

Abstract
Ribonuclease H (RNase H), an enzyme that cleaves an RNA sequence base-paired with a complementary DNA sequence, is proposed to be the mediator of antisense phosphorothioate oligonucleotide (S-oligo) lethality in a cell. To understand the role of RNase H in the killing of the parasitic protozoan Leishmania by antisense S-oligos, we expressed an episomal copy of the Trypanosoma brucei RNase H1 gene inside L. amazonensis promastigotes and amastigotes that constitutively express firefly luciferase. Our hypothesis was that S-oligo-directed degradation of target mRNA is facilitated in a cell that has higher RNase H activity. Increased inhibition of luciferase mRNA expression by anti-luciferase S-oligo and by anti-miniexon S-oligo in these stably transfected promastigotes overexpressing RNase H1 was correlated to the higher activity of RNase H in these cells. The efficiency of killing of the RNase H overexpressing amastigotes inside L. amazonensis-infected macrophages by anti-miniexon S-oligo was higher than in the control cells. Thus, RNase H appears to play an important role in the antisense S-oligo-mediated killing of Leishmania. Chemical modification of S-oligos that stimulate RNase H and/or co-treatment of cells with an activator of RNase H may be useful for developing an antisense approach against leishmaniasis. The transgenic Leishmania cells overexpressing RNase H should be a good model system for the antisense-mediated gene expression ablation studies in these parasites.
Keywords: Leishmania, Amastigote, Promastigote, Differentiation, Ribonuclease H, Antisense phosphorothioate oligonucleotide
1. Introduction
The etiological agent for the human parasitic disease leishmaniasis is the protozoon of the genus Leishmania. This obligate intracellular parasite of mammalian macrophages has a digenetic life cycle [1]. These cells are exposed to a variety of environmental factors during their transmission from sandfly vectors to the human body, their establishment of infection in human macrophages, and their propagation as amastigotes inside macrophage phagolysosomes [1]. In its life cycle, Leishmania alternates between an insect vector and a vertebrate host. The parasite lives as a motile, flagellated promastigote in the gut of various sandfly vectors, characterized by its elongated shape (10–12 μm) and a single, long anterior flagellum. When delivered into the mammalian body by the bite of an infected sandfly, the promastigotes are engulfed by macrophages by a receptor-mediated process [1]. The promastigotes end up in the phagolysosomes of macrophages where they transform into non-motile amastigotes [1].
Among the many distinguishing molecular and biochemical features [2,3] that differentiate this parasite from its mammalian host is a 39 nt conserved sequence of RNA post-transcriptionally added at the 5′-ends of all leishmanial mRNAs and related cells [4]. This unique 5′-end sequence (miniexon) is methylated to form the ‘cap4’ structure, which is essential for the entry of ribosomes to translate the mRNA [4,5]. We and others [6–9] have targeted this miniexon sequence with antisense phosphorothioate oligonucleotides to ablate the translation of mRNAs of the parasite resulting in the demise of the parasite and survival of the host cells. Adoption of a macrophage scavenger receptor-mediated delivery system for the antisense oligonucleotide to Leishmania-infected macrophage phagolysosomes yielded better efficacy of amastigote killing [9]. Although we have clues from other eukaryotic systems, we do not yet have any information about the mechanism of antisense action in Leishmania. Since RNase H is implicated [10] in the modality of antisense phosphorothioate action in ablation of gene expression in other cells, we decided to study the effect of overexpression of this enzyme on the efficacy of antisense oligos in these cells.
RNase H is a widely distributed enzyme found in organisms ranging from retroviruses to mammals [11–16]. This enzyme hydrolyzes RNA in RNA/DNA heteroduplexes [16]. RNase H isotypes vary significantly in molecular size and associated functional activities. On the other hand, the nuclease properties of all RNase Hs are similar. All RNase Hs require a divalent cation for activity [16]. Thus far, three classes of RNase Hs have been discovered in bacterial cells. They are RNase H1, H2, and H3 [17]. RNase H1 and H3 share similar divalent cation preferences, and RNase H2 and H3 have significant similarity at the amino acid sequence level [16,17]. RNase H2 is the most ubiquitous of the three [16]. No single bacterium has been found to have all three RNase H isotypes [16]. Escherichia coli RNase H1, which is involved in DNA replication [18–23], is the best characterized among the prokaryotic enzymes. The key amino acids involved in divalent cation binding, substrate binding, and catalysis have been identified and are highly conserved in this family of enzymes [22,24–26]. In retroviral replication, RNase H acts as a prosthetic domain of the reverse transcriptase and helps convert the retroviral RNA into double-stranded DNA by generating RNA primers during second strand cDNA synthesis [24,27]. Retroviral RNase H domains share homology with E. coli RNase H1 [24]. Thus far, mammalian cells have been shown to have two isotypes of RNase H [12–16,25,26,28,29]. These two isotypes differ from each other by Mg2+ and Mn2+ requirements and sensitivity towards sulfhydryl reagents [16].
Although RNase H is likely to be present in Leishmania, it has not been shown in these cells until this study. RNase H has been cloned from organisms related to Leishmania [30–34]. Along with the universal RNase H catalytic domain in these protozoal enzymes, there is also a double-stranded RNA binding domain that is typical to eukaryotic RNase H [33,34]. The latter domain is absent in E. coli RNase H and, hence, is not needed for the functions of this enzyme in this cell. The physiological role of this domain of RNase H in eukaryotes is not clearly understood [33,34]. A single gene seems to be responsible for at least two forms of the Crithidia fasciculata RNase H1 [32,33]. Using the renaturation gel assay for RNase H activity, species of 38 and 45 kDa are obtained, both of which disappear in strains whose RNH1 gene is deleted [32,33].
In this paper, we provide evidence that RNase H may be involved in antisense phosphorothioate oligonucleotide-mediated gene expression ablation in pathogenic Leishmania promastigotes and amastigotes.
2. Materials and methods
2.1. Leishmania amastigotes and promastigotes
The promastigotes of L. amazonensis were grown at 25° in Medium M199 with 10% HIFBS [9]. The amastigotes of L. amazonensis were obtained from tail-base lesions of infected Balb/C mice [35]. The amastigotes were also isolated from infected, cultured mouse macrophages (J774G8 cells) [9]. Axenic amastigotes of L. amazonensis were grown in axenic medium at pH 4.5 and at 33° [36,37]. Amastigotes were allowed to transform into promastigotes at 25° in Medium M199 containing 10% HIFBS. Leishmania and J774G8 cells were gifts from Prof. K-P. Chang of the Chicago Medical School.
2.2. Oligonucleotides
The following phosphorothioate ODNs (S-oligos) were used in this study: anti-miniexon antisense oligo ASM, 5′-CTGATACTTATATAGCG-3′ [9], the corresponding sense oligo SSM, 5′-CGCTATATAAGTATCAG-3′ [9], anti-luciferase antisense oligo LUAS, 5′-ATGCCCATA-CTGTTGAG-3′, and the corresponding sense oligo LUS, 5′-CTCAACAGTATGGGCAT-3′. The following phosphodiester ODNs were used for PCR amplifications: TbRHF, 5′-CCTTCTCTGCTCGCTTTTTAAC-3′;TbRHR, 5′-TGT-CTGTGCCACGTTAGCC-3′; Mex2, 5′-GGATCCAGT-TTCTGTACTTTATTG-3′ and LUAS, 5′-ATGCCCATA-CTGTTGAG-3′. The nucleotide sequence of firefly luciferase was obtained from the nucleotide sequence of pGL3-Basic plasmid (Promega), GenBank Accession Number U47295. The sense and antisense primers were designed using MacVector software (Oxford Molecular Inc.).
2.3. Amplification, cloning, and sequence verification of the Trypanosoma brucei RNase H1 gene
The RNase H1 gene of T. brucei was amplified from the genomic DNA templates of the procyclic form of the parasite. Primers for the amplification (TbRHF and TbRHR) were designed using the MacVector program (Oxford Molecular Inc.) from the published T. brucei RNase H1 gene sequence [31]. These primers encompass a 1018 bp region of the T. brucei RNase H1 gene, of which 99 bp are upstream of the translational start site, 905 bp are coding sequence, and 14 bp are downstream of the translational stop site. The amplification reaction was carried out following standard PCR protocol [38] using Pfu DNA polymerase (Stratagene). The PCR reaction product was gel-purified [38] and incubated with AmpliTaq DNA polymerase and dATP for 30 min at 65° to add 3′-A overhangs. This PCR product was then cloned into the pCRII-TOPO vector following the TOPO-TA-cloning protocol (Invitrogen). The nucleotide sequence of the insert of the recombinant phage-mid pCR-TbRH1 was verified by automated sequencing in a 310 Genetic Analyzer (PE Biosystems).
2.4. Development of T. brucei RNase H1 constructs for stable expression of this enzyme in L. amazonensis promastigotes
The insert of the phagemid pCR-TbRH1 (see above) was cut out with KpnI and XhoI and was subcloned at the KpnI/XhoI sites of pGL3-Basic plasmid to get the pGL3-TbRH plasmid. This T. brucei RNase H1 gene insert was then cut out of the pGL3-TbRH plasmid with BamHI and BglII and subcloned into the BamHI site of pX63-Neo plasmid ([4,39], a gift from Prof. S. Beverley) to obtain pXNeo-TbRH1F and pXNeo-TbRH1R clones (Fig. 1, B and C). These plasmids are E. coli/Leishmania shuttle vectors and thus contain all the elements needed for replication in both E. coli and Leishmania [4,39]. In Leishmania, genes are thought to be transcribed from the plasmid DNA without requiring any specific promoter elements [4], but the appropriate export of the message from the nucleus to the cytosol and its translation into protein are thought to be dependent upon proper trans-splicing and polyadenylation of the message. The presence of a trans-splicing signal at the 5′-flank of the pre-mRNA, which is a poly-pyrimidine stretch followed by an ‘AG’ sequence, is critical for trans-splicing [4,39]. There are two such trans-splicing sites in pX63-Neo: one in front of the ‘Neo’ gene (confers G418-resistance to Leishmania) and the other is in front of the unique BamHI site [39]. Thus, in Leishmania cells transfected with the plasmid constructs, the Neo gene and the TbRH1 gene cloned at the BamHI site are expected to be expressed. The orientation of the insert was determined by nucleotide sequencing with a T3 primer [38].
Fig. 1.

Amplification and cloning of T. brucei RNase H1 in pX63-Neo. (A) Ethidium bromide-stained agarose gel showing the PCR amplification product of T. brucei RNase H1 (lane 2); lane 1, 1-kb ladder molecular size marker (GIBCO-BRL). (B) Map of the pXNeo-TbRH1F plasmid. (C) Map of the pXNeo-TbRH1R plasmid. Amp, Neo and TbRH1 indicate the beta-lactamase, neomycin phosphotransferase, and T. brucei ribonuclease H1 genes, respectively. Arrowheads before the genes indicate Leishmania trans-splicing signals. These trans-splicing signals are needed for the expression of the genes cloned behind them. In these plasmids, nucleotide sequences 0–821, 4752–5710, and 6619–7166 are originated from the Leishmania DHFR locus [39]. The TbRH1 gene sequence is 822–1925. The rest of the sequences are from bacterial plasmids [39]. The orientations of the genes with respect to their promoter (Amp gene) or trans-splicing signal (Neo or TbRH1 genes) are indicated as solid arrows.
2.5. Development of a firefly luciferase construct for the stable expression of this enzyme in L. amazonensis promastigotes
The firefly luciferase gene was cleaved from pGL3-Basic plasmid and was cloned, along with a hygromycin-resistance marker gene, into pBluescript KS(+) (Stratagene) to construct the pXHyg-Luci phagemid (Fig. 3A). Briefly, a 2 kb DNA fragment containing a 1 kb upstream sequence (US) of the L. major dihydrofolate reductase gene, cloned in front of the hygromycin-resistance marker gene, was cut out of the pX63-Hyg plasmid ([4,39], a gift from Prof. S. Beverley) and cloned into the SalI/BamHI sites of pBlue-script KS(+) to create the phagemid pBS-HygDhfr. In another experiment, a 580 bp KpnI/SacI fragment containing the US of the L. donovani leishmanolysin (gp63) gene was cloned at the KpnI/SacI sites, in front of the firefly luciferase gene in proper orientation, in pGL3-Basic to create the pGL3bK1 plasmid. Then, the pBHygDhfr plasmid was cut by digestion with KpnI/SalI, and the resulting KpnI/SalI fragment, containing gp63 US in front of firefly luciferase gene, was cloned [38] to create the pXHyg-Luci plasmid (Fig. 3A).
Fig. 3.

Stable expression of firefly luciferase in L. amazonensis promastigotes overexpressing T. brucei RNase H1. (A) Map of the plasmid pXHyg-Luci; Amp, Hyg, and Luciferase indicate the beta-lactamase, hygromycin phosphotransferase, and firefly luciferase genes. Arrowheads before the genes indicate Leishmania trans-splicing signals. These trans-splicing signals are needed for the expression of the genes cloned behind them. The orientations of the genes with respect to their promoter (Amp gene) or trans-splicing signal (Hyg or Luciferase genes) are indicated as solid arrows. (B) Nucleotide sequence (5′ to 3′) (453 bp) of the miniexon end of the 5′-RACE product from the mRNA of the recombinant cells using Mex2 and LUAS as primers. The Mex2 sequence and the luciferase mRNA translation start site (AUG) are boldfaced and underlined. (C) Expression of luciferase activity in the recombinant promastigotes and amastigotes. LU = light units. Results are means ± SEM (N = 6). The difference in luciferase activity between the amastigote and promastigote cell extracts was not statistically significant.
2.6. Transfection of Leishmania cells
Late-log phase cells were washed and resuspended in high ionic strength electroporation buffer (20 mM HEPES, pH 7.2, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 6 mM glucose) to a final concentration of 3.3 × 108 cells/mL [40,41]. Cells (0.3 mL) were mixed in a 0.2-cm electroporation cuvette (Bio-Rad) with plasmid DNA (25 μg/108 cells) and were exposed to an exponential discharge of 2250 V/cm from a 500 μF capacitor (Gene Pulsar, Bio-Rad) [40]. Electroporated cells were incubated on ice for 10 min, transferred into culture medium (10 mL), and incubated at 25° for 20–24 hr before growth in selective medium. Cells were selected in increasing concentrations of antibiotic, starting from 1 to 200 μg/mL.
2.7. RT–PCR
Poly(A)+ RNAs were isolated from L. amazonensis promastigotes using the PolyATtract® mRNA Isolation System (Promega). The RNA was treated with RNase-free DNase (Promega) to remove contaminating DNA. Complementary DNAs were synthesized from these RNAs (1 μg) using Superscript II reverse transcriptase (GIBCO-BRL) and oligo(dT) primers in a total volume of 20 μL, following a recommended protocol (GIBCO-BRL). An aliquot (1 μL) of these cDNAs was used for PCR amplification using AmpliTaq DNA polymerase (Qiagen) [38]. For the amplification of T. brucei RNase H1 cDNA, primers TbRHF and TbRHR were used. For 5′-RACE (rapid amplification of cDNA ends) analyses of T. brucei RNase H1 mRNA and firefly luciferase mRNA isolated from transgenic Leishmania cells, the miniexon-specific sense primer Mex2 and gene specific antisense primer (TbRHR or LUAS, see above) were used. The PCR products were analyzed by agarose gel electrophoresis, stained with ethidium bromide, and photographed [38]. In some experiments, the RT–PCR products were gel-purified, cloned into the pCRII-TOPO vector (Invitrogen), and sequenced using T7 and SP6 primers [38].
2.8. Luciferase assay
The luciferase assay was performed using the Luciferase Reporter Assay System (Promega). G418/Hygromycin-resistant cells (promastigotes or amastigotes) were incubated with the phosphorothioate oligonucleotide (LUS or LUAS) for 20 hr in complete culture medium under appropriate growth conditions. Cells were washed with ice-cold PBS and lysed with Passive Lysis Buffer (Promega, 100 μL/107 cells) at room temperature for 15 min on a rocker platform. The lysates were centrifuged at 16,000 g for 10 min. Aliquots (20 μL) of the supernatant were added to the substrate LARII (Promega, 100 μL) and mixed, and luminescence was assayed for 10 sec in a Turner Designs model 20/20 Luminometer [42].
2.9. Assay for RNase H activity in Leishmania cell extracts
[32P]Poly(rA):poly(dT) was used as the substrate of RNase H [43]. E. coli RNA polymerase (6 μL, 10 units/μL, Amersham Pharmacia Biotech) was added to a mixture containing 5 A260 units poly(dT), 5% glycerol, 50 mM Tris · HCl, pH 8.0, 5 mM MgCl2, 1 mM MnCl2, 4 mM dithiothreitol, 30 μM ATP, and 100 μCi [α-32P]ATP (3000 Ci/mmol, Amersham) in a total volume of 3 mL. The reaction mixture was extracted twice with an equal volume of water-saturated phenol (GIBCO-BRL). The pooled phenol phases were extracted with an equal volume of 50 mM Tris · HCl, pH 8.0. The aqueous phases were pooled and extracted with an equal volume of chloroform/isoamyl alcohol (24/1). Absolute ethanol was added to the aqueous phase to a final concentration of 35%, and the resulting solution was loaded onto a 2-mL column of cellulose (CF11, Sigma). The column was pre-equilibrated with a 35:65 mixture of ethanol:‘buffer A’ (50 mM Tris · HCl, pH 7.5/0.1 M NaCl, 50 mM EDTA). After sample loading, the column was washed extensively with this ethanol:buffer A mixture (~50 mL) until all unbound radioactivity was removed (<1000 cpm/10 μL wash). Then the bound nucleic acid polymer was eluted from the column with 5 mL of buffer A, and 500-μL fractions were collected in microfuge tubes. Fractions with high radioactivity (generally tubes 3–7) were pooled, and radioactivity was measured by scintillation counting [38]. A typical preparation usually gave us ~125,000 cpm/10 μL. The hybrid solution was divided into aliquots and stored at −20° for later use. Leishmania promastigotes or amastigotes were washed in PBS and lysed in the Passive lysis buffer (Promega, 108 cells/mL) for 15 min on ice. The lysate was cleared by centrifugation at 16,000 g for 10 min at 4°, and the supernatant was assayed for RNase H activity. Protein concentration was measured using the Bradford reagent (Bio-Rad). For the RNase H solution assay, the cell lysate (1 μg protein), in 10 mM Tris · HCl, pH 8.0, 10 mM MgCl2, 50 mM KCl, and 0.1 mM dithiothreitol, was incubated with the substrate (5 μL) at 37° for 30 min in a total volume of 25 μL. The reaction was terminated by the addition of EDTA (50 mM, final concentration) and glycogen (1 μg/μL, final concentration). The unreacted substrates and proteins were precipitated with trichloroacetic acid (5%, final concentration, 10 min at 4°) and were centrifuged at 16,000 g for 10 min at 4° [43]. The radioactivity in the supernatant was determined by liquid scintillation counting [38], and the data were used as a measure of RNase H activity.
2.10. Preparation of MBSA-coated liposomes
BSA was maleylated with maleic anhydride [9]. Di-palmitoyl phosphatidylethanolamine (Sigma Chemical Co.) (100 mg, dissolved in 0.15 M NaCl by ultrasonication) was coupled to MBSA (30 mg) by a 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (100 mg)-mediated reaction [44,45], in a total volume of 3 mL. DMPC was used to prepare liposomes for encapsulation of ASM or SSM [9]. Briefly, DMPC (10 mg) was made into vesicles with cholesterol (9 μmol) and dicetyl phosphate (1.2 μmol) in 1 mL HBS (HEPES buffer, 25 mM; NaCl, 0.15 M; sometimes contained oligonucleotide, 50 μmol/mL). Typically, 0.1 to 0.2 μmol oligonucleotides became trapped in 1 mg of liposomal phospholipids using this procedure. The amount of phospholipids in each liposomal preparation was assayed by estimating its phosphate content [9]. For coating the liposomes with phospholipid-tailed MBSA (which binds via the phospholipid part), 200 μg of the protein was incubated with 1 mg of liposomes in HBS for 20 hr at 4°. Bound MBSA was separated from unbound molecules by centrifugation at 100,000 g for 1 hr at 4°, as described previously [9].
2.11. Assay of leishmanicidal activity of ASM
Monolayers of J774G8 cells in 24-well plastic tissue culture plates were incubated at 37° with L. amazonensis promastigotes (parasite-to-macrophage ratio of 10:1) in RPMI 1640 medium containing 20% HIFBS for 16 hr [9]. The infected macrophages were then incubated in the growth medium at 35° for 48 hr. The growth medium was then replaced with fresh medium, and the test compound was added. After incubation of the parasite-laden macrophages at 35° for 20 hr, the number of amastigotes per 100–200 macrophages were counted microscopically, as described [9]. Axenic amastigotes were harvested by centrifugation (3000 g, 10 min, 4°), resuspended (106 cells/mL) in growth medium, and incubated with or without the oligonucleotide in a 24-well plastic tissue culture plate (500 μL/well) for 20 hr with gentle rocking (100 rpm). Aliquots (10 μL) were withdrawn from the wells, and the cells were counted in a hemocytometer [9].
2.12. Statistical procedures
Each value is presented as the mean ± SEM. The statistical significance of a difference between two series of data was tested by determining the P value [46]. If the P value was less than 0.05, the difference was considered significant [46].
3. Results
3.1. Overexpression of T. brucei RNase H in L. amazonensis
The RNase H1 gene has been cloned from T. brucei by genetic complementation in RNase H negative E. coli [31]. Primers were designed from the published [31] (GenBank Accession Number U74470) nucleotide sequence of the T. brucei RNase H1 (TbRH1) gene. The gene was amplified by PCR using T. brucei genomic DNA as the template (Fig. 1A). A DNA polymerase with a proofreading function was used in the amplification to avoid any inadvertent mutation. The PCR product was sequenced to confirm that there was no change in nucleotide sequence. Two plasmid constructs were generated with the TbRH1 gene cloned into the BamHI/BglII sites of the Leishmania/E. coli shuttle vector pX63-Neo (Fig. 1, B and C). The L. amazonensis promastigotes were electroporated individually with those plasmids. G418-selected clones were checked for the presence of plasmid DNA and the expression of RNase H1 mRNA. As shown in Fig. 2A, the primers used only amplified DNA of the expected size from L. amazonensis cells transfected with the TbRH1 gene. RT–PCR also showed expression of the TbRH1 gene in cells transfected with pXNeo-TbRH1F plasmid and not in those transfected with pXNeo-TbRH1R plasmid, as expected (Fig. 2A).
Fig. 2.

Expression of T. brucei RNase H1 in L. amazonensis. (A) Evidence for the presence of T. brucei RNase H1 DNA and mRNA in recombinant L. amazonensis. Lane 1, 1-kb ladder DNA marker (GIBCO); lanes 2 and 3, PCR products from plasmid DNA templates isolated from TbRH1− (lane 2) and TbRH1+ (lane 3) cells; lanes 4–8, RT–PCR data. Lanes 4–6 represent RT–PCR products from RNA(−), RT(−), and AmpliTaq(−) reaction controls; lanes 7 and 8, RT–PCR products from RNA isolated from TbRH1+ and TbRH1− cells, respectively. PCR amplifications were done with TbRHF and TbRHR primers. The identity of the amplified products was confirmed by nucleotide sequencing. The faint DNA band in lane 2 was a PCR artifact. (B) Nucleotide sequence (5′ to 3′) of the miniexon end (261 bp) of the 5′-RACE product with Mex2 and TbRHR. The Mex2 sequence and the RNase H1 mRNA translation start site (AUG) are boldfaced and underlined. (C) Overexpression of RNase H activity in transgenic Leishmania. The plasmids that were used to transfect L. amazonensis cells are shown on the x-axis. RNase H activity in the cell extracts is expressed as nanograms of substrate degraded per microgram of cell extract protein per hour. Results are means ± SEM (N = 3). The difference in RNase H activity between amastigote and promastigote cell extracts was statistically significant (P < 0.01).
Like other trypanosomatids, genes are transcribed in Leishmania as homo- or hetero-polycistronic RNAs, which then are trans-spliced and polyadenylated to produce mono-cistronic translatable messages [2–4]. Although a few candidates for RNA polymerase II promoters have been reported in Leishmania and T. brucei [47–49], their identification remains controversial [3]. Trans-splicing seems to be regulated by a pyrimidine-rich region upstream of the coding sequence, which guides the miniexon to attach to a nucleotide next to an ‘AG’ sequence near this regulatory region [5]. The smaller ribosomal subunit seems to be entering the cap4 structure at the 5′-end of the mature mRNA and, by the scanning mechanism [50], finds the AUG codon, usually following the Kozak rule (−3 is a purine and +4 is a guanine nucleotide) [50]. The plasmid pX63-Neo has two strong trans-splicing signals: one before the ‘Neo ’ gene and the other before the BamHI site [39]. Thus, the mRNA transcribed from the TbRH1 gene in L. amazonensis should have been trans-spliced in the expected site. We checked the trans-splicing site of the TbRH1 mRNA by 5′-RACE analysis. The first strands of the cDNAs were synthesized using oligo(dT) primers. This ensured that only mature transcripts would participate in the cDNA synthesis reaction. The nucleotide sequence of the miniexon end of the PCR product showed that trans-splicing occurred at the expected site as directed by the trans-splicing signal in the plasmid (Fig. 2B).
Thus, our PCR data showed that the TbRH1 gene cloned behind the trans-splicing signal in pX63-Neo was expressed as a polyadenylated and trans-spliced mRNA. Although the antisense strand of the DNA is also known to be transcribed in Leishmania [51], we did not detect any mature antisense transcript of the TbRH1 gene in L. amazonensis promastigotes transfected with the pXNeo-TbRH1R plasmid (data not shown). When we transformed the TbRH1+ promastigotes into amastigotes in axenic culture medium and analyzed them for expression of the TbRH1 gene, a similar expression pattern of the polyadenylated and trans-spliced TbRH1 transcripts was observed (data not shown).
Finally, we evaluated whether RNase H activity in the Leishmania cell extract increases with the episomal expression of the TbRH1 gene in these cells. Using a poly(dT): [32P]poly(rA) substrate, we detected significant RNase H activity in the cell-free extract of L. amazonensis promastigotes and axenic amastigotes (Fig. 2C). This RNase H is optimally active at 37° and requires Mg2+. Episomal expression of the TbRH1 gene in these cells increased the RNase H activity 2- to 3-fold over that present in the cell (Fig. 2C). The axenic amastigotes of L. amazonensis had 3-to 4-fold higher RNase H activity than the promastigotes (Fig. 2C). However, episomal expression of TbRH1 in these amastigotes also boosted the RNase H activity about 2-fold (Fig. 2C). Further characterization of Leishmania RNase H will offer a better explanation for these observations.
3.2. Development of stable transfectants of L. amazonensis promastigotes expressing firefly luciferase
The G418-resistant transgenic L. amazonensis promastigotes expressing the TbRH1 gene were electroporated with a firefly luciferase construct, pXHyg-Luci, which contains the hygromycin-resistant gene (Fig. 3A). Like pX63-Neo, pXHyg-Luci also has two strong trans-splicing signals: one in front of the Hygr gene (imported from pX63-Hyg [39]) and the other just before the luciferase gene (from upstream of the L. donovani stationary phase-specific gp63 gene) (Fig. 3A). The gp63 upstream sequence has been shown by others to direct the expression of the luciferase gene in transfected Leishmania [5]. The hygromycin/G418-resistant (200 μg/mL each) L. amazonensis promastigotes were analyzed for luciferase transcripts by RT–PCR. Oligo(dT) primers were used in the reverse transcription of mRNA to cDNA. The 5′-end of the luciferase cDNA was then amplified with a miniexon-specific sense primer (Mex2) and a luciferase-specific antisense primer (LUAS). As shown in Fig. 3B, the luciferase gene was trans-spliced at the expected splicing site. Our data thus suggest that in these stable transfectants of L. amazonensis promastigotes, firefly luciferase is episomally expressed as a trans-spliced and polyadenylated transcript. We transformed these transgenic promastigotes into amastigotes. As expected, amastigotes also expressed the mature luciferase transcript (data not shown). Cell-free lysates from transgenic Leishmania promastigotes and amastigotes contained significant luciferase activity (Fig. 3C). These cells continued to express TbRH1 mRNA and to display enhanced RNase H activity in both the promastigotes and the axenic amastigotes, even after several passages in G418-free growth medium (over a period of 3 weeks).
3.3. Increased inhibition of luciferase mRNA expression by anti-luciferase antisense phosphorothioate ODN in promastigotes and axenic amastigotes overexpressing RNase H
We tested whether an anti-luciferase antisense phosphorothioate ODN ablates luciferase activity more efficiently in the transgenic TbRH1+ L. amazonensis axenic amastigotes (which have pXNeo-TbRH1F plasmid) than in the TbRH1− amastigotes (which have pXNeo-TbRH1R plasmid). L. amazonensis amastigotes growing in axenic culture medium containing 200 μg/mL each of G418 and hygromycin were incubated with the anti-luciferase antisense S-oligo (LUAS) for 20 hr. Control incubations were performed with sense S-oligo (LUS) in parallel. The effect of LUS on cell growth was insignificant. In the presence of LUAS, the cells expressing T. brucei RNase H demonstrated less luciferase activity than those without the T. brucei enzyme (Fig. 4). The antisense S-oligo was more effective in amastigotes than in promastigotes, perhaps because of higher RNase H activities in the former (Fig. 2C).
Fig. 4.
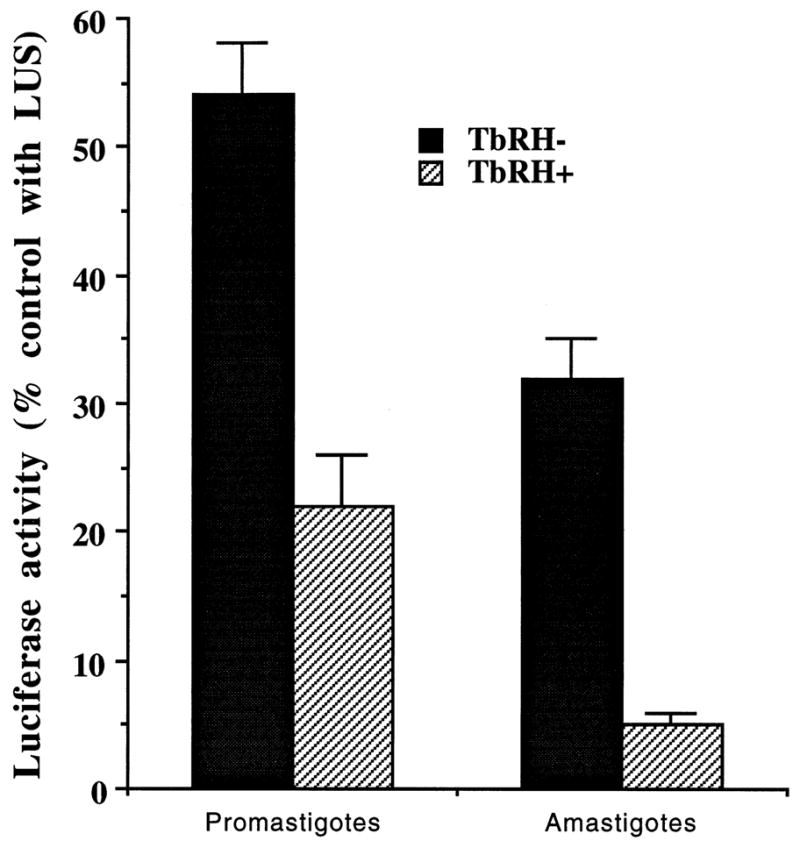
Inhibition of the expression of luciferase activity in RNase H overexpressing L. amazonensis promastigotes and amastigotes by anti-luciferase. Results are means ± SEM (N = 6). Luciferase activities in the controls were 5326 ± 159 and 3648 ± 221 light units/106 cells for promastigotes and amastigotes, respectively. The differences in luciferase activities between TbRH1+ and TbRH1− cell extracts were statistically significant (P < 0.05).
3.4. Increased efficacy of antileishmanial antisense phosphorothioate oligonucleotides in the stable transfectants
We tested whether overexpression of RNase H in Leishmania amastigotes affects their sensitivity against anti-miniexon antisense S-oligo (ASM). Initially, we tested the effect of ASM on the growth of TbRH1− and TbRH1+ L. amazonensis amastigotes cultured in axenic medium. Control incubations were done with the S-oligo with sense nucleotide sequence (SSM) in parallel. SSM at 10 or 20 μM had no significant effect on the growth of the amastigotes. At both concentrations tested, ASM killed the axenic amastigotes more efficiently when the cells expressed the T. brucei RNase H (Fig. 5). We then tested whether ASM was more efficient in killing TbRH1+ amastigotes inside infected macrophages. We previously developed a targeting strategy to deliver S-oligo to the phagolysosomes of Leishmania-infected J774G8 macrophages [9]. The S-oligo was encapsulated inside liposomes, and the liposomes were then coated with phosphatidylethanolamine-anchored MBSA. MBSA is an effective ligand for macrophage scavenger receptor type II, which is lysosomotropic [44,45]. When offered to Leishmania-infected macrophages, ASM encapsulated-MBSA-coated liposomes readily bind to the macrophage scavenger receptor. The liposomes are endocytosed, the resulting endosome fuses with parasitophorous vesicles, and the drug is released following digestion of the liposomes by hydrolases. When targeted this way, the parasite cells are exposed to high concentrations of ASM [9]. In this study, ASM killed the TbRH1+ amastigotes more efficiently than the TbRH1− amastigotes (Fig. 6). In contrast, SSM had no significant effect on the growth of amastigotes.
Fig. 5.

Inhibition of growth of RNase H-overexpressing axenic amastigotes of L. amazonensis by anti-miniexon antisense S-oligo. Results are means ± SEM (N = 4). TbRH1+ and TbRH1− amastigotes incubated in parallel with 20 μM SSM had 2.3 × 107 and 2.2 × 107 cells/mL of culture medium, respectively. The numbers of amastigotes per milliliter of culture, when incubated without S-oligo or with 10 μM SSM, were very similar, and the differences were statistically insignificant. The differences of antisense activities of ASM between TbRH1+ and TbRH1− cells were statistically significant (P < 0.05).
Fig. 6.
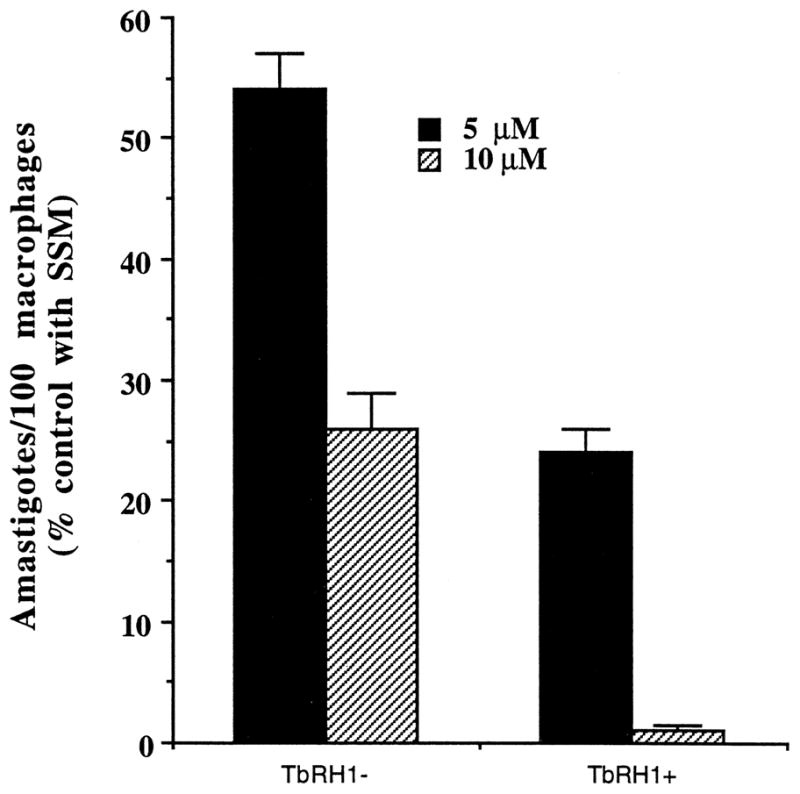
Killing of RNase H-overexpressing L. amazonensis amastigotes by anti-miniexon antisense S-oligo inside macrophage phagolysosomes. Results are means ± SEM (N = 4). Macrophages incubated in parallel with 10 μM SSM had 546 ± 32 and 539 ± 22 amastigotes/100 cells, respectively, for TbRH1+ and TbRH1− cells. The numbers of amastigotes/100 macrophages incubated without S-oligo or with 5 μM SSM were very similar, and the differences were statistically insignificant. The differences of antisense activities of ASM between TbRH1+ and TbRH1− cells were statistically significant (P < 0.01).
4. Discussion
Our aim was to understand, optimize, and evaluate the feasibility of the antisense approach against the expression of leishmanial genes. We previously demonstrated the practicality of the antisense approach against leishmaniasis [9]. We used anti-miniexon ODN as our test compound. We packaged the ODN inside MBSA-coated liposomes (MBSA-liposomes) and offered the liposomes to Leishmania-infected macrophages or mice. This modality of drug delivery proved to be very effective as the drug is quickly and selectively dispatched to macrophages without exposing other biomolecules of the host, thus reducing the possibility of secondary reactions [10].
Although antisense S-oligo worked effectively against Leishmania, we do not know the mode of action of the ODN in this parasite. Nucleotide sequence specificity of the effective S-oligo indicates that the mode of action involves hybridization of the antisense S-oligo to the complementary sense sequence of its target mRNA. The heteroduplex formed between the sense RNA and the antisense DNA may result in inhibition of ribosome binding, capping, polyadenylation, trans-splicing, and/or transport of the mRNA from the nucleus to the cytosol, depending upon the structural distortion caused by the heteroduplex [10]. RNase H is implicated in aiding this process [10]. RNase H recognizes and binds to the heteroduplex and cleaves its RNA component. Originally, it had been thought that antisense oligonucleotides produced their effect by blocking or sterically hindering their target mRNAs. It was soon recognized [10], however, that RNase H may play a prominent role in the mode of action of S-oligos [10].
In this study, using antisense phosphorothioate oligos designed against two different targets, we demonstrated that the efficacy of these antisense S-oligos is enhanced in Leishmania promastigotes and amastigotes overexpressing RNase H. We expressed the T. brucei RNase H1 gene in Leishmania. This gene has all the conserved features for RNase H1 including an N-terminal RNA binding subdomain, conserved RNase H1 catalytic residues, and other invariant amino acid residues conserved at the C-terminus of the enzyme molecule [34]. The recombinant protein expressed from this gene has been shown recently to have RNase H activity by a solution assay using [α-32P]RNA: DNA duplex as a substrate [34]. We also have shown in this study that Leishmania cells transfected with the T. brucei RNase H1 gene had higher RNase H activity than the control cells transfected with the gene cloned in the reverse orientation (with respect to the trans-splicing signal). Thus, T. brucei RNase H1 appeared to be expressed as an active enzyme inside the recombinant Leishmania cells.
We used S-oligos because these molecules are known to mediate their action in an RNase H-dependent manner as opposed to other types of chemical modifications, e.g. morpholino antisense oligomers, which mediate their actions via RNase H-independent mechanisms [52]. The specificity of the antisense S-oligos used in this study was documented using appropriate control S-oligos that did not have any significant target ablation effects. We previously documented the specificity of ASM against Leishmania cell growth using additional control oligonucleotides [9].
The study described herein indicates a possible pivotal role for RNase H of Leishmania promastigotes and amastigotes in the antisense phosphorothioate ODN-mediated killing of these cells. Thus, conceptually, co-treatment of Leishmania with an activator of RNase H and an antisense oligonucleotide should accentuate the efficacy of the latter and provide an efficient chemotherapy against leishmaniasis.
Acknowledgments
We thank Prof. Robert C. Crouch of the National Institutes of Health and Dr. Andrew Campbell of Providence for the suggestions on RNase H assays, Dr. Minu Chaudhuri for T. brucei genomic DNA, and Mrs. Angelika K. Parl for technical assistance. This work was supported by the National Institutes of Health, USA, Grants 5-R01AI42327–03 and 2S06GM08037–24 to G.C.
Abbreviations
- RNase H
ribonuclease H
- S-oligo
phosphorothioate-modified oligodeoxyribonucleotide
- LUAS
anti-luciferase antisense S-oligo
- LUS
S-oligo complementary to LUAS
- ASM
S-oligo antisense to Leishmania miniexon
- SSM
S-oligo complementary to ASM
- HIFBS
heat-inactivated fetal bovine serum
- TbRH1
Trypanosoma brucei RNase H1 gene
- RACE
rapid amplification of cDNA ends
- MBSA
maleylated bovine serum albumin
- ODN
oligodeoxyribonucleotide
- PCR
polymerase chain reaction
- DMPC
dimyristoyl phosphatidylcholine
References
- 1.Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112:2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- 2.Myler PJ, Audleman L, deVos T, Hixson G, Kiser P, Lemley C, Magness C, Rickel E, Sisk E, Sunkin S, Swartzell S, Westlake T, Bastien P, Fu G, Ivens A, Stuart K. Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc Natl Acad Sci USA. 1999;96:2902–6. doi: 10.1073/pnas.96.6.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donelson JE, Gardner MJ, El-Sayed NM. More surprises from Kinetoplastida. Proc Natl Acad Sci USA. 1999;96:2579–81. doi: 10.1073/pnas.96.6.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clayton CE, Ha S, Rusche L, Hartmann C, Beverley SM. Test of heterologous promoters and intergenic regions in Leishmania major. Mol Biochem Parasitol. 2000;105:163–7. doi: 10.1016/s0166-6851(99)00172-3. [DOI] [PubMed] [Google Scholar]
- 5.Ramamoorthy R, Donelson JE, Wilson ME. 5′ Sequences essential for trans-splicing of msp (gp63) RNAs in Leishmania chagasi. Mol Biochem Parasitol. 1996;77:65–76. doi: 10.1016/0166-6851(96)02581-9. [DOI] [PubMed] [Google Scholar]
- 6.Ramazeilles C, Mishra RK, Moreau S, Pascolo E. Antisense phosphorothioate oligonucleotides: selective killing of the intracellular parasite Leishmania amazonensis. Proc Natl Acad Sci USA. 1994;91:7859–63. doi: 10.1073/pnas.91.17.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Compagno D, Lampe JN, Bourget C, Kutyavin IV, Yurchenko L, Lukhtanov EA, Gorn VV, Gamper HB, Toulme JJ. Antisense oligonucleotides containing modified bases inhibit in vitro translation of Leishmania amazonensis mRNAs by invading the miniexon hairpin. J Biol Chem. 1999;274:8191–8. doi: 10.1074/jbc.274.12.8191. [DOI] [PubMed] [Google Scholar]
- 8.Mishra RK, Moreau C, Ramazeilles C, Moreau S, Bonnet J, Toulme JJ. Improved leishmanicidal effect of phosphorothioate antisense oligonucleotides by LDL-mediated delivery. Biochim Biophys Acta. 1995;1264:229–37. doi: 10.1016/0167-4781(95)00145-7. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhuri G. Scavenger receptor-mediated delivery of anti-mini-exon antisense phosphorothioate oligonucleotide to Leishmania-infected macrophages. Selective and efficient elimination of the parasite. Biochem Pharmacol. 1997;53:385–91. doi: 10.1016/s0006-2952(96)00763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook PD. Medicinal chemistry strategies for antisense research. In: Crooke ST, Lebleu B, editors. Antisense research and applications. Boca Raton: CRC Press; 1993. pp. 149–203. [Google Scholar]
- 11.Hausen P, Stein H. Ribonuclease H. An enzyme degrading the RNA moiety of DNA-RNA hybrids. Eur J Biochem. 1970;14:278–83. doi: 10.1111/j.1432-1033.1970.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 12.Eder PS, Walder JA. Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J Biol Chem. 1991;266:6472–9. [PubMed] [Google Scholar]
- 13.Busen W. The subunit structure of calf thymus ribonuclease HI as revealed by immunological analysis. J Biol Chem. 1982;257:7106–8. [PubMed] [Google Scholar]
- 14.Kane CM. Renaturase and ribonuclease H: a novel mechanism that influences transcript displacement by RNA polymerase II in vitro. Biochemistry. 1988;27:3187–96. doi: 10.1021/bi00409a010. [DOI] [PubMed] [Google Scholar]
- 15.Frank P, Albert S, Cazenave C, Toulme JJ. Purification and characterization of human ribonuclease HII. Nucleic Acids Res. 1994;22:5247–54. doi: 10.1093/nar/22.24.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Lima WF, Crooke ST. Properties of cloned and expressed human RNase H1. J Biol Chem. 1999;274:28270–8. doi: 10.1074/jbc.274.40.28270. [DOI] [PubMed] [Google Scholar]
- 17.Ohtani N, Haruki M, Morikawa M, Crouch RJ, Itaya M, Kanaya S. Identification of the genes encoding Mn2+ -dependent RNase HII and Mg2+ -dependent RNase HIII from Bacillus subtilis: classification of RNases H into three families. Biochemistry. 1999;38:605–18. doi: 10.1021/bi982207z. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura H, Oda Y, Iwai S, Inoue H, Ohtsuka E, Kanaya S, Kimura S, Katsuda C, Katayanagi K, Morikawa K, Miyashiro H, Ikehara M. How does RNase H recognize a DNA · RNA hybrid? Proc Natl Acad Sci USA. 1991;88:11535–9. doi: 10.1073/pnas.88.24.11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–9. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- 20.Kanaya S, Katsuda-Nakai C, Ikehara M. Importance of the positive charge cluster in Escherichia coli ribonuclease HI for the effective binding of the substrate. J Biol Chem. 1991;266:11621–7. [PubMed] [Google Scholar]
- 21.Kanaya S, Kohara A, Miura Y, Sekiguchi A, Iwai S, Inoue H, Ohtsuka E, Ikehara M. Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J Biol Chem. 1990;265:4615–21. [PubMed] [Google Scholar]
- 22.Kanaya S. Enzymatic activity and protein stability of E. coli ribonuclease HI. In: Crouch RJ, Toulme JJ, editors. Ribonuclease H. Paris: INSERM; 1998. pp. 1–38. [Google Scholar]
- 23.Kogoma T, Foster PL. Physiological functions of Escherichia coli RNase HI. In: Crouch RJ, Toulme JJ, editors. Ribonuclease H. Paris: INSERM; 1998. pp. 39–66. [Google Scholar]
- 24.Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC and RNase H. Structure. 1995;3:131–4. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- 25.Wu H, Lima WF, Crooke ST. Molecular cloning and expression of cDNA for human RNase H. Antisense Nucleic Acid Drug Dev. 1998;8:53–61. doi: 10.1089/oli.1.1998.8.53. [DOI] [PubMed] [Google Scholar]
- 26.Cerritelli SM, Crouch RJ. Cloning, expression, and mapping of ribonucleases H of human and mouse related to bacterial RNase HI. Genomics. 1998;53:300–7. doi: 10.1006/geno.1998.5497. [DOI] [PubMed] [Google Scholar]
- 27.Hughes SH, Arnold E, Hostomsky Z. RNase H of retroviral reverse transcriptase. In: Crouch RJ, Toulme JJ, editors. Ribonuclease H. Paris: INSERM; 1998. pp. 195–224. [Google Scholar]
- 28.Frank P, Braunshofer-Reiter C, Wintersberger U, Grimm R, Busen W. Cloning of the cDNA encoding the large subunit of human RNase HI, a homologue of the prokaryotic RNase HII. Proc Natl Acad Sci USA. 1998;95:12872–7. doi: 10.1073/pnas.95.22.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itaya M. Isolation and characterization of a second RNase H (RNase HII) of Escherichia coli K-12 encoded by the rnhB gene. Proc Natl Acad Sci USA. 1990;87:8587–91. doi: 10.1073/pnas.87.21.8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell AG, Ray DS. Functional complementation of an Escherichia coli ribonuclease H mutation by a cloned genomic fragment from the trypanosomatid Crithidia fasciculata. Proc Natl Acad Sci USA. 1993;90:9350–4. doi: 10.1073/pnas.90.20.9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hesslein DGT, Campbell AG. Molecular cloning and expression of a ribonuclease H from the kinetoplastid, Trypanosoma brucei. Mol Biochem Parasitol. 1997;86:121–6. doi: 10.1016/s0166-6851(97)90014-1. [DOI] [PubMed] [Google Scholar]
- 32.Ray DS, Hines JC. Disruption of the Crithidia fasciculata RNH1 gene results in the loss of two active forms of ribonuclease H. Nucleic Acids Res. 1995;23:2526–30. doi: 10.1093/nar/23.13.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crouch RJ, Cerritelli SM. Ribonuclease H of Saccharomyces cerevisae, Schizosaccharomyces pombe, Crithidia fasciculata and Neurospora crassa. In: Crouch RJ, Toulme JJ, editors. Ribonuclease H. INSERM; Paris: 1998. pp. 79–100. [Google Scholar]
- 34.Kobil JH, Campbell AG. Trypanosoma brucei RNase HI requires its divergent spacer subdomain for enzymatic function and its conserved RNA binding motif for nuclear localization. Mol Biochem Parasitol. 2000;107:135–42. doi: 10.1016/s0166-6851(00)00182-1. [DOI] [PubMed] [Google Scholar]
- 35.Chaudhuri G, Chaudhuri M, Pan A, Chang KP. Surface acid proteinase (gp63) of Leishmania mexicana. A metallo-enzyme capable of protecting liposome-encapsulated proteins from phagolysosomal degradation by macrophages. J Biol Chem. 1989;264:7483–9. [PubMed] [Google Scholar]
- 36.Sarr Y, Ransford A, Waldman E, Mazareb S, Amin-Spector S, Plumblee J, Turco SJ, Zilberstein D. Characterization of developmentally regulated activities in axenic amastigotes of Leishmania donovani. Mol Biochem Parasitol. 1998;95:9–20. doi: 10.1016/s0166-6851(98)00062-0. [DOI] [PubMed] [Google Scholar]
- 37.Bates PA, Robertson CD, Tetley L, Coombs GH. Axenic cultivation and characterization of Leishmania mexicana amastigote-like forms. Parasitology. 1992;105:193–202. doi: 10.1017/s0031182000074102. [DOI] [PubMed] [Google Scholar]
- 38.Ausubel M, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York: John Wiley; 1994. [Google Scholar]
- 39.Cruz A, Coburn CM, Beverley SM. Double-targeted gene replacement for creating null mutants. Proc Natl Acad Sci USA. 1991;88:7170–4. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seay MB, Heard PL, Chaudhuri G. Surface Zn-proteinase as a molecule for defense of Leishmania mexicana amazonensis promastigotes from cytolysis inside macrophage phagolysosomes. Infect Immun. 1996;64:5129–37. doi: 10.1128/iai.64.12.5129-5137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beverley SM, Clayton C. Transfection of Leishmania and Trypanosoma brucei by electroporation. Methods Mol Biol. 1993;21:333–48. doi: 10.1385/0-89603-239-6:333. [DOI] [PubMed] [Google Scholar]
- 42.Sharan C, Hamilton NM, Parl AK, Singh PK, Chaudhuri G. Identification and characterization of a transcriptional silencer upstream of the human BRCA2 gene. Biochem Biophys Res Commun. 1999;265:285–90. doi: 10.1006/bbrc.1999.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cazenave C, Mizrahi V, Crouch RJ. Methods - rnh gene and ribonuclease H activity analysis. In: Crouch RJ, Toulme JJ, editors. Ribonuclease H. Paris: INSERM; 1998. pp. 251–65. [Google Scholar]
- 44.Chaudhuri G, Mukhopadhyay A, Basu SK. Selective delivery of drugs to macrophages through a highly specific receptor. An efficient chemotherapeutic approach against leishmaniasis. Biochem Pharmacol. 1989;38:2995–3002. doi: 10.1016/0006-2952(89)90007-5. [DOI] [PubMed] [Google Scholar]
- 45.Mukhopadhyay A, Chaudhuri G, Arora SK, Sehgal S, Basu SK. Receptor-mediated drug delivery to macrophages in chemotherapy of leishmaniasis. Science. 1989;244:705–7. doi: 10.1126/science.2717947. [DOI] [PubMed] [Google Scholar]
- 46.Campbell MJ, Machin D. Medical statistics: a commonsense approach. 2. New York: John Wiley; 1994. [Google Scholar]
- 47.Wong AK, Curotto de Lafaille MA, Wirth DF. Identification of cis-acting gene regulatory element from the lemdr1 locus of Leishmania enriettii. J Biol Chem. 1994;269:26497–502. [PubMed] [Google Scholar]
- 48.Lee MG. An RNA polymerase II promoter in the hsp70 locus of Trypanosoma brucei. Mol Cell Biol. 1996;16:1220–30. doi: 10.1128/mcb.16.3.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dresel A, Clos J. Transcription of the Leishmania major Hsp70-I gene locus does not proceed through the non-coding region. Exp Parasitol. 1997;86:206–12. doi: 10.1006/expr.1997.4161. [DOI] [PubMed] [Google Scholar]
- 50.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 51.Kapler GM, Beverley SM. Transcriptional mapping of the amplified region encoding the dihydrofolate reductase-thymidylate synthase of Leishmania major reveals a high density of transcripts, including overlapping and antisense RNAs. Mol Cell Biol. 1989;9:3959–72. doi: 10.1128/mcb.9.9.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489:141–58. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]