRα induced significant protection from solvent exchange in the regions 347-353 (H7) and 419-432 (H10-H11) of the RXRα subunit compared to RXRα alone (Fig. 1b and Supplementary Table 1c(i–2)). Consistent with the HDX profiles, high affinity interaction between the two subunits of the heterodimer was also observed using AlphaScreen (Supplementary Fig. 1a). The HDX data indicate that the dimerization interface between VDR and RXRα involves, at a minimum, residues 317-325 of VDR and residues 347-353 and 419-432 of RXRα. This suggests that the arrangement of the LBDs within the RXRα-VDR heterodimer closely resembles that of RXRα-PPAR 23 and RXRα-RAR 24, in which the dimer interface consists of helices H7, H9, H10, and H11, and loops L8-9 and L9-10.
Figure 1. The interactions along the dimer interface of the RXR-VDR heterodimer.
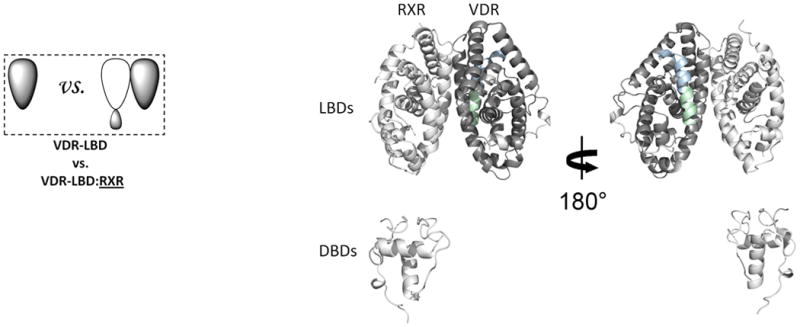
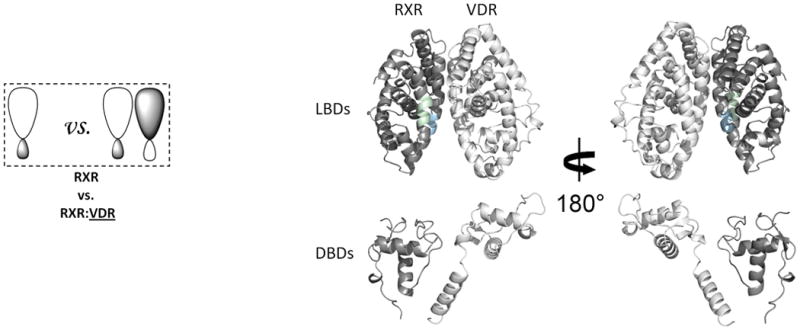
(a) The addition of RXR induced conformational changes in VDR LBD. The average differential HDX of VDR LBD vs. VDR LBD:RXR (Supplementary Table 1b(i–1)) mapped onto VDR LBD-RXR complex docking model. (b) The addition of VDR induced conformational changes in RXR. The average differential HDX of RXR vs. RXR:VDR (Supplementary Table 1c(i–2)) mapped onto RXR-VDR heterodimer docking model. Note: The uniform color legends indicating the differential HDX between two states were used throughout the entire manuscript and they were shown in Supplementary Table 1. The regions in the structural model colored in white mean they are not covered in this study.
Impact of 1,25D3 on RXRα-VDR
We next examined the impact of ligand binding (1,25D3) on the conformational dynamics and stability of the RXRα-VDR heterodimer (Fig. 2a). Conditions used for ligand addition were saturating, due to the high affinity of 1,25D3 for the heterodimer (Kd ∼0.42nM, determined using a thermal stability binding assay; Supplementary Fig. 1c). The addition of 1,25D3 to the heterodimer resulted in a large magnitude change within the VDR LBD (Fig. 2a and Supplementary Table 1b(ii)), yet ligand binding did not impact the binding efficiency of RXRα to VDR (Supplementary Fig. 1a). The perturbations in the HDX profile of the VDR subunit upon 1,25D3 binding were similar to those observed upon 1,25D3 binding to the VDR LBD alone19. There was a strong correlation between the regions of VDR that were protected from solvent exchange upon binding 1,25D3 and the regions of VDR known to directly interact with ligand25. The 1,25D3-VDR cocrystal structure shows that H12 (412-419), a region of the receptor critical for coactivator binding and activation, made direct Van der Waals contacts with the ligand as well as several intermolecular interactions with neighboring residues that directly interact with the 1,25D3. Consistent with the structure, ligand binding afforded robust reduction in HDX within H12. Moreover, residues 134-150 (H1), 225-244 (H3), 273-329 (H5-H7) and 391-403 (H11), regions of VDR within the ligand binding pocket (LBP), displayed significant perturbation in HDX, which can all be attributed to hydrogen bonds between hydroxyl groups of 1,25D3 and interacting residues within the LBP19. Interestingly, despite being remote from the LBP, ligand binding increased solvent exchange in the DBD of VDR suggesting that ligand binding can directly impact the conformation of the DBD and potentially modulate its function.
Figure 2. Ligand-induced domain-domain interactions within the RXR-VDR heterodimer complex.
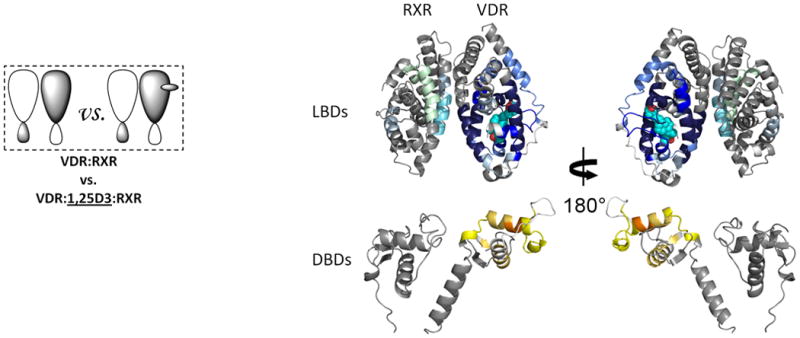
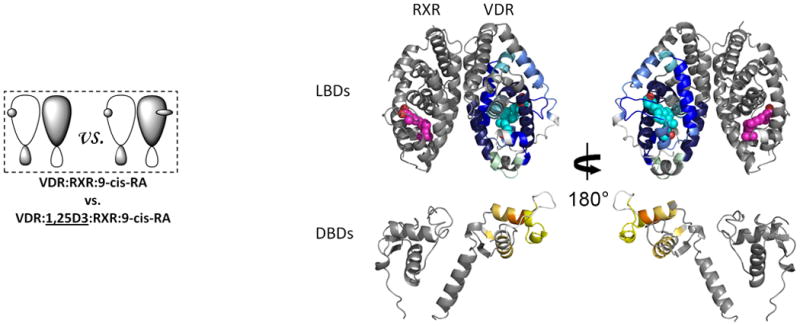
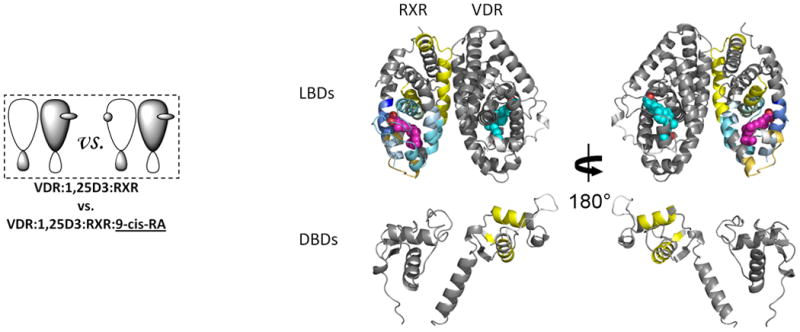
Differential HDX data mapped onto RXR-VDR docking model demonstrates (a) 1,25D3 (Supplementary Tables 1b(ii),and 1c(ii)) (b) 9-cis-RA (Supplementary Tables 1b(iii) and 1c(iii)) induced conformational changes of the RXR-VDR heterodimer complex as shown by comparing the deuterium incorporation of both receptors in the absence presence of ligands. (c) 1,25D3 induced conformational changes of the RXR-VDR heterodimer complex bound by 9-cis-RA (Supplementary Tables 1b(iv) and 1c(iv)). (d) 9-cis-RA induced conformational changes of the RXR-VDR heterodimer complex when bound to 1,25D3 (Supplementary Tables 1b(v) and 1c(v)).
To elucidate the mechanism driving the allosteric communication between coreceptors, we investigated the change in dynamics of RXRα upon binding of 1,25D3 to the heterodimer. The HDX profile of RXRα was largely unaffected by 1,25D3 binding with the exception of three regions. Significant reduction in HDX was observed for residues 347-362 (H7) and 419-425 (H10), regions within the heterodimer interface (Fig.2a and Supplementary Table 1c(ii)), suggesting that 1,25D3 binding favors coreceptor interaction to stabilize the heterodimer26. The third region, residues 271-279 (H3) of RXRα, is remote of the heterodimer interface and displayed reduced HDX, suggesting a 1,25D3-dependent allosteric communication between coreceptors.
Impact of 9-cis-RA on RXRα-VDR
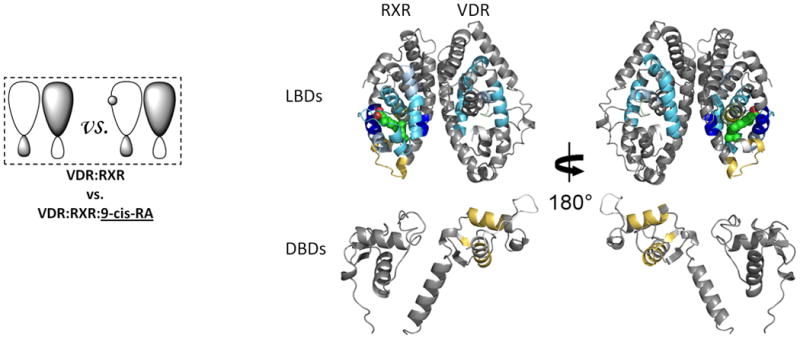
Ligand binding to RXR has been implicated in allosteric regulation of several RXR coreceptors27-28. Thus, we probed the impact of 9-cis-RA binding on the conformational dynamics of the RXRα-VDR heterodimer (Fig. 2b). Binding of 9-cis-RA afforded decreased HDX in the RXRα subunit (Supplementary Table 1c(iii)) similar to that observed for binding to the RXRα LBD alone13 and to intact receptor alone (data not shown). The HDX profile correlates well with the cocrystal structure of the 9-cis-RA:RXRα29. Three regions of RXRα displayed protection to HDX upon 9-cis-RA binding; 249-279 (H3), 309-362 (H5-H7, including β turn), and 429-438 (H10). These regions are contained within the LBP and involved in direct interaction with the ligand. The 9-cis-RA:RXRα structure shows that while ligand does not make direct contacts with H12 or the H11-H12 loop, H12 is repositioned by ligand suggesting it is stabilized indirectly by several hydrophobic interactions29. Contrary to the crystal structure, we observe increased HDX in H12. This observation is similar to HDX studies on ERα where estradiol (E2) binding does not impact HDX within H12, yet the cocrystal structure of E2:ERα shows a similar ligand-dependent repositioning of H12 in the absence of direct contact between H12 and ligand16,30. In contrast to the effects of 1,25D3 on the VDR DBD, no statistically significant changes in HDX were observed in the RXRα DBD upon binding 9-cis-RA.
As anticipated, allosteric modulation of coreceptor was observed upon 9-cis-RA binding to heterodimer (Fig. 2b and Supplementary Table 1b(iii)). Regions containing residues 309-329 (H7) of the VDR dimer interface displayed reduced HDX consistent with enhanced interaction between the coreceptors upon ligand binding. In addition, significant decrease in HDX was observed around residues 234-244 (H3) and 273-279 (H5) of VDR which are remote of the dimer interface. An interaction between these two regions has been shown to produce a gain of function in some NRs31. Unexpectedly, while 9-cis-RA binding did not perturb HDX within the RXRα DBD, it did induce a subtle but statistically significant increase in HDX within the VDR DBD.
Having identified components within the coreceptors that are altered upon dimerization and ligand binding, we examined the impact of VDR ligand binding to the heterodimer already bound by RXR ligand and vice-versa. The HDX profile of 1,25D3 binding in the presence of 9-cis-RA (Fig. 2c and Supplementary Table 1b(iv)), was similar to that observed upon binding 1,25D3 alone (Fig. 2a and Supplementary Table 1b(ii)) with only the magnitude of protection to exchange being attenuated in some regions. These regions with attenuated protection are identical to those perturbed upon binding 9-cis-RA alone and thus this attenuation comes from the allosteric effects of 9-cis-RA binding. Interestingly, perturbation in HDX within RXRα induced by 1,25D3 binding was blunted by the presence of 9-cis-RA (Fig. 2c and Supplementary Table 1c(iv)). Similarly, the presence of 1,25D3 blunts perturbation in HDX in VDR induced by 9-cis-RA binding (Fig. 2d and Supplementary Table 1b(v)). Perhaps more intriguing, both 1,25D3 and 9-cis-RA induced HDX perturbation in the DBD of VDR and this perturbation was not affected by the presence of the other ligand (Fig. 2; Supplementary Table 1b(ii),(iv); Supplementary Table 1b(iii),(v)). Thus, binding of either ligand to the heterodimer appears to destabilize the DBD of VDR and this effect is independent of the order of addition of ligand.
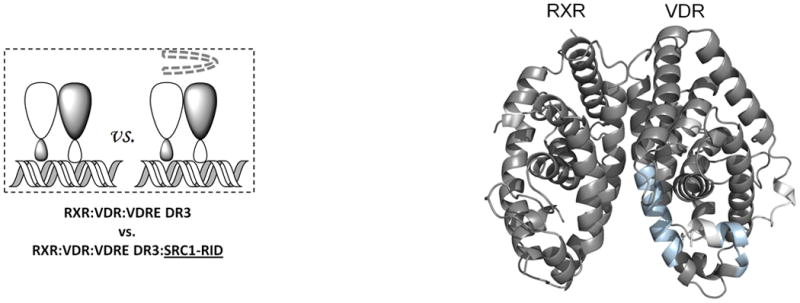
DNA Binding Modulates AF-2 in RXRα-VDR
Heterodimeric RXRα-VDR must bind to specific nucleotide sequences known as vitamin D response elements (VDREs) in genomic DNA to activate VDR target genes. HDX studies were performed to probe the influence of DNA binding on the conformational dynamics of the heterodimer in the presence or absence of ligands (Fig. 3). In these studies, the VDRE used contains two copies of the consensus half-site AGGTCA separated by 3 base pairs, forming a DR3 element. This element, VDRE DR3, was determined to have high binding affinity to the heterodimer complex (Kd=0.76 nM, Supplementary Fig. 1c) and under HDX analysis conditions, the concentration of DNA was saturating. DNA binding to apo heterodimer perturbed HDX in regions of both coreceptors that directly interact with DNA (Fig. 3a; Supplementary Tables 1b(vi) and 1c(vi)). Interestingly, stronger protection to solvent exchange was observed in the VDR DBD upon DNA binding as compared to the RXRα DBD (Supplementary Fig. 2a), suggesting that VDR makes more base and phosphate backbone interactions than RXRα, which is similar to that observed in the crystal structures of PPARγ-RXRα on PPRE DR17 and TR-RXR DBDs on TRE DR422. RXRα residues 169-192 and VDR residues 17-36 and 82-92 showed protection to HDX upon DNA binding (Supplementary Fig. 2b). These regions are not expected to engage DNA but would be at the interface of the two DBDs if the RXRα DBD resides upstream of the VDR DBD. This polarity is in agreement with biochemical studies32 and the proposed structure of RXRα-VDR DBDs on VDRE DR322. The recognition helix of both DBDs showed decreased HDX (Supplementary Fig. 2c), confirming that this conserved helix is inserted into the major groove in registration with the AGGTCA half site. Furthermore, the VDR hinge (CTE portion: residues 93-108) was also highly protected to HDX upon DNA binding (Supplementary Fig. 2d), suggesting perhaps that the hinge makes extensive interactions with DNA similar to that observed within the PPARγ–RXRα7 and Rev-Erb structures33 where PPARγ and Rev-Erb have their CTEs deeply embedded into the minor groove. However, other interpretations are possible to explain the stabilization of CTE such as intra- and inter- molecular interactions between the coreceptors. Unexpectedly, DNA binding impacted the HDX profile of the LBD portions of both coreceptors (Supplementary Fig. 3a; Supplementary Tables 1b(vi) and 1c(vi)). VDR residues 309-333 and 366-383 (H7-8 and H9-10) and RXRα residues 354-367 and 419-432 (H7 and H10) were stabilized upon DNA binding. These regions are within or directly adjacent to the dimerization interface of the coreceptors suggesting that DNA binding modulates both DBD-DBD and LBD-LBD interactions.
Figure 3. The interactions between RXR and VDR when bound to DNA.
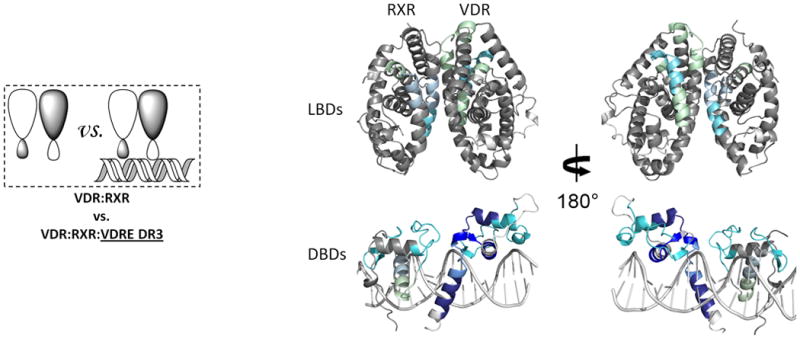
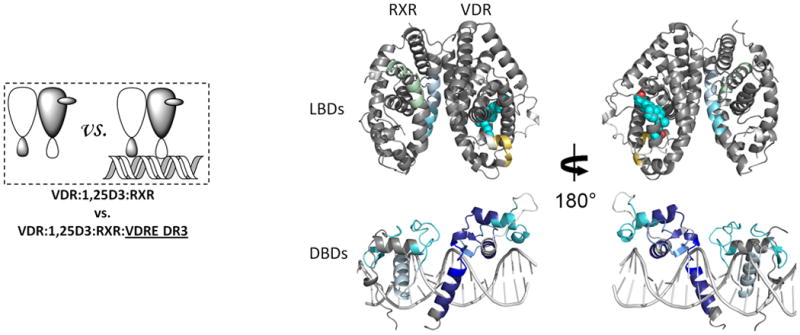
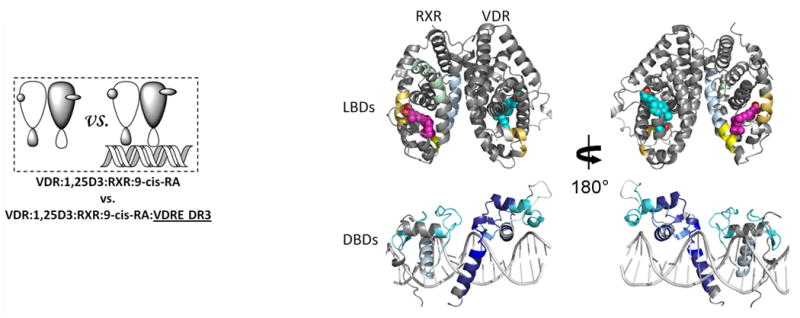
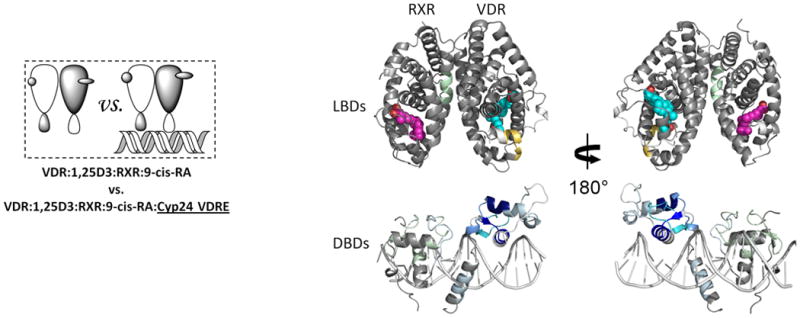
Differential HDX data mapped onto RXR-VDR docking model of DBD and LBD domains when bound to different ligands and DNA response elements. (a) The interactions between RXR and VDR on VDREdr3 in absence of ligands (Supplementary Tables 1b(vi) and 1c(vi)). (b) The interactions between RXR and VDR on VDREdr3 in presence of 1,25D3 only (Supplementary Tables 1b(vii) and 1c(vii)). (c) The interactions between RXR and VDR on VDREdr3 in presence of 1,25D3 and 9-cis-RA (Supplementary Tables 1b(viii) and 1c(viii)). (d) The interactions between RXR and VDR on Cyp24vdre in presence of 1,25D3 and 9-cis-RA (Supplementary Tables 1b(ix) and 1c(ix)).
The addition of 1,25D3 did not alter the HDX profile of RXRα bound to DNA (Fig. 3b, Supplementary Fig. 3b and Supplementary Table 1c(vii)), suggesting that although ligand binding stabilizes heterodimer formation, the enhanced interaction between coreceptors observed upon DNA binding is ligand-independent. DNA binding did not further reduce HDX on the VDR side of the heterodimer interface, H7-H8, (Fig. 3b, Supplementary Fig. 3b and Supplementary Table 1b(vii)) as HDX in this region was dramatically slowed upon 1,25D3 binding, making further reduction in HDX impossible to detect within the experimental time scale (Fig. 2a and Supplementary Table 1b(ii))19. Although the addition of 1,25D3 had minimal impact on the binding efficiency of heterodimer to VDRE DR3 (Supplementary Fig. 1b), the magnitude of protection to HDX within the DBD of VDR after DNA binding was greater in the presence of 1,25D3, which is consistent with our earlier observation that the 1,25D3 bound VDR DBD is more destabilized than the apo VDR DBD. Perhaps most surprising, H12 of VDR, which is remote from DNA binding and heterodimer interface, was destabilized suggesting that DNA binding directly influences AF2 conformation of VDR. Addition of 9-cis-RA to heterodimer in the presence of 1,25D3 did not further perturb the HDX profile of DNA bound DBDs nor the LBD of VDR (Fig. 3c, Supplementary Fig. 3c, Supplementary Tables 1b(viii) and 1c(viii)), but did have a subtle impact on the HDX profile of the LBD of RXRα (the difference between Supplementary Table 1c(vii) and (viii)) with two regions, H3 residues 271-279 and loop between H10-11 residues 428-438, showing decreased stability upon addition of 9-cis-RA (Fig. 3c, Supplementary Fig. 3c and Supplementary Table 1c(viii)). It has been proposed that the coactivator-binding surface on nuclear receptors is a cleft (AF2 surface) formed by H3, H3′ and H4 at the top and by the AF2 helix, H12, at the bottom23. These results demonstrate that DNA binding alters the conformational dynamics and stability of AF2 of both coreceptors, an observation that suggests DNA binding can directly influence coactivator recognition and binding.
Having observed VDRE DR3-induced perturbation of HDX in a number of important functional regions of the heterodimer, the experiment was repeated using a native VDRE, Cyp24 VDRE, which contains only one AGGTCA consensus half-site (Fig. 3d). Cyp24 VDRE, like VDRE DR3, stabilized the VDR DBD while destabilizing part of AF2 (Fig. 3d and Supplementary Table 1b(ix)). The most striking difference between HDX profiles induced by these two different VDREs was observed in the hinge of VDR (residues 93-108). Previous biochemical studies have implicated this region in DNA recognition34 where the specific sequence of DNA modulates the flexibility of the hinge and in turn assists DNA interaction. The magnitude of perturbation to HDX upon binding to Cyp24 VDRE was large on the VDR DBD yet minimal on the RXRα DBD suggesting that the AGGTCA half-site was occupied by VDR. Reduced stability of the heterodimer when bound to Cyp24 VDRE versus VDRE DR3 was reflected in reduced HDX protection in the RXR DBD (residues 130-197) and dimer interface (H7, residues 354-367 and H10, residues 419-432) (Fig. 3d and supplementary Table 1c(ix)). Furthermore, AF-2 of RXRα displayed no perturbation in HDX when bound to Cyp24 VDRE compared to VDRE DR3 binding. It is important to note that the heterodimer interacts with both Cyp24 VDRE and VDRE DR3 with similar efficacy (Supplementary Fig. 1b,c) demonstrating that the differential HDX induced by these two response elements is related to the difference in nucleotide sequence and not affinity suggesting that the specific sequence of the response element drives the alterations in conformational dynamics of the coreceptors.
Ligand and DNA modulate coactivator interaction
Ligand binding alters the conformational landscape of NRs creating a binding surface to facilitate interaction with coregulatory proteins35 such as SRC1. SRC1 interacts with nuclear receptors through an interaction domain that contains three conserved helical NR box motifs of the consensus LXXLL sequence36. The nature of the recognition of NR boxes by NRs has been examined in crystal structures where a lysine residue in H3–H4 and a glutamate residue in H12 define a “charge clamp” that allows the orientation and placement of the NR box into the coactivator binding AF2 surface37. Differential HDX was performed to examine the ligand-dependency of SRC1 interaction with the heterodimer (Fig. 4). Unlike many previous studies, a large fragment of SRC1 receptor interaction domain (SRC1 RID), containing three NR boxes, was used. SRC1 interaction with the heterodimer bound to both 1,25D3 and 9-cis-RA protected VDR residues 411-419 (H12) and RXRα residues 271-279 (H3) and 433-451 (H10-H12) from HDX (Fig. 4a, Supplementary Tables 1b(x) and 1c(x)). The significantly reduced HDX of VDR H12 due to the interaction with SRC1 RID (Supplementary Fig. 4a) correlates well with many biochemical and crystallographic studies that demonstrate the contribution of H12 in coactivator binding38. Unlike H12, no significant protection to HDX was observed in H3-H4 of VDR which contains the other side of the charge clamp. This can be explained by the large stabilization of H3 induced already by 1,25D3 binding (Supplementary Table 1b(ii)) 19 making it unlikely to detect a further reduction in HDX. The coactivator binding surface of RXRα can also be inferred from the HDX data and it is similar to that observed in PPAR-RXRα 23 and RAR/RXRα heterodimers 24. It is interesting that as indicated in these structures the loop between H10 and H11 (residues 428-438) does not directly interact with coactivator, although this region is important in the formation of the hydrophobic groove facilitating coactivator binding. HDX analysis revealed that this region is protected to solvent exchange (Supplementary Fig. 4b) demonstrating its involvement in coactivator interaction. In addition to the “charge clamp,” it has been observed that coactivator binding drives a concerted reorientation of the side chains of Phe437, Phe277 (H3) and Phe450 (H12) to form an “aromatic clamp” bringing these residues in close contact with the NR box39. In addition, Phe437 and Phe438, which are on the edge of the AF2 surface, are required for transcriptional activity as RXRα40.
Figure 4. Ligand dependency of SRC1-RID binding to RXR-VDR heterdimer complex.
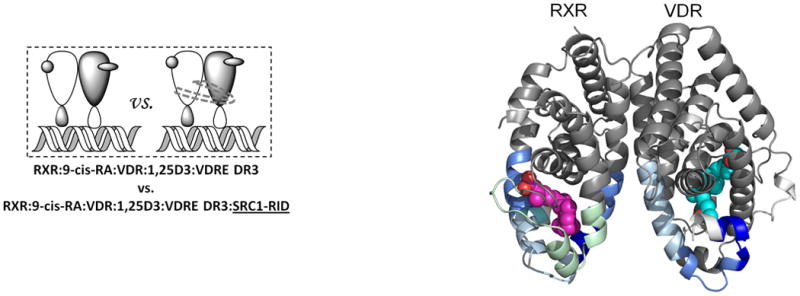
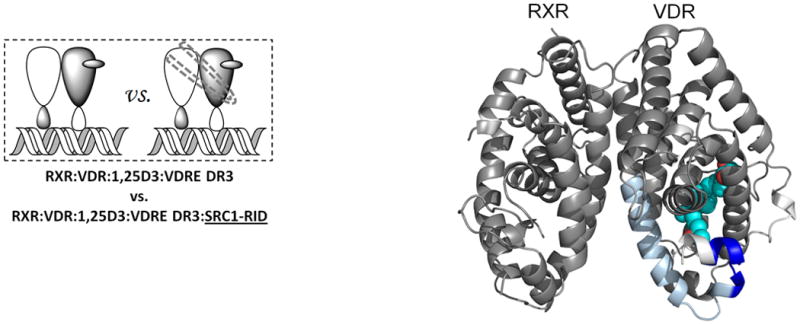
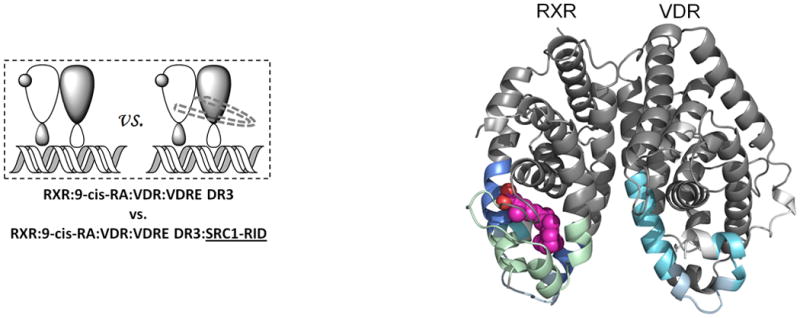
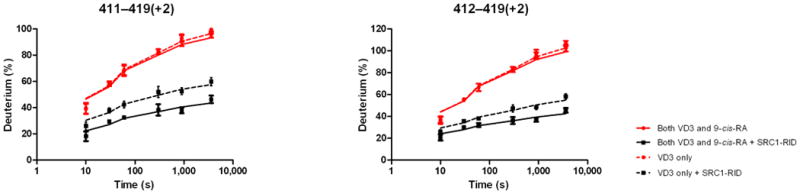
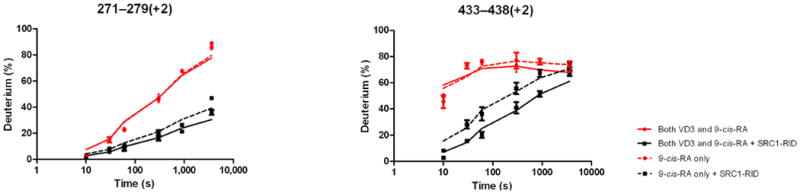
Differential HDX data mapped onto RXR-VDR docking model of LBD domains in presence of various ligands and SRC1-RID. (a) In presence of both 1,25D3 and 9-cis-RA (Supplementary Tables 1b(x) and 1c(x)). (b) In presence of 1,25D3 only (Supplementary Tables 1b(xi) and 1c(xi)). (c) In presence of 9-cis-RA only (Supplementary Tables 1b(xii) and 1c(xii)). (d) In absence of both ligands (Supplementary Tables 1b(xiii) and 1c(xiii)). Comparison of differential HDX dynamics of the peptides (residues 411-419 and 412-419) from VDR helix 12 (e) and the peptides (residues 271-279 and 433-438) from RXR H3 and H10-11 (f) induced by SRC1-RID binding. Solid lines represent the deuterium incorporation of the peptides from the heterodimer bound to both 1,25D3 and 9cis-RA in the presence or absence of SRC1-RID and the dotted lines represent the deuterium incorporation of the peptides from the heterodimer bound to either 1,25D3 (e) or 9-cis-RA (f) only in the presence or absence of SRC1-RID. The value in parentheses represents the charge state of the peptide ions. Data were the mean ± s.d. of triplicate individual measurements.
Either 1,25D3 or 9-cis-RA could drive coactivator interaction to its cognate coreceptor (Fig. 4b, Fig. 4c, Supplementary Tables 1b(xi),(xii) and 1c(xi),(xii)) and as expected, no interaction was observed in the absence of both ligands (Figure 4d, Supplementary Tables 1b(xiii) and 1c(xiii)). These results suggest that each coreceptor can interact with SRC1 RID independently (Supplementary Fig. 4). For this to be possible each coreceptor must interact with a unique NR box on one SRC1 molecule (1:1 SRC1:heterodimer) or each coreceptor bind a unique SRC1 molecule (2:1 SRC1:heterodimer). The HDX would support a model of synergistic binding of one molecule of SRC1 spanning both coreceptors (1:1 SRC1:heterodimer), as the HDX protections induced by SRC1 RID interaction with heterodimer when bound to either ligand alone was significantly enhanced in the presence of both ligands (Fig. 4e,f). To provide additional evidence for this model, cell-based functional assays were performed to examine the additive effects of 1,25D3 and 9-cis-RA on heterodimer activation. Mutations of SRC1 RID in each of the three NR boxes (LXXAA) were made individually as it is known that these specific NR box mutations abrogate coactivator:receptor interaction. Cotransfection of HEK293T cells with wt VDR, RXRα, VDRE DR3:Luc reporter, and either wt SRC1 or mutant NR box 1, 2 or 3 SRC1 were treated with ligands. In the presence of 1,25D3 only, the NR box 3 mutant alters activity of the heterodimer whereas only the NR box 1 mutant reduced activity of the heterodimer in the presence of 9-cisRA, and either NR box 1 or box 3 mutants reduced receptor activity in the presence of both ligands (Supplementary Fig. 5). This data supports that the cognate ligand of each coreceptor drives interaction with a distinct NR box within SRC1. In addition, the activity of the heterodimer was monitored using a Cyp24 reporter gene assay where treatment with 1,25D3 or 9-cis-RA alone robustly activated the heterodimer, 12-fold and 9-fold over the control, respectively (Supplementary Fig. 6). Moreover, there was an additive effect on Cyp24 reporter gene activation in the presence of both compounds. These data are consistent with an earlier report that either ligand could activate the RXRα-VDR heterodimer and that they may function synergistically41. Taken together, the data suggest two ligands together may synergistically activate the heterodimer by facilitating a concerted interaction between both coreceptors with one molecule of SRC1.
To further this analysis, regions within the heterodimer displaying protection to HDX upon interaction with SRC1 RID were mutated to evaluate their role in receptor activity. These specific regions contain residues implicated in “charge clamp” formation necessary for coactivator binding. Therefore, point mutations in VDR were generated and their impact on receptor activity was determined in the presence of 1,25D3, 9-cis-RA, or both ligands. Mutation of either Lys246 or Glu420 resulted in a large decrease in the ability of 1,25D3 to activate the mutant receptor while the Lys240 mutant had very little effect (Supplementary Fig. 7). Interestingly, the Glu420 mutation also reduced the ability of 9-cis-RA to activate the reporter gene while neither of the VDR lysine mutations had any impact on 9-cis-RA dependent activation. Likewise, point mutations were generated in RXRα at Lys274, Lys284, Glu434 and Glu453 located within the putative RXRα “charge clamp.” Mutations at Lys274, Lys284 and Glu434 reduced the ability of 1,25D3 or 9-cis-RA to activate the heterodimer (Supplementary Fig. 8). Combined, these data suggest that the coactivator binding surface of each coreceptor is important for activation of the heterodimer, and further supports the notion that coactivator interacts simultaneously with both coreceptors.
Finally, since DNA binding destabilized H12 of VDR, and H3, H10-H11 of RXRα, and these regions are involved in SRC1 interaction, differential HDX was used to probe the impact of DNA binding on heterodimer:SRC1 interaction. Protection to HDX in H12 of VDR upon interaction with SRC1 was reduced in presence of DNA (Supplementary Fig. 9a). This finding is consistent with the fact that DNA binding destabilizes H12 of VDR. In contrast, SRC1 binding to RXRα was further enhanced by DNA binding (Supplementary Fig. 9b). Thus, DNA binding alters the conformation of the NR heterodimer presumably altering SRC1 recruitment.
Discussion
Structural studies have provided significant details concerning the mechanism of action of NRs; however, the field is still very limited in its ability to examine the structural characteristics of an intact full length nuclear receptor. Using HDX, we were able to probe the conformational dynamics of an intact RXRα-VDR heterodimer revealing details of how an NR functions beyond those previously characterized. Our results indicate that there is extensive allosteric communication throughout the heterodimer, which is more extensive than previously suggested. For example, binding of either 1,25D3 or 9-cis-RA to the heterodimer leads to dynamic changes in the stability of the DBD of VDR. These results suggest that the ligand itself may alter the DNA binding properties of this NR heterodimer. Furthermore, it is reasonable to hypothesize that different classes of ligands may differentially alter the DBD and thus provide unique pharmacological profiles in terms of target gene activities. This hypothesis is particularly intriguing given the known ability to design “selective” NR modulator ligands that display tissue and promoter gene selectivity. This allosteric communication is ligand dependent and bidirectional as we observe that binding to DNA (VDRE DR3) results in significant alterations in the conformation of the LBDs of both the RXRα and VDR components of the heterodimer. These conformational changes are quite relevant to the function of the NR since they occur primarily within regions of VDR and RXRα that are critical for interaction with coactivators. Interestingly, the effects of DNA binding targeted unique components of the coactivator binding sites on VDR vs. RXRα with the effects on VDR primarily occurring in H12 while H3 and the loop between H10-11 were altered in RXRα. These DNA-dependent alterations in the LBD were shown to have differential effects on the interaction of SRC1 with the heterodimer suggesting that the DNA can directly modulate the coactivator binding activity of the NR and its larger dimer complex.
Recently, Yamamoto and colleagues proposed that the DNA response element functions as a sequence-specific allosteric ligand that modulates the activity of the receptor 42; suggesting that allosteric signals are relayed from the DBD to the LBD. Our results provide direct structural evidence for DNA-dependent allosteric communication between the DBD and LBD of an intact NR as well as between heterodimer partners. A recent study of the crystal structure of a full-length nuclear receptor complex on DNA provides some insight into the mechanism in which DBD-LBD communication may be occurring by demonstrating the LBD of PPARγ and the DBD of RXRα make direct contacts with one another and that these contacts are required for normal receptor function7.
Interestingly, we found that the ligand binding appeared to correlate with increased deuterium incorporation in the DBD of VDR indicating increased flexibility in the DBD. This long range ligand-induced flexibility in the DBD may engender the receptor for DNA binding. A “fly-casting” mechanism43 in protein-DNA interactions suggests that a relatively unstructured protein molecule can have a greater capture radius for a specific DNA recognition than the equivalent folded state with restricted conformational freedom44. In fact, however, crystallography suggests that nuclear receptor DBDs are highly structured, neither extended nor disordered, and the modest increased flexibility induced by ligand binding does not appear sufficient to reel in DNA. A more plausible explanation would be that the increased flexibility observed within the DBD upon ligand binding enables the DBD to rapidly explore vast nonspecific DNA sequences in search of a specific VDRE sequence, a model that was advanced by von Hippel.45
We also show that different classes of DNA response elements differentially affect the conformation of this heterodimer. The natural VDRE sequence derived from the Cyp24 gene induced distinct conformational changes in the heterodimer relative to those using a consensus DR3 element of differing nucleotide composition. H12 dynamics were altered in VDR, but no alterations in the coactivator binding regions of RXRα were detected. This distinction could be expected to lead to large differences in coactivator binding kinetics when bound to different types of response elements. These data clearly indicate that the sequence of the DNA response element can indeed relay information to the LBD that alters its conformation. This is particularly intriguing since there is considerable evidence that a particular NR can behave differently at distinct target genes presumably in part due to binding to different classes of DNA response elements 42,46-47. Our results suggest that DNA sequence-dependent alterations in LBD conformation can lead to significant changes in cofactor preference, which may be one mechanism by which this can occur.
Methods
Reagents
His-hVDR LBD (residues 118-425, Δ[165-215]) was expressed in E. coli and purified via a three step purification Ni-NTA/Refolding/Q Sepharose FastFlow (QFF) chromatography. Full length WT His-hVDR and WT Flag-hRXRα were expressed in Baculovirus system and purified by Ni-NTASEC or Flag/SEC, respectively. His-hSRC1-RID (627-786) variant 1 (NM_003743) was expressed in E. coli and purified using His-Trap (GE Healthcare). The final protein buffer was 50mM Tris (pH 8.0), 150mM NaCl, 10% glycerol, and 2mM DTT. The purity for each protein was > 95% and was verified using SDS-PAGE, Western Blot and MALDI mass spectrometry.
The VDRE DR3 5′-CGTAGGTCAATCAGGTCACGTCGT-3′ and Cyp24 VDRE 5′-CTAGCTCCCGAGGTCAGCGACGGCGCAGG-3′ were purchased from Integrated DNA Technologies. Heterodimer complex was formed by mixing VDR and RXRα at 1:1 molar ratio (final concentration ∼10 μM) and was confirmed by gel shift assays. Vitamin D3 (Sigma) and 9-cis-retinoid acid (Sigma) were added at a 10 fold molar excess to heterodimer. SRC1-RID was added in 2× molar ratio to heterodimer and oligonucleotide (VDRE DR3 or Cyp24 VDRE) was mixed with the protein complex as needed (1.5× molar ratio).
HDX
Differential HDX experiments were performed as previously described with a few modifications48. Exchange reactions were carried out at 4 °C and were quenched by mixing with 3 M Urea, 1% TFA at 1 °C. Protein was passed across a pepsin column (2mm × 2cm) at 200 ul/min and digested peptides were captured onto a 2mm × 1cm C8 trap column (Agilent) and desalted. Peptides were separated across a 2.1mm × 5cm C18 column (1.9μL Hypersil Gold, Thermo Scientific) with linear gradient of 4%-40% CH3CN, 0.3 % formic acid, over 5 min. Protein digestion and peptide separation were performed at 1°C. Mass spectrometric data were acquired with a measured resolving power of 65,000 at m/z 400. Three replicates were performed for each on-exchange time point. Peptide Identification was achieved using MSMS with a linear ion trap mass spectrometer (LTQ, ThermoFisher). Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis. The MSMS *.raw data files were submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptides included for HDX analysis had a MASCOT score > 20 and the MSMS spectra were manually inspected. The MASCOT search was also performed against a decoy (reverse) sequence and ambiguous identifications were ruled out. The intensity weighted average m/z value (centroid) of each peptide isotopic envelopes were calculated with HD Desktop49. The deuterium level was calculated as described previously50. Corrections for back-exchange were made based on an estimated 70% deuterium recovery and accounting for the known 80% deuterium content of the on-exchange buffer.
Site-directed Mutagenesis
The pSPORT-RXRα, pSPORT-VDR, pSPORT-SRC1 mutant constructs were made using QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The mutant primers were used to amplify mutant plasmids from wild type using PfuUltra HF DNA polymerase. The PCR products were treated with Dpn I to select for mutation containing synthesized DNA and then transformed into XL1-Blue supercompetent cells. Positive clones were picked up and grew overnight in LB media. The plasmid were isolated using QIAprep Spin Miniprep Kit (Qiagen) and verified by sequencing.
Cell-based luciferase reporter assays
Luciferase reporter assays were performed by co- transfecting HEK293T cells with pSport6 VDR, pSport6 RXRα and a luciferase reporter driven by the Cyp24 promoter containing a single VDR response element (Switchgear Genomics) in a 1:1:1 ratio. Reverse transfections were performed in batch using 3×106 cells with 7μg of total DNA and FuGene6 (Roche) in a 1:3 DNA to lipid ratio. Following transfection, cells were incubated for 16 hours and replated in 384 well plates at a density of 10,000 cells/well. Six hours after replating, cells were treated with either DMSO, 3nM 1,25D3, 1μM 9-cis RA or both for 16 hours prior to developing the assay by addition of Brite-lite plus. For mutagenesis experiments, the protocol was identical except wild-type VDR and RXRα were replaced with constructs for VDR mutants (K240A, K246A, E420A) or RXRα mutants (K274A, K284A, E434A, E453A) where indicated. For assays involving SRC1 mutations, HEK293T cells were cotransfected with constructs for pSport6 VDR, pSport6 RXRα, the Cyp24 luciferase reporter and constructs for wild-type SRC1 or mutants to NR box 1(Mut1), 2 (Mut2) or 3 (Mut3). Transfections were performed in batch, and cells were dispensed in 384 well format and treated with 3nM 1,25D3, 1μM 9-cisRA, both or vehicle only as previously described.
Thermshift binding assay (TSBA)
TSBA was performed as previously described51 to determine estimated binding affinities of RXR with VDR, RXR-VDR with VD3, DNA interaction with RXR-VDR, and RXR:VDR:VD3 with SRC1. TSBA data were fit to equations describing the fluorescence intensity in protein denaturation curves52-53 using nonlinear regression least squares fit to the data in Graphpad Prism to obtain fitted values of ΔHu and Tm, which were then used to estimate ΔCpu from the data. Ligand binding affinity estimates at Tm and a reference temperature (25°C) were calculated from equations previously described54-55. Proteins concentrations were 1 μM (ligand free sample) and ligand (protein, DNA or VD3) concentrations were 10 μM (ligand bound sample).
AlphaScreen assay
Experiments were performed under subdued lighting at room temperature with an sssay buffer (100 mM NaCl, 25 mM Hepes, 0.1% BSA, pH 7.4, 0.1% DMSO). For titration of biotinylated oligonucleotides, a mix of His-VDR, Flag-RXR, 1,25D3, flag-acceptor and streptavidin-donor beads was made and increasing concentrations (230 pM to 500 nM) of either Biotin-VDRE DR3 or Biotin-CYP24 VDRE were added and incubated for 2h at room temperature. Unlabeled oligonucleotides were used for the competition experiments. Assay plates were read on PerkinElmer Envision 2104 and data analyzed using GraphPad Prism software (La Jolla, CA).
Supplementary Material
Acknowledgments
We are grateful for support from Mark Southern and Scooter Willis for software analyzing the HDX data. This work was supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Mental Health [Grant U54-MH074404: H. Rosen PI], the National Institute of General Medical Sciences [Grant R01-GM084041: P. Griffin PI], the National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01-DK080201:T. Burris PI] and the James and Esther King Biomedical Research Program, Florida Department of Health [Grant 10KN-09:D. Kojetin PI].
Footnotes
Author Contributions: J.Z., M.J.C., K.R.S., J.A.D. and P. R. G. conceived the project and designed research; J.Z, L.L.B, Y.W., S.A.B., B.D.P., R.D.G-O, J.B.B., M.I., and D.J.K. performed research; J.Z, M.J.C., K.R.S, L.L.B, Y.W., S.A.B., B.D.P., R.D.G-O, J.B.B., M. I., D.J.K., T.P.B., J.A.D, and P.R.G. analyzed data; and J.Z., K.R.S., T.P.B., S.A.B. and P.R.G. wrote the paper with contributions from all authors.
Competing interest statement: The authors declare no competing financial interests.
Methods: Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
References
- 1.Gennari L, Merlotti D, De Paola V, Martini G, Nuti R. Update on the pharmacogenetics of the vitamin D receptor and osteoporosis. Pharmacogenomics. 2009;10:417–433. doi: 10.2217/14622416.10.3.417. [DOI] [PubMed] [Google Scholar]
- 2.Narvaez CJ, Matthews D, Broun E, Chan M, Welsh J. Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology. 2009;150:651–661. doi: 10.1210/en.2008-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall TG, Lee RE, Marshall FE. Common angiotensin receptor blockers may directly modulate the immune system via VDR, PPAR and CCR2b. Theor Biol Med Model. 2006;3:1. doi: 10.1186/1742-4682-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krishnan AV, et al. Tissue-selective regulation of aromatase expression by calcitriol: implications for breast cancer therapy. Endocrinology. 2010;151:32–42. doi: 10.1210/en.2009-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kostner K, et al. The relevance of vitamin D receptor (VDR) gene polymorphisms for cancer: a review of the literature. Anticancer Res. 2009;29:3511–3536. [PubMed] [Google Scholar]
- 6.Dilworth FJ, Chambon P. Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene. 2001;20:3047–3054. doi: 10.1038/sj.onc.1204329. [DOI] [PubMed] [Google Scholar]
- 7.Chandra V, et al. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iacob RE, et al. Conformational disturbance in Abl kinase upon mutation and deregulation. Proc Natl Acad Sci U S A. 2009;106:1386–1391. doi: 10.1073/pnas.0811912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamuro Y, et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu YH, Burke JE, Li S, Woods VL, Jr, Dennis EA. Localizing the membrane binding region of Group VIA Ca2+-independent phospholipase A2 using peptide amide hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2009;284:23652–23661. doi: 10.1074/jbc.M109.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruning JB, et al. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Hamuro Y, et al. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J Biomol Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- 13.Yan X, Broderick D, Leid ME, Schimerlik MI, Deinzer ML. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43:909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- 14.Yan X, et al. Deuterium exchange and mass spectrometry reveal the interaction differences of two synthetic modulators of RXRalpha LBD. Protein Sci. 2007;16:2491–2501. doi: 10.1110/ps.073019707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chalmers MJ, Busby SA, Pascal BD, Southern MR, Griffin PR. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J Biomol Tech. 2007;18:194–204. [PMC free article] [PubMed] [Google Scholar]
- 16.Dai SY, et al. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc Natl Acad Sci U S A. 2008;105:7171–7176. doi: 10.1073/pnas.0710802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai SY, et al. Unique Ligand Binding Patterns between Estrogen Receptor alpha and beta Revealed by Hydrogen-Deuterium Exchange. Biochemistry. 2009 doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JH, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, et al. Hydrogen/Deuterium Exchange Reveals Distinct Agonist/Partial Agonist Receptor Dynamics within the intact Vitamin D Receptor/Retinoid X Receptor Heterodimer. Structure. 2010 doi: 10.1016/j.str.2010.07.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 22.Rastinejad F, Perlmann T, Evans RM, Sigler PB. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–211. doi: 10.1038/375203a0. [DOI] [PubMed] [Google Scholar]
- 23.Gampe RT, Jr, et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 24.Bourguet W, et al. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–298. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 25.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 26.Carlberg C, Polly P. Gene regulation by vitamin D3. Crit Rev Eukaryot Gene Expr. 1998;8:19–42. doi: 10.1615/critreveukargeneexpr.v8.i1.20. [DOI] [PubMed] [Google Scholar]
- 27.Schulman IG, Li C, Schwabe JW, Evans RM. The phantom ligand effect: allosteric control of transcription by the retinoid X receptor. Genes Dev. 1997;11:299–308. doi: 10.1101/gad.11.3.299. [DOI] [PubMed] [Google Scholar]
- 28.Willy PJ, Mangelsdorf DJ. Unique requirements for retinoid-dependent transcriptional activation by the orphan receptor LXR. Genes Dev. 1997;11:289–298. doi: 10.1101/gad.11.3.289. [DOI] [PubMed] [Google Scholar]
- 29.Egea PF, et al. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–2601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Simisky J, Tsai FT, Geller DS. A critical role of helix 3-helix 5 interaction in steroid hormone receptor function. Proc Natl Acad Sci U S A. 2005;102:2707–2712. doi: 10.1073/pnas.0409663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemon BD, Freedman LP. Selective effects of ligands on vitamin D3 receptor- and retinoid X receptor-mediated gene activation in vivo. Mol Cell Biol. 1996;16:1006–1016. doi: 10.1128/mcb.16.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Q, Khorasanizadeh S, Miyoshi Y, Lazar MA, Rastinejad F. Structural elements of an orphan nuclear receptor-DNA complex. Mol Cell. 1998;1:849–861. doi: 10.1016/s1097-2765(00)80084-2. [DOI] [PubMed] [Google Scholar]
- 34.Miyamoto T, et al. The role of hinge domain in heterodimerization and specific DNA recognition by nuclear receptors. Mol Cell Endocrinol. 2001;181:229–238. doi: 10.1016/s0303-7207(01)00483-x. [DOI] [PubMed] [Google Scholar]
- 35.Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene. 2000;246:9–21. doi: 10.1016/s0378-1119(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 36.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 37.Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 38.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 39.Pogenberg V, et al. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J Biol Chem. 2005;280:1625–1633. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 40.Kersten S, Dong D, Lee W, Reczek PR, Noy N. Auto-silencing by the retinoid X receptor. J Mol Biol. 1998;284:21–32. doi: 10.1006/jmbi.1998.2168. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Martinez R, Castillo AI, Steinmeyer A, Aranda A. The retinoid X receptor ligand restores defective signalling by the vitamin D receptor. EMBO Rep. 2006;7:1030–1034. doi: 10.1038/sj.embor.7400776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci U S A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy Y, Onuchic JN, Wolynes PG. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J Am Chem Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 45.von Hippel PH. Biochemistry. Completing the view of transcriptional regulation. Science. 2004;305:350–352. doi: 10.1126/science.1101270. [DOI] [PubMed] [Google Scholar]
- 46.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature. 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]
- 48.Chalmers MJ, et al. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- 49.Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD desktop: an integrated platform for the analysis and visualization of H/D exchange data. J Am Soc Mass Spectrom. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 52.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005;44:5258–5266. doi: 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- 53.Marky LA, Breslauer KJ. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers. 1987;26:1601–1620. doi: 10.1002/bip.360260911. [DOI] [PubMed] [Google Scholar]
- 54.Pantoliano MW, et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 55.Bruylants G, Wouters J, Michaux C. Differential scanning calorimetry in life science: thermodynamics, stability, molecular recognition and application in drug design. Curr Med Chem. 2005;12:2011–2020. doi: 10.2174/0929867054546564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.