

Abstract
In pre-hearing mice, vesicle exocytosis at cochlear inner hair cell (IHC) ribbon synapses is triggered by spontaneous Ca2+ spikes. At the onset of hearing, IHC exocytosis is then exclusively driven by graded potentials, and is characterized by higher Ca2+ efficiency and improved synchronization of vesicular release. The molecular players involved in this transition are still unknown. Here we addressed the involvement of synaptotagmins and otoferlin as putative Ca2+ sensors in IHC exocytosis during postnatal maturation of the cochlea. Using cell capacitance measurements, we showed that Ca2+-evoked exocytosis in mouse IHCs switches from an otoferlin-independent to an otoferlin-dependent mechanism at postnatal day 4. During this early exocytotic period, several synaptotagmins (Syts), including Syt1, Syt2 and Syt7, were detected in IHCs. The exocytotic response as well as the release of the readily releasable vesicle pool (RRP) was, however, unchanged in newborn mutant mice lacking Syt1, Syt2 or Syt7 (Syt1−/−, Syt2−/− and Syt7−/− mice). We only found a defect in RRP recovery in Syt1−/− mice which was apparent as a strongly reduced response to repetitive stimulations. In post-hearing Syt2−/− and Syt7−/− mutant mice, IHC synaptic exocytosis was unaffected. The transient expression of Syt1 and Syt2, which were no longer detected in IHCs after the onset of hearing, indicates that these two most common Ca2+-sensors in CNS synapses are not involved in mature IHCs. We suggest that otoferlin underlies highly efficient Ca2+-dependent membrane-membrane fusion, a process likely essential to increase the probability and synchrony of vesicle fusion events at the mature IHC ribbon synapse.
Introduction
Before the onset of hearing, cochlear inner hair cells (IHCs) exhibit spontaneous Ca2+ spiking activity that triggers synaptic exocytosis (Kros et al., 1998; Beutner et al., 2001; Marcotti et al., 2003; Tritsch and Bergles, 2010) and postsynaptic trains of action potentials (APs) in cochlear ganglion neurons (Lippe, 1994; Jones et al., 2007; Tritsch et al., 2007). In mature IHCs, exocytosis is then driven by sound-evoked fast graded receptor potentials. In both immature and mature IHCs, neurotransmitter release is triggered by Ca2+ influx flowing through L-type Ca2+ channels (Moser and Beutner, 2000; Glowatzki and Fuchs, 2002; Brandt et al., 2003), and the efficiency of brief Ca2+ currents in evoking release increases with cell maturation (Beutner and Moser, 2001; Johnson et al., 2005). Synchronization of the multivesicular release process (Glowatzki and Fuchs, 2002) also improves with IHC maturation (Grant et al., 2010), which suggests that concomitant, as yet poorly understood changes take place in the IHC synaptic machinery.
Like in most neurosecretory cells, synaptic exocytosis in IHCs is thought to involve interactions of SNARE (soluble N-ethylmaleimide-sensitive-factor attachment protein receptor) complex proteins attached to synaptic vesicles (synaptobrevin) with the target plasma membrane (SNAP-25 and syntaxin 1) (Safieddine and Wenthold, 1999). Synaptotagmins (Syts), a large family of transmembrane proteins containing tandem Ca2+-binding C2-domains, confer Ca2+ sensitivity to SNARE-dependent vesicle fusion in the CNS (Chapman, 2008), but their implication in IHC synaptic exocytosis is still unclear. Indeed, the transcripts encoding Syt1 and Syt2, the major Ca2+ sensors for fast synchronized transmitter release in central neurons (Geppert et al., 1994; Sun et al., 2007), have not been detected in mature IHCs (Safieddine and Wenthold, 1999). Therefore, otoferlin, a six C2-domain transmembrane protein (Yasunaga et al., 1999), has been proposed as a major Ca2+ sensor required for Ca2+-evoked exocytosis in IHCs (Roux et al., 2006).
The synaptic active zone of IHCs has been proposed to be organized in independent Ca2+ nanodomains, where the activity of one or few Ca2+ channels is sufficient to activate release of a nearby vesicle (Brandt et al., 2005), allowing the synapse to function in a linear regime (Johnson et al., 2005; Keen and Hudspeth, 2006; Goutman and Glowatzki, 2007). Recently, Syt4, which does not bind Ca2+ and is not established as a Ca2+ sensor (von Poser et al., 1997), has been shown to be required for the linear Ca2+ dependence of exocytosis (Johnson et al., 2010). Notably, this latter study also reported the expression of Syt1 and Syt2 in immature and mature IHCs, respectively, but the functional implication of these findings has not yet been investigated. To clarify the respective roles of these putative Ca2+ sensors at the IHC ribbon synapse along the course of cochlear maturation, we studied their expression and analyzed Ca2+-dependent exocytosis in IHCs from Otof−/−, Syt1−/−, Syt2−/− and Syt7−/− mutant mice that lack otoferlin, Syt1, Syt2 and Syt7, respectively (Geppert et al., 1994; Roux et al., 2006; Sun et al., 2007).
Materials and Methods
Electrophysiological recordings
Experiments were performed on mice, both male and female, obtained by interbreeding of heterozygous mutant mice (Otof+/− or Syt+/− animals) and genotyped using PCR. Recordings and analyses were performed in blind before knowing the genotype of the mice. All experiments were performed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC). All cares were taken to minimize animals' pain. Whole-cell recordings were performed on excised apical coils of organs of Corti as previously described (Beurg et al., 2008). Dissections were done in a solution containing the following (in mm): 135 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 glucose, 2 Na-pyruvate, 10 HEPES, pH 7.4.
Borosilicate patch electrodes (World Precision Instruments) were coated with ski wax (SWIX) to minimize electrode capacitance. Pipette resistances in solution were 2–3 MΩ. Experiments were done at room temperature (20−22°C).
Recordings in otoferlin-null mice were acquired with an Optopatch amplifier (Cairn Research Ltd) digitized using pClamp10 software and a Digidata 1320 A (Molecular Devices). Analysis was performed using Clampfit (Molecular Devices) and Igor software (Wavemetrics). Recordings in Syt1−/− and Syt7−/− mice were performed using an Axopatch 200A digitized using JClamp (SciSoft) and a Micro1401 Interface (Cambridge Electronic Design). Recordings in Syt2−/− mice were performed using an EPC-10 patch-clamp amplifier and the Patchmaster software (HEKA). Current recordings were corrected for linear leak conductance measured near −80 mV. All voltages were corrected for liquid junction potential. For cell capacitance experiments, extracellular solution contained the following (in mm): 115 NaCl, 6 KCl, 5 CaCl2, 1 MgCl2, 30 TEA, 2 Na pyruvate, 8 glucose, 10 Na-HEPES, pH 7.4. Tetrodotoxin (1 μm) and apamin (1 μm) were added to the extracellular solution. Pipette solution for cell membrane capacitance (Cm) experiments contained the following (in mm): 142 CsCl, 1.5 MgCl2,5 TEA, 1 EGTA, 5 creatine phosphate, 10 Cs-HEPES, pH 7.2. ATP was omitted to minimize activation of purinergic receptors, reported in IHCs after P4 (Tritsch and Bergles, 2010). No noticeable difference was observed in changes in cell membrane capacitance (ΔCm) measurements performed in the presence or absence of the nucleotide (data not shown). When EGTA was increased to 5 mm, osmolarity was kept constant to 290 mOsm/L by reduction of CsCl. For experiments in Syt2−/− mice, dissections were done in a solution containing the following (in mm): 143 NaCl, 6 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 8 glucose, 2 Na-pyruvate, 10 Na-HEPES, pH 7.4. Extracellular recording solution was (in mm): 119 NaCl, 6 KCl, 5 CaCl2, 1 MgCl2, 27 TEA, 2 Na pyruvate, 8 glucose, 10 Na-HEPES, pH 7.4. Tetrodotoxin (1 μm) and apamin (1 μm) were also added to the extracellular solution for Cm measurements. Pipette solution for Cm experiments in Syt2−/− mice contained the following (in mm): 140 Cs-gluconate, 4 MgATP, 0.3 Na2GTP,20 TEA-Cl, 0.5 EGTA, 5 creatine phosphate, 10 HEPES pH 7.2.
For action potential and afferent fiber recordings, the internal solution contained the following (in mm): 140 KCl, 3.5 MgCl2, 0.1 CaCl2, 5 EGTA, HEPES 5, pH 7.2.
Capacitance measurement.
Changes in cell membrane capacitance (ΔCm) were used to monitor fusion of synaptic vesicles during exocytosis. Cm was measured according to the Lindau-Neher technique (Lindau and Neher, 1988), using the tracking circuitry of the Optopatch (experiments on otoferlin-null mice) or implemented in the JClamp (Scisoft, New Haven, CT) (Syt1-null or Syt7-null mice recordings), or the Patchmaster softwares (Syt2-null mice). A 20 mV amplitude sine wave from a holding potential of −80 mV was used; for recording of Syt2-null mice, the sine wave had an amplitude of 30 mV from a holding potential of −90 mV. The resulting maximal depolarizations to ∼−60 mV were sufficiently small to avoid activation of Ca2+ current (ICa). The command sine wave (781.3–1600 Hz) was blanked during the duration of the voltage step and resumed immediately upon repolarization to capture the capacitance before and after the pulse. ΔCm was estimated as the mean Cm measured 150 ms after the end of the voltage step (to discard capacitance changes due to ICa tail currents) over a period of 50 ms and the prepulse Cm averaged over 50 ms. Only cells with stable series resistance below 10 MΩ (uncompensated) were included in the study.
Afferent fiber recordings.
EPSCs were recorded using the whole-cell patch-clamp technique, digitized at 50 kHz, and low pass filtered at 5–10 kHz. Patch pipettes had a resistance of ∼10 MΩ.
Curve fitting and statistical analyses.
Curve fitting and analysis were done using Igor Pro (WaveMetrics). The criterion for statistical significance was chosen to be p < 0.05 using unpaired Student's t test. Variability is reported as mean ± SEM.
Single-cell reverse transcription PCR
Inner hair cells were collected from excised organs of Corti (of P1 and P7 mice). Single-cell reverse transcriptase (RT)-PCR was done as previously described (Michalski et al., 2007). Synthesis of cDNA was performed using the SuperScript II Reverse Transcriptase kit (Invitrogen). For each cell, we used the extracellular fluid as a negative control. A multiplex nested PCR was performed using specific primers for the tested synaptotagmin (Syt) transcripts (see supplemental Table, available at www.jneurosci.org as supplemental material, for primer sequence) and for two control transcripts, namely myosin VIIa [Myo7A, expressed in both outer hair cells (OHCs) and IHCs] and prestin (only in OHCs) (same primers reported by Michalski et al., 2007). PCR products were sequenced to confirm their specificity. The PCR consisted of 20 amplification cycles (40 s at 95°C, 45 s at 58°C, and 50 s at 72°C). Amplified DNA fragments had a molecular weight ranging between 300 and 500 bp. The nested PCR was performed in a 25 μl mix containing 2 μl of the first PCR mix and only one set of inner primers at a time through 35 cycles (40 s at 95°C, 45 s at 58°C, and 50 s at 72°C).
Twenty IHCs were collected for each developmental stage. Only IHCs that showed the myosin VIIa-positive and prestin-negative expression profiles were taken into account for result analysis (n = 13). The number of Syt-positive cells was then counted for each Syt and compared statistically to the number of extracellular fluid control samples (n = 20) that abnormally scored positive for the presence of cDNA amplification, using the χ2 test (with Yates correction for small samples) to check for significance (p < 0.05).
Whole-mount immunohistochemistry
Cochleae were fixed with 4% paraformaldehyde in PBS for 1 h at 4°C, and the external bone was removed. The organ of Corti was isolated and the tectorial membrane was removed. The tissue samples were permeabilized with 0.3% Triton X-100 in PBS containing 20% normal goat serum for 1 h at room temperature. After PBS washes, they were incubated with the primary antibodies, diluted in PBS containing 10% goat serum (or horse serum) and 0.1% Triton X-100, overnight at 4°C. Omission of the primary antibody was used as a negative control. Primary antibodies were used at the following dilutions: anti-Syt1 (1: 200, rabbit polyclonal, Synaptic Systems; 1:500 mouse monoclonal (C141.1), Synaptic Systems; 1:500 rabbit polyclonal, Abcam), anti-Syt7 (1:200, rabbit polyclonal, Synaptic Systems), anti-Syt2 (znp-1 1:200, mouse monoclonal, ZIRC; or 1:200, rabbit polyclonal against Syt2, Synaptic Systems), anti-neurofilament NF-200 (1:1000, mouse monoclonal, a gift from Dr. D. Dahl, Harvard Medical School, Boston, MA), anti-synaptophysin (1:200 mouse monoclonal, Sigma; 1:200 rabbit polyclonal, Synaptic Systems), anti-CtBP2 (1:200, goat polyclonal, Santa Cruz Biotechnology). Tissue samples were washed in PBS, and incubated with secondary antibodies for 2 h at room temperature. Dilutions of secondary antibodies were as follows: goat anti-rabbit IgG coupled to Alexa Fluor 488 (1:500); goat anti-mouse IgG antibody conjugated with Cy3 fluorophore (Jackson ImmunoResearch Laboratories); goat anti-mouse IgG coupled to Alexa Fluor 488 (1:500), chicken anti-goat IgG coupled to Alexa Fluor 488 (1:1000). The nuclear dye DAPI was added to aid in hair cell visualization. The samples were analyzed using a confocal laser scanning microscope LSM510 Meta (Zeiss, Pasteur Institute, Imagepole) or a Leica confocal upright microscope (Leica DMR TCS SP2 AOBS, Bordeaux Imaging Center). The images were taken with a step size of 0.4 μm.
Specificity of anti-Syt antibodies was determined by the lack of residual Syt-immunostaining in the organ of Corti of the respective Syt-null mutant mice. The persistence of the IHCs immunostaining observed in Syt2-null mice when using anti-Syt2 polyclonal antibody (from Synaptic Systems) indicated the nonspecificity of this labeling (data not shown). Therefore, the Syt2 distribution in the organ of Corti was only documented using the znp-1 monoclonal antibody. Similarly, the distribution of Syt7 could not be reliably analyzed since the immunostaining of the organ of Corti obtained with the anti-Syt7 polyclonal antibody (Synaptic Systems) was still detected in Syt7−/− mutant mice (data not shown). In contrast, the specificity of the IHC immunolabeling that was observed in wild-type mice using polyclonal anti-Syt1 (Synaptic Systems) and monoclonal anti-Syt2 (ZIRC) antibodies could be established by the lack of staining in the Syt1−/− or Syt2−/− mutant mice that lack Syt1 and Syt2, respectively (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The same Syt1 immunostaining was observed in the organ of Corti from wild-type mice using any of the three antibodies directed against this protein (data not shown).
Results
Exocytosis in IHCs from early postnatal mice is otoferlin independent
Otoferlin is critical for Ca2+-triggered synaptic exocytosis in IHCs from P6 onwards (Roux et al., 2006). However, IHC exocytosis has not been explored at earlier developmental stages. We thus characterized the Ca2+-dependent exocytosis in IHCs from Otof−/− mice and control littermates from postnatal days 1–8 (P1–P8). To this purpose, we monitored ΔCm in response to voltage activation of ICa. Electrophysiological data obtained from Otof+/+ and Otof+/− IHCs were pooled and referred to as “controls”, after checking that there was no statistical difference between the two groups (unpaired t test, p < 0.01).
In P1 control and Otof−/− IHCs, substantial ΔCm responses could be recorded (Fig. 1A,B). In P1 Otof−/− IHCs, the mean ΔCm evoked by a 100 ms voltage-step to −10 mV (ICa peak) was 17.5 ± 1.7 fF (n = 25), a value similar to that found in control IHCs (14.9 ± 3.1 fF, n = 12; p = 0.12) (Fig. 1C). These ΔCm responses were abolished by 50 μm nifedipine (data not shown, n = 2), indicating that this otoferlin-independent exocytosis is also triggered by L-type Ca2+ channels. The ΔCm responses in Otof−/− IHCs dramatically decreased at P4, and became undetectable from P5 on (Fig. 1C,F), as previously reported (Roux et al., 2006). Notably, a gradual increase of otoferlin immunolabeling in IHCs was observed during the first week of postnatal development: the staining was weak at P0, maximal at P6 (Fig. 1E) and remained constant thereafter (Roux et al., 2006). In Otof−/− IHCs, the biophysical properties of Ca2+ current (kinetics, voltage activation) and developmental increase in Ca2+ channel density were similar to those of control mice (data not shown and Fig. 1B–D). During the first postnatal week, we observed a parallel increase of the ICa density and the triggered membrane capacitance (ΔCm) in control IHCs (Fig. 1A,C,D), while the Ca2+ efficiency of exocytosis, defined here as (ΔCm/ICa) remained identical (Fig. 1F), in agreement with previous results showing that the Ca2+ efficiency only starts to increase after P6 (Beutner and Moser, 2001; Johnson et al., 2005). Up to P3, the Ca2+ efficiency of exocytosis in Otof−/− IHCs was similar to that in control IHCs (Fig. 1F).
Figure 1.
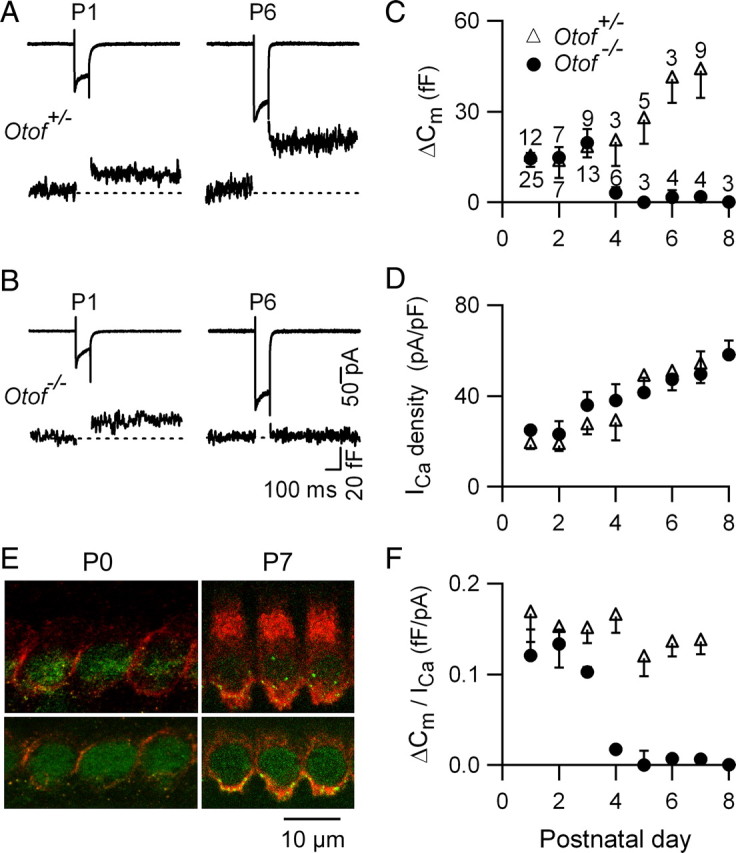
Ca2+-evoked exocytosis in IHCs during early postnatal development: an otoferlin-independent process before P4. A, B, Examples of ICa and corresponding ΔCm responses in P1 and P6 control and Otof−/− IHCs. Cells were stimulated by a 100 ms voltage step from a holding potential of −80 mV to −10 mV. C, D, ΔCm response average (C) and corresponding ICa density (D) from IHCs at different postnatal stages. E, Otoferlin (red) and CTBP2 (green) immunolabelings in IHCs from P0 and P7 mice. Note the increase in otoferlin immunoreactivity at P7. F, Ca2+ efficiency (ΔCm/ICa) of exocytosis as a function of development for the cells tested in C and D.
Spontaneous firing activity (sensitive to nifedipine) was present with similar event frequencies in P1–P3 IHCs from Otof−/− and control mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This spontaneous IHC spiking is likely to be at the origin of the EPSCs and spontaneous APs that we recorded in afferent fibers from Otof−/− and control mice, with similar amplitudes and event frequencies (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results suggest that exocytosis triggered by spontaneous APs in P1–P3 IHCs does not require otoferlin either.
We further characterized the otoferlin-independent synaptic exocytosis in early postnatal IHCs by studying the synaptic transfer function, that is, the relationship between Ca2+ entry and exocytosis. The voltage-evoked ΔCm responses (100 ms step ranging from −60 to −10 mV) were plotted against the corresponding ICa amplitudes (Fig. 2A,B). The Ca2+ dependence of transmitter release in central synapses, notably at the large calyx of Held nerve terminals, is described by a nonlinear model of cooperative Ca2+ binding to the sensor (Schneggenburger and Neher, 2000; Wölfel and Schneggenburger, 2003). Considering that Ca2+-evoked vesicle fusion (ΔCm) in IHCs is also intrinsically cooperative (Beutner et al., 2001), the data were fitted using the equation ΔCm = g [ICa]N, with ICa varying both as a function of open Ca2+ channels (number increasing with depolarization) and Ca2+ flux per channel (the Ca2+ driving force decreasing with depolarization). In IHCs from both control and Otof−/− P1–P3 mice, the synaptic transfer function displayed a mean power N = 1.7 ± 0.2 (parameters fitted separately to each cell) (Fig. 2B). Moreover, the otoferlin-independent (P1–P3) and otoferlin-dependent (P7) synaptic transfer functions were similar in the control mice (N = 1.6 ± 0.5 for P7 IHCs, n = 7; p = 0.8) (Fig. 2C). These results suggest that otoferlin-dependent and otoferlin-independent exocytosis in immature IHCs display comparable Ca2+ dependencies, notably with a similar cooperative index.
Figure 2.

Properties of otoferlin-independent exocytosis in immature IHCs. A, ICa–voltage relationship and corresponding ΔCm in IHCs from control (triangles) and Otof−/− (circles) P1 mice. B, Corresponding synaptic transfer function plot (ΔCm against ICa amplitude, up to ICa peak). Lines are power function fits with N = 1.6 and N = 1.8 in control (dashed line) and Otof−/− (solid line) IHCs. C, Synaptic transfer function for IHCs from P7 control mice (power fit: N = 1.6), plotted on log scales. D, Kinetics of vesicle pool depletion in P1 IHCs. ΔCm is plotted as a function of the stimulus duration for a step to −10 mV, from a holding potential of −80 mV. Solid lines correspond to a single exponential fit of RRP (inset, between 0 and 200 ms) and dashed lines to another single exponential fit of SRP (500–3000 ms). Fit parameters were for RRP, τ = 48 and 63 ms, ΔCm(max) = 20.8 fF and 16.2 fF, and for SRP, τ = 1363 and 1739 ms, ΔCm(max) = 167 fF and 138 fF in control (black) and Otof−/− (red) IHCs, respectively. E, Kinetics of exocytosis in the presence of 5 mm EGTA in P1 IHCs. Note the block of SRP in the presence of EGTA. Fit values were as follows: τ = 77 ms, ΔCm(max) = 13 fF (control) and τ = 45.5 ms, ΔCm(max) = 11 fF (Otof−/−). F, Absence of fast exocytosis in IHCs from Otof−/− P7 mice, while residual slow vesicular release was still observed. Fit values for IHCs from control mice τ = 29 ms, ΔCm(max) = 56 fF for RRP, and τ = 1000 ms, ΔCm(max) = 526 fF for SRP. Black and red lines are fits for IHCs from control and Otof−/− mice, respectively.
We compared the dynamics of vesicle pool depletion in IHCs from Otof−/− and control newborn mice (P1) by using a constant depolarizing step (from −80 to −10 mV) of duration increasing from 10 to 3000 ms (Fig. 2D–F). Both in control and Otof−/− mice, data points (ΔCm vs time) were best fitted by two exponentials, suggesting the presence of two vesicular pools: a readily releasable pool (RRP) and a slowly releasable pool (SRP) that may arise from vesicles docked at the active zones and vesicles located further from the Ca2+ channels, respectively. The RRP component of control and Otof−/− P1 IHCs, spanning between 10 and 200 ms, was fitted using a single-exponential with time constants τ = 48 and 63 ms, and ΔCm(max) = 21 fF and 16 fF, respectively (Fig. 2D, inset) (not statistically different, with p = 0.5, in comparing individual cell fit values). Assuming a ΔCm of 37 aF per vesicle (Lenzi et al., 1999), RRP sizes were estimated at 562 and 434 vesicles, respectively. For longer stimuli, control and Otof−/− P1 IHCs displayed similar SRP components (τ = 1363 and 1739 ms, maximal ΔCm = 167 fF and 138 fF, respectively; Fig. 2D).
In P7 Otof−/− IHCs, we confirmed the absence of vesicular release from the RRP (no ΔCm <500 ms voltage-steps), but found a residual slow exocytosis for longer stimuli (Fig. 2F), as previously reported (Roux et al., 2006).
To address the issue of the relative spatial organization of the Ca2+ channels and Ca2+ sensor, we measured the sensitivity of the RRP and SRP components to various intracellular Ca2+ buffer concentrations. Raising the intracellular concentration of the slow Ca2+ buffer EGTA from 1 to 5 mm, a way to limit intracellular Ca2+ spread to within the microdomains, suppressed the SRP-release but not the RRP-release in IHCs from both control and Otof−/− P1 mice (Fig. 2E). This indicates that Ca2+ channels and non-otoferlin Ca2+ sensors triggering RRP vesicular release work in close vicinity in P1 IHCs. This result also suggests that SRP release results from vesicles located farther from the Ca2+ channels, or, alternatively, might reflect a Ca2+-dependent recovery of the RRP. Furthermore, repetitive exocytosis of the RRP (evoked by a train of 50 ms depolarizing steps) was similar in control (see Fig. 4G) and Otof−/− P1 IHCs (data not shown), with an average cumulative ΔCm response of 21.8 ± 0.5 fF (n = 6) and 18.4 ± 4.1 fF (n = 4), respectively, which suggests that the recruitment of synaptic vesicles to maintain a constant RRP in P1 IHCs is not affected in the absence of otoferlin. In summary, all parameters of exocytosis tested in P1–P2 Otof−/− mice were unchanged compared with their control littermates.
Figure 4.
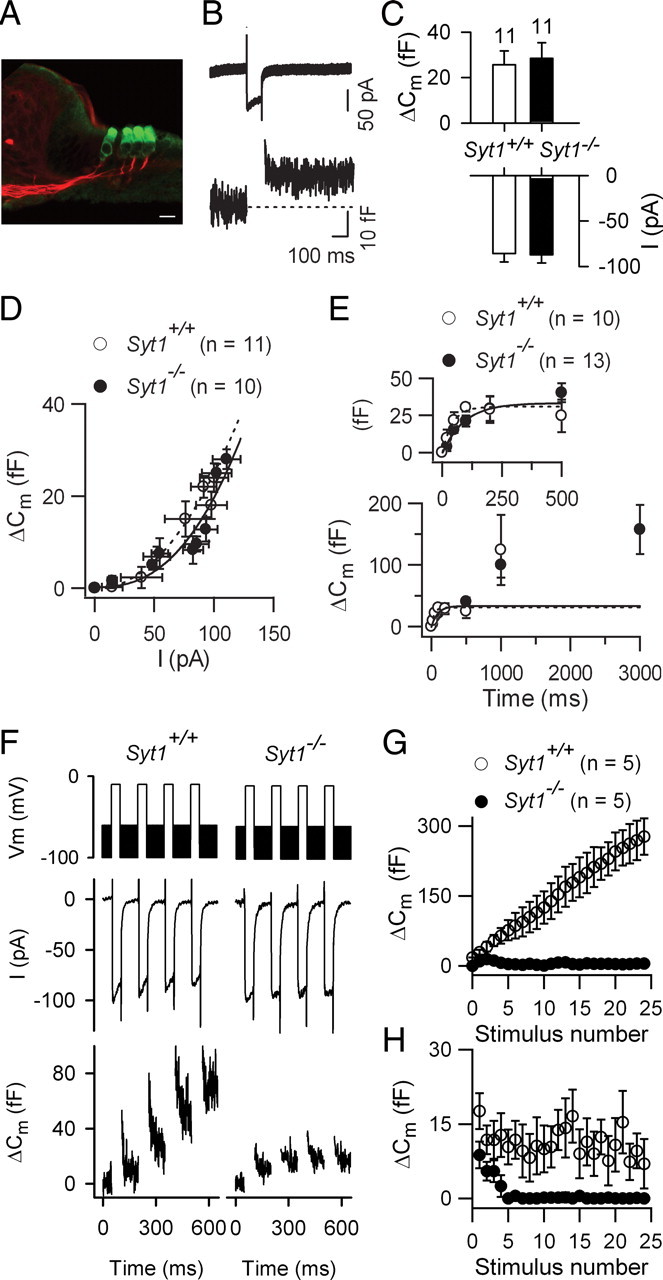
Ca2+-dependent exocytosis in IHCs from Syt1−/− newborn mice. A, The organ of Corti of Syt1−/− P0 mice has a normal arrangement of hair cells (green, anti myosin-VIIA labeling) and afferent nerve fibers (red, neurofilament labeling). Scale bar, 10 μm. B, Example of ICa with its corresponding ΔCm response in an IHC from a Syt1−/− P0 mouse. C, Histograms showing the mean ICa peak amplitude and its corresponding ΔCm for Syt1−/− and Syt1+/+ IHCs (100 ms voltage step to −10 mV). D, Average ΔCm plotted against ICa elicited by 100 ms voltage steps ranging from −60 to −10 mV (in 5 mV increments). Lines correspond to a power function fit with N = 2.0 and 2.1 in Syt1+/+ (dashed line) and Syt1−/− (solid line) IHCs, respectively. E, Kinetics of Ca2+-dependent exocytosis. ΔCm is plotted against stimulus duration at a constant voltage (−10 mV). Lines are single exponential fits for Syt1−/− (solid line) and Syt1+/+ (dashed line) IHCs. F–H, Recruitment of vesicles from the fast synaptic vesicular pool. ICa and corresponding ΔCm elicited by a train of 25 successive 50 ms voltage steps to −10 mV, separated by 100 ms time intervals. F, Examples are shown for the first four steps. G, Average cumulative ΔCm responses obtained for 25 consecutive voltage steps. H, Plot of individual ΔCm measured after each voltage step.
Expression of synaptotagmins in IHCs
The persistence of Ca2+-dependent exocytosis in IHCs from P0–P3 Otof−/− mice raises the question of the molecular identity of the Ca2+ sensors involved at early postnatal stages. Since several Syts are regarded as Ca2+ sensors regulating exocytosis in the CNS, we used single-cell RT-PCR to detect Syt transcripts in IHCs. We investigated the expression of eight different Syts, Syt1, 2, 3, 5, 6, 7, 9 and 10, all known for their Ca2+-binding properties (Südhof, 2002), in P1 and P7 IHCs. Six different Syts were found to be expressed in P1 IHCs, of which Syt 1, 2, 6, and 7 displayed the highest expression levels. In P7 IHCs, Syt1, Syt6 and Syt7 still were strongly expressed, whereas the expression of Syt2 was markedly decreased (Fig. 3G).
Figure 3.

Syts expression in cochlear hair cells. Confocal microscopy images of whole mounts of the organ of Corti at P1, P6, P8, and P15, double-labeled either for Syt1 and synaptophysin (Syphy) (A, F), or for Syt1 and Syt2 (B–E). DAPI staining (blue) was used to stain cell nuclei. Dashed lines outline IHCs and OHCs (based on the reconstructed z-stack, data not shown). A–D, Left and right panels are cross-section views in the x,z and y,z plans, respectively. At P1 and P6, a Syt1 staining can be seen along the membrane of IHCs (arrows in A and B, top) and in the terminals of efferent fibers stained for Syphy (A, middle, arrowheads). The merge image (A, bottom) shows an overlapping of Syt1 and Syphy stainings at the efferent fibers below IHCs (arrowheads). No Syphy labeling was seen in IHCs. Syt2 labeling is predominantly cytoplasmic (B, C, middle), compared with that of Syt1, which is mainly located along the basolateral membrane of the cell (B, C, top). At P8, a Syt1 staining is still present in IHCs and OHCs (D, top), while Syt2 is no longer detected in these cells (D, middle panels). E, F, At P15, neither Syt2 nor Syt1 are detected in IHCs and OHCs (E, left and middle). Both proteins are, however, still present in the efferent fibers stained for Syphy (F, middle). Scale bars, 5 μm. G, Single cell RT-PCR was performed in IHCs harvested at P1 and P7. Only IHCs expressing the proper control transcripts (positive for myosin VIIA, and negative for prestin) were computed in the histograms. Data were expressed as a percentage of positive samples [(number of samples containing the Syt transcript/total number of samples tested)*100)]. Asterisks indicate IHCs with significant positive levels of Syt transcripts (χ2 test, p < 0.05). Transcripts for Syts 1, 2, 6, 7 and Syts 1, 6, 7 were the most abundant Syt transcripts in P1 (n = 13) and P7 (n = 13) IHCs, respectively. Note the drastic decrease in Syt2 mRNA expression at P7.
These results were further substantiated by immunolabeling experiments and confocal imaging of the organ of Corti before (P1, P6, P8) and after (P15 and P21) the onset of hearing. Whole-mount preparations were double-immunostained for Syt1 or Syt2, and for the synaptic vesicle protein synaptophysin. This latter protein is a marker of lateral efferent presynaptic terminals, that form axodendritic synapses with afferent dendrites below IHCs, and medial efferent synapses contacting OHCs (Gil-Loyzaga and Pujol, 1988; Safieddine and Wenthold, 1999). From P1 to P6, Syt1 and Syt2 labelings were present in both IHCs and OHCs (Fig. 3A–C), but the two proteins displayed different subcellular distributions as shown by double-labeling experiments (Fig. 3B,C). The strongest Syt1-immunolabeling was observed along the basolateral membrane that lines the IHC presynaptic region (Fig. 3A,B), whereas the Syt2-immunolabeling was mainly cytoplasmic (Fig. 3B,C). In addition, Syt1 and Syt2 displayed different temporal profiles in the IHCs, as expected from our single-cell RT-PCR results. The Syt2-immunolabeling was no longer observed in IHCs from P8 onward, while Syt1 could still be detected up to P10 through all the cochlear spiral (Fig. 3D; supplemental Fig. 3, available at www.jneurosci.org as supplemental material), but vanished at P15 in the IHCs of the cochlear middle turn (Fig. 3E,F) and at P21 in those of the apical turn (data not shown). Finally, at all stages investigated (from P1 to P21), Syt1 and Syt2 immunolabelings were detected in the efferent, synaptophysin-immunoreactive, nerve fibers (Fig. 3A–F; data not shown).
In Otof−/− IHCs, Syt1 and Syt2 had the same distributions and developmental patterns, as in control littermates (data not shown). Furthermore, Syt2 was present in P0 Syt1−/− IHCs and Syt1 was present in P0 and P6 Syt2−/− IHCs (data not shown), suggesting that the lack of one Ca2+ sensor does not influence the expression of the other.
Syt1−/− IHCs exhibit a RRP release unchanged but a diminished RRP recovery
We next assessed the role of Syt1 in IHC exocytosis by measuring Cm changes in response to ICa in P0 Syt1−/− mice. The short postnatal survival of these mice (∼24 h) (Geppert et al., 1994) restricted measurements to newborn mice. No morphological anomalies were observed in the IHCs and OHCs of the Syt1−/− newborn mice. Indeed, we found presynaptic ribbons and proper innervation contacting the hair cells, with no decrease in the number of cochlear ganglion neurons (Fig. 4A).
In Syt1−/− P0 IHCs, ICa-evoked ΔCm responses were similar to those in Syt1+/+ (wild-type) IHCs (Fig. 4B,C), with a mean amplitude of 25.6 ± 6.1 fF (n = 11) and 28.5 ± 8.8 fF (n = 11), respectively (p = 0.9) for a 100 ms voltage-step close to ICa peak. The corresponding ICa amplitudes were 87 ± 9 pA and 86 ± 9 pA (p = 0.8) (Fig. 4C). The synaptic transfer function in Syt1−/− IHCs was evaluated as described above, by plotting ΔCm against ICa (100 ms voltage-step ranging from −60 mV to −10 mV, from a holding potential of −80 mV) (Fig. 4D). In wild-type and Syt1−/− IHCs, the synaptic transfer function was best fitted using N = 2.1 ± 0.6 (n = 11) and N = 2.0 ± 0.2 (n = 10), respectively (p = 0.4), indicating that exocytosis evoked in the absence of Syt1 has an unchanged cooperative index in P0 IHCs. The kinetics of RRP exocytosis (−10 mV voltage-steps of various durations) were also similar in Syt1−/− mice and control littermates (τ = 60 ± 20 ms and 65 ± 14 ms, respectively; p = 0.8; Fig. 4E). This suggested that release from the RRP was unchanged in newborn Syt1−/− mice.
We observed, however, that depolarizing stimuli had to be separated by at least 1 min intervals to trigger reproducible ΔCm responses in Syt1−/− IHCs. This long time requirement after each Ca2+ stimulation suggested that the recovery of RRP vesicles might be affected in Syt1−/− IHCs. To further investigate the vesicle recruitment and refilling of the RRP, we stimulated IHCs with a train of brief (50 ms) voltage steps spaced by 100 ms at the holding potential of −80 mV, during which sine wave for Cm measurements was applied (Fig. 4F, top). In wild-type mice, a 50 ms-depolarizing train evoked a linear increase of the cumulative ΔCm values (Fig. 4F,G), indicating that the RRP was able to be rapidly refilled in-between successive stimuli. In contrast, in Syt1−/− IHCs, the RRP recruitment was affected after the second test pulse, despite a stable ICa amplitude (Fig. 4F,G). The ΔCm at each step, when plotted against the cumulative number of stimuli, showed a constant value in wild-type IHCs, while this value rapidly decreased to zero after a few steps in Syt1-null IHCs (Fig. 4H). In summary, these results indicated that in P0 Syt1−/− IHCs, the RRP release is normal but the process of RRP recovery is strongly affected.
Ca2+-dependent exocytosis is not affected in IHCs from Syt2−/− or Syt7−/− mice
Syt2−/− mice generally survive up to 3 weeks, but exhibit severe motor dysfunction after the second week (Pang et al., 2006). Given the selective expression of Syt2 in IHCs during the first few postnatal days (Fig. 3), we first studied exocytosis in P2–P3 Syt2−/− IHCs (Fig. 5), but we failed to detect deficits in exocytosis. In Syt2+/+ or Syt2+/− control mice, a 100 ms voltage step to −10 mV evoked a Ca2+ current (ICa) of 172 ± 19 pA, which elicited a ΔCm step of 6.8 ± 1.3 fF (n = 17). In Syt2−/− IHCs, a same voltage stimuli elicited an average ΔCm response of 6.3 ± 1.3 fF (n = 10), which was indistinguishable from the one measured in the control mice (p > 0.5). The Ca2+ current was 125 ± 23 pA (n = 10); which was slightly, but not significantly smaller than the one measured in control mice (p = 0.13). In plots of the exocytotic ΔCm response versus the length of the depolarizing pulses, the release kinetics was not substantially modified in Syt2−/− mice compared with control littermates (Fig. 5C). Using a train of brief (50 ms) voltage steps to −10 mV (close to peak of ICa), a sustained vesicular release, similar to controls, could be observed in IHCs from P2–P3 Syt2−/− mice, indicating that the RRP was rapidly refilled between voltage pulses (Fig. 5D).
Figure 5.

Ca2+-evoked exocytosis in IHCs from P2–P3 Syt2−/− mice. A, Examples of Ca2+-evoked exocytosis in P2–P3 Syt2+/+ and Syt2−/− IHCs. Cells were stimulated by a 100 ms voltage step to −10 mV from a holding potential of −90 mV. B, Bar graph of average and individual ICa peak amplitudes and their corresponding ΔCm values for Syt2+/+ (n = 17) and Syt2−/− (n = 10) IHCs. C, Kinetics of Ca2+-dependent exocytosis. Only cells with large ΔCm responses were included for the analysis in this graph. D, Average cumulative ΔCm responses from P2–P3 Syt2+/+ and Syt2−/− IHCs elicited using 50 ms repetitive voltage steps to −10 mV (interstep intervals of 100 ms).
Since Syt2 has been recently proposed to underlie the Ca2+ dependence of vesicle release in mature IHCs (Johnson et al., 2010), we also investigated the properties of exocytosis in near-mature, P15–P17 IHCs from Syt2−/− mice (Fig. 6). Again, we did not detect any obvious exocytotic defect. In Syt2−/− P15–P17 IHCs, 100 ms voltage steps to ICa peak evoked ΔCm with a mean amplitude of 27.1 ± 2.3 fF for ICa peak of 183 ± 11 pA (n = 15). These values were similar to those obtained in the wild-type littermates [27.4 ± 4.7 fF and 182 ± 23 pA, n = 8 (p > 0.5 for both comparisons)]. In P15–P17 IHCs from Syt2−/− mice, both the kinetics of exocytosis (Fig. 6C) and vesicle recruitment (Fig. 6D) were similar to those in the control littermates. Furthermore, the synaptic transfer function showed similar linear relationships in P15–P17 wild-type and Syt2−/− mice (Fig. 6E).
Figure 6.
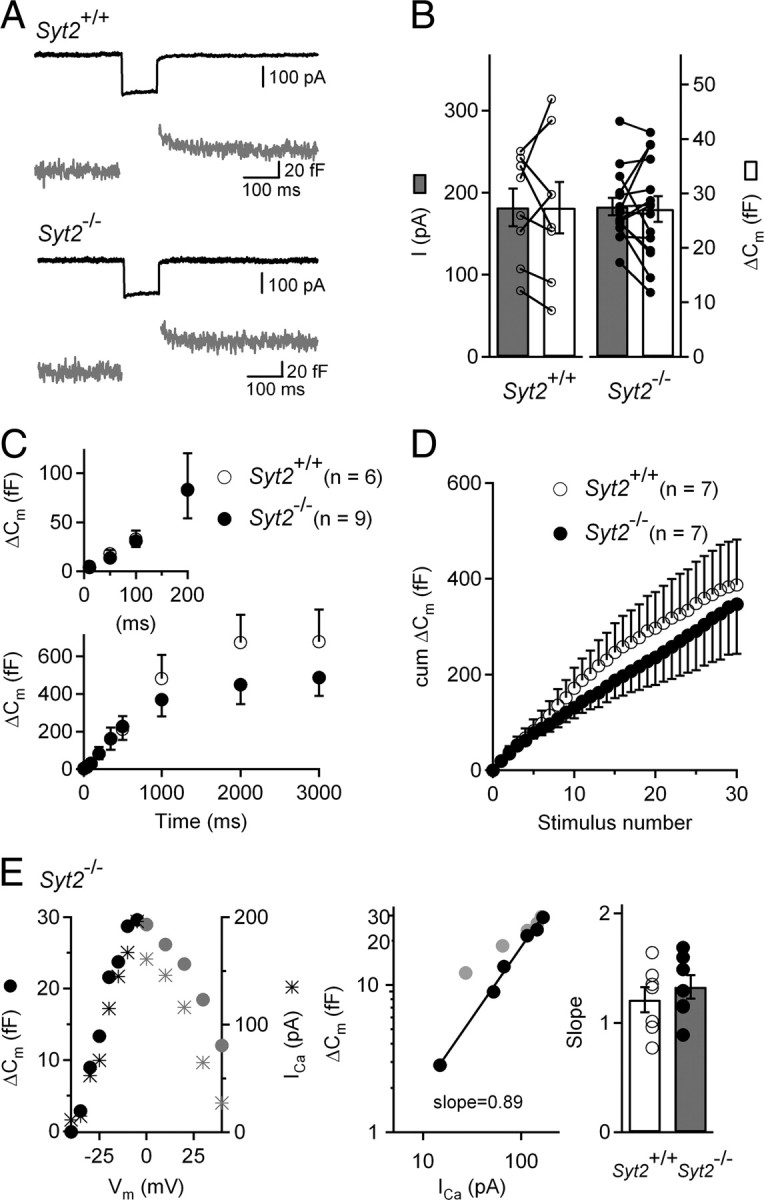
Ca2+-evoked exocytosis in IHCs from P15–P17 Syt2−/− mice. A, Examples of Ca2+-evoked exocytosis in P15–P17 Syt2+/+ and Syt2−/− IHCs. Cells were stimulated by a 100 ms voltage step from a holding potential of −90 to −10 mV. B, Bar graph of average and individual ICa peak amplitudes and their corresponding ΔCm values, for Syt2+/+ (n = 8) and Syt2−/− (n = 15) IHCs. C, Kinetics of Ca2+-dependent exocytosis. ΔCm is plotted against stimulus duration for pulses to −10 mV. Note that the Syt2+/+ and Syt2−/− data points are overlaid in inset. D, Average cumulative ΔCm responses from P15–P17 Syt2+/+ and Syt2−/− IHCs elicited by 50 ms repetitive voltage steps to −10 mV (interstep interval of 100 ms). E, Left, ΔCm and ICa peak amplitude for an IHC from a P16 Syt2−/− mouse, plotted for different voltages following a 100 ms voltage step from a holding potential of −90 mV. Gray points indicate voltages beyond the ICa peak which were not taken into account to calculate the ΔCm-ICa slope. Middle, Corresponding ΔCm versus ICa plot. The logarithmized data were fitted by a line, giving a slope of 0.89. Right, Average and individual slope values for P15–P17 Syt2+/+ (n = 7) and Syt2−/− (n = 6) IHCs.
In Syt7−/− mice, we did not find any defect of Ca2+-evoked exocytosis (RRP component) induced by ICa activation in immature (P2–P8) or post-hearing onset (P15–P30) IHCs (Table 1, Fig. 7). In these mice, Ca2+-evoked exocytosis underwent a normal postnatal maturation, with an increase in Ca2+ efficiency and a linearization of its Ca2+ dependency after hearing onset. A train of short depolarizations was also able to induce sustained vesicular release, which is indicative of a normal RRP refilling (data not shown).
Table 1.
Ca2+-dependent exocytosis is not affected in Syt7−/− IHCs
| Age | RRP component |
|||
|---|---|---|---|---|
| Genotype | ΔCm (max) (fF) | ΔCm /ICa (fF pA−1) | Ca2+ dependency N power | |
| P2–P3 | Syt7+/+ | 12.5 ± 1.9 (n = 3) | 0.13 ± 0.02 (n = 3) | – |
| Syt7−/− | 11 ± 0.2 (n = 3) | 0.12 ± 0.02 (n = 3) | 2.1 ± 0.2 (n = 2) | |
| P6–P8 | Syt7+/+ | 20 ± 4 (n = 6) | 0.12 ± 0.02 (n = 6) | 2.2 ± 0.2 (n = 4) |
| Syt7−/− | 24.3 ± 6 (n = 8) | 0.17 ± 0.04 (n = 8) | 2.2 ± 0.1 (n = 6) | |
| P15–P30 | Syt7+/+ | 66 ± 11 (n = 6) | 0.28 ± 0.05 (n = 6) | 0.9 ± 0.1 (n = 5) |
| Syt7−/− | 69 ± 11 (n = 5) | 0.37 ± 0.08 (n = 5) | 1.1 ± 0.2 (n = 5) | |
Figure 7.

Comparative Ca2+ thresholds triggering exocytosis in IHCs during postnatal development. A, Comparative IHC synaptic transfer functions (ΔCm vs ICa) plotted on a double log scale, for P1-Otof+/− (open triangle), P1-Otof−/− (black circle) (data from Fig. 2B), P0-Syt1−/− (gray circle; data from Fig. 4D), P7-Otof+/− (green triangle), adult (P15–P30) Syt7+/+(open square) and adult (P16–P30) Syt7−/− (blue square) IHCs. The ICa thresholds necessary to evoke significant ΔCm (>5 fF) were extrapolated by fitting (line) the slope of the initial Cm increase. B, Synaptic vesicular release in adult IHCs has a higher Ca2+ sensitivity (4- to 5-fold) than in P0–P1 and P7 IHCs. This increase in Ca2+ sensitivity in mature IHCs is most likely not due to otoferlin on its own, since P7 IHCs, which still have low Ca2+ sensitivity of exocytosis, already require otoferlin for synaptic vesicular release.
Discussion
The here uncovered switch from an otoferlin-independent to an otoferlin-dependent exocytosis, reveals an as yet unknown maturation step of the IHC synapse that takes place around P4 in the mouse. At this developmental transition, the spontaneous Ca2+-driven spiking activity of IHCs rises to its highest frequency (Marcotti et al., 2003), and is generated both intrinsically and extrinsically through ATP-dependent excitation of IHCs (Tritsch and Bergles, 2010). The rise in IHC spike frequency and resulting increase in glutamate release, are supposed to be important for IHC synaptic maturation (Seal et al., 2008) and for proper maturation of the central auditory pathway (Rubel and Fritzsch, 2002; Leake et al., 2006; Leao et al., 2006). Notably, this period of postnatal cochlear development corresponds to an intense period of synaptic reorganization, refinement and retraction of type I and type II afferent neurites contacting IHCs (Lenoir et al., 1980; Huang et al., 2007). Our results show that before P4, immature IHCs make use of other Ca2+ sensors than otoferlin to drive Ca2+-dependent exocytosis. In that respect, it is noteworthy that a ΔCm response has been recorded in IHCs from hypothyroid rats, in which otoferlin could not be detected (Brandt et al., 2007; Sendin et al., 2007). The otoferlin-independent exocytosis phase reported here may account for this observation. Indeed, it has been shown that the maturation of the auditory neuroepithelium is delayed in thyroid hormone-deficient mice (Sendin et al., 2007).
Before P3, a period during which IHC exocytosis is otoferlin-independent, Syt1, Syt2 or Syt7 were found to be dispensable for vesicle fusion, indicating that other, as yet unknown Ca2+ sensors, are involved in IHCs. Interestingly, we found that in response to repetitive stimulation, IHC exocytosis was seriously affected in Syt1−/− mice, suggesting that Syt1 is involved in the recruitment of synaptic vesicles to the RRP, possibly by ensuring fast Ca2+-dependent vesicle replenishment of the ribbon synapse. In CNS nerve terminals, Syt1 is also known to regulate the rate of endocytosis of synaptic vesicles that have undergone fusion (Poskanzer et al., 2003; Nicholson-Tomishima and Ryan, 2004). The replenishment defect of the RRP observed in Syt1−/− IHCs could therefore result from an impaired clearance of recently exocytosed vesicles from release sites (Hosoi et al., 2009).
The absence of Syt1 immunolabeling in IHCs after the onset of hearing, however, suggests that this Ca2+ sensor is not involved in mature IHCs. Furthermore, post-hearing-onset IHCs from Syt2−/− or Syt7−/− mice display normal exocytosis, indicating that Syt2 and Syt7 are also not essential for exocytosis in mature IHCs. This result is consistent with the absence of Syt2 in IHCs beyond the first postnatal week. Since several Syt isoforms are expressed in immature IHCs, we cannot exclude at present that the lack of a strong phenotype in single Syt-null mice may be due to functional redundancy between Syts, although we note that a redundancy between Syt isoforms has not been shown so far in other synapses. Complexins, that are also known to bind to the SNARE complex and regulate fast exocytosis in conjunction with Syts in most neurosecretory synapses, are also dispensable at the mature IHC ribbon synapse (Strenzke et al., 2009). Together, these results suggest that the mature IHC makes use of an unconventional synaptic machinery, which includes otoferlin, to control exocytosis. The recent report of a temperature-sensitive form of deafness caused by an otoferlin mutation, that manifests a sudden hearing impairment provoked by fever in otherwise normal hearing children, further argues in favor of otoferlin playing a critical role at the synapses of mature IHCs (Marlin et al., 2010).
Surprisingly, exocytosis of P7 immature IHCs, while otoferlin-dependent, still showed a low Ca2+ efficiency compared with mature IHCs (Fig. 7). Indeed, the Ca2+ threshold (minimum ICa required to evoke a release of vesicles >5 fF) was similar in P1 IHCs (otoferlin not required) and P7 IHCs (otoferlin required), while it is much lower in post-hearing onset (P15–P30), mature IHCs (Fig. 7). Likewise, the slopes of the synaptic transfer function were steep for all genotypes at P7 or younger (N ∼2), while they were less steep (N ∼1) in P15–P30 wild-type mice (Fig. 7A). One might hypothesize that as yet undefined otoferlin partners (such as specific SNAREs, or other Syts) are lacking to set a linear, highly Ca2+-sensitive, synaptic function in P6 IHCs. Synaptotagmin 4, present in mature IHCs only, might be one of the otoferlin partners, since it has been shown to be required for the linearization of the synaptic transfer function (Johnson et al., 2010). A different distribution of Ca2+ channels at the synaptic active zones between immature and mature IHCs is also expected to change of the synaptic transfer function. In P6 IHCs, Ca2+ channels are not exclusively located at the active zone (Zampini et al., 2010), and the extrasynaptic location of some Ca2+ channels may contribute to the lower “Ca2+efficiency” of exocytosis in immature IHCs.
Otoferlin is a member of the ferlin protein family, which includes fer-1, dysferlin and myoferlin. When defective, dysferlin and myoferlin are responsible for muscular dystrophies (Bansal and Campbell, 2004). In Caenorhabditis elegans, Fer-1 is required for Ca2+-dependent vesicle fusion in spermatids during spermiogenesis (Achanzar and Ward, 1997; Washington and Ward, 2006). Based on the muscular phenotypes of the dysferlin and myoferlin knock-out mice, a role of these proteins in Ca2+-dependent membrane repair processes and myoblast membrane fusion, respectively, has been proposed (Bansal et al., 2003; Doherty et al., 2005). Otoferlin, whose C2 domains bind to SNARE proteins in a Ca2+-dependent manner (Roux et al., 2006), is critically involved in membrane vesicle fusion in IHCs from ∼P4–P5 onwards. In an in vitro fusion assay, otoferlin accelerates membrane fusion with higher Ca2+ affinity than Syt1 (C. Johnson and E. R. Chapman personal communication). In vestibular hair cells, otoferlin has been shown to be essential for a high affinity Ca2+ sensor function that allows fast exocytosis during brief cell depolarizations and low Ca2+ stimuli (Dulon et al., 2009). The requirement for otoferlin at the mature IHC synapse is likely due to a constraint specific to the auditory ribbon synapse organization, and/or its mode of exocytosis that involves multivesicular release (Glowatzki and Fuchs, 2002). It is possible that the larger number of C2 domains in otoferlin than in Syt1 (six vs two) enables more complex interactions between otoferlin and SNARE proteins, perhaps controlling the supply of vesicles to the release sites, as recently suggested in pachanga mice (Pangrsic et al., 2010) and/or allowing a variety of vesicle fusion modes. In the mature IHC, which does not contain Syt1 or Syt2, the proportion of coordinated multivesicular fusion events at the ribbon synapse has been shown much higher than in Syts-expressing IHCs from pre-hearing animals (Grant et al., 2010). We thus suggest that otoferlin has a synchronizing effect on the multivesicular fusion events, an effect that could be attenuated by the coexistence of Syts, notably Syt1, in immature IHCs.
Footnotes
This work was supported by grants from the European Commission FP6 Integrated Project EuroHear (LSHG-CT-2004-512063), the French National Research Agency (ANR-07-Neuroscience), Louis-Jeantet for Medecine Foundation (C.P.), the Fondation Voir & Entendre, and National Institutes of Health Grant R01-MH06186 (E.R.C.). E.R.C. is an Investigator of the Howard Hughes Medical Institute. N.M. was supported by the EMBO (European Molecular Biology Organization) Long Term Fellowship (ALTF 724-2008). We thank Norma Andrews at the University of Maryland for providing us with the Syt7-null mice, and Roberto Adachi and Thomas C. Südhof for providing us with Syt2-null mice. We also thank Robert Fettiplace for his scientific support and advice, Huisheng Liu for his help on Syt1 and Syt7 projects, Jean-Pierre Hardelin for critical reading of the manuscript, and Lance Rodenkirch and Mireille Montcouquiol for their help with confocal microscopy.
References
- Achanzar WE, Ward S. A nematode gene required for sperm vesicle fusion. J Cell Sci. 1997;110:1073–1081. doi: 10.1242/jcs.110.9.1073. [DOI] [PubMed] [Google Scholar]
- Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C, Dulon D. Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J Neurosci. 2008;28:1798–1803. doi: 10.1523/JNEUROSCI.4653-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001;21:4593–4599. doi: 10.1523/JNEUROSCI.21-13-04593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577–11585. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt N, Kuhn S, Münkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J Neurosci. 2007;27:3174–3186. doi: 10.1523/JNEUROSCI.3965-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon D, Safieddine S, Jones SM, Petit C. Otoferlin is critical for a highly sensitive and linear calcium-dependent exocytosis at vestibular hair cell ribbon synapses. J Neurosci. 2009;29:10474–10487. doi: 10.1523/JNEUROSCI.1009-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Gil-Loyzaga P, Pujol R. Synaptophysin in the developing cochlea. Int J Dev Neurosci. 1988;6:155–160. doi: 10.1016/0736-5748(88)90040-8. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002;5:147–154. doi: 10.1038/nn796. [DOI] [PubMed] [Google Scholar]
- Goutman JD, Glowatzki E. Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci U S A. 2007;104:16341–16346. doi: 10.1073/pnas.0705756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant L, Yi E, Glowatzki E. Two modes of release shape the postsynaptic response at the inner hair cell ribbon synapse. J Neurosci. 2010;30:4210–4220. doi: 10.1523/JNEUROSCI.4439-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Holt M, Sakaba T. Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron. 2009;63:216–229. doi: 10.1016/j.neuron.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Huang LC, Thorne PR, Housley GD, Montgomery JM. Spatiotemporal definition of neurite outgrowth, refinement and retraction in the developing mouse cochlea. Development. 2007;134:2925–2933. doi: 10.1242/dev.001925. [DOI] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563:177–191. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Rüttiger L, Münkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, Knipper M, Marcotti W. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci. 2010;13:45–52. doi: 10.1038/nn.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Leake PA, Snyder RL, Stakhovskaya O, Bonham B. Spontaneous discharge patterns in cochlear spiral ganglion cells before the onset of hearing in cats. J Neurophysiol. 2007;98:1898–1908. doi: 10.1152/jn.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen EC, Hudspeth AJ. Transfer characteristics of the hair cell's afferent synapse. Proc Natl Acad Sci U S A. 2006;103:5537–5542. doi: 10.1073/pnas.0601103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rüsch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- Leake PA, Hradek GT, Chair L, Snyder RL. Neonatal deafness results in degraded topographic specificity of auditory nerve projections to the cochlear nucleus in cats. J Comp Neurol. 2006;497:13–31. doi: 10.1002/cne.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao RN, Sun H, Svahn K, Berntson A, Youssoufian M, Paolini AG, Fyffe RE, Walmsley B. Topographic organization in the auditory brainstem of juvenile mice is disrupted in congenital deafness. J Physiol. 2006;571:563–578. doi: 10.1113/jphysiol.2005.098780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M, Shnerson A, Pujol R. Cochlear receptor development in the rat with emphasis on synaptogenesis. Anat Embryol (Berl) 1980;160:253–262. doi: 10.1007/BF00305106. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM. Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 1999;19:119–132. doi: 10.1523/JNEUROSCI.19-01-00119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflügers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- Lippe WR. Rhythmic spontaneous activity in the developing avian auditory system. J Neurosci. 1994;14:1486–1495. doi: 10.1523/JNEUROSCI.14-03-01486.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Rusch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol. 2003;552:743–761. doi: 10.1113/jphysiol.2003.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlin S, Feldmann D, Nguyen Y, Rouillon I, Loundon N, Jonard L, Bonnet C, Couderc R, Garabedian EN, Petit C, Denoyelle F. Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem Biophys Res Commun. 2010;394:737–742. doi: 10.1016/j.bbrc.2010.03.062. [DOI] [PubMed] [Google Scholar]
- Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin JP, Sato M, Petit C. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007;27:6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson-Tomishima K, Ryan TA. Kinetic efficiency of endocytosis at mammalian CNS synapses requires synaptotagmin I. Proc Natl Acad Sci U S A. 2004;101:16648–16652. doi: 10.1073/pnas.0406968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Müller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;7:869–876. doi: 10.1038/nn.2578. [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Marek KW, Sweeney ST, Davis GW. Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature. 2003;426:559–563. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Rubel EW, Fritzsch B. Auditory system development: primary auditory neurons and their targets. Annu Rev Neurosci. 2002;25:51–101. doi: 10.1146/annurev.neuro.25.112701.142849. [DOI] [PubMed] [Google Scholar]
- Safieddine S, Wenthold RJ. SNARE complex at the ribbon synapses of cochlear hair cells: analysis of synaptic vesicle- and synaptic membrane-associated proteins. Eur J Neurosci. 1999;11:803–812. doi: 10.1046/j.1460-9568.1999.00487.x. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR, Edwards RH. Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron. 2008;57:263–275. doi: 10.1016/j.neuron.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendin G, Bulankina AV, Riedel D, Moser T. Maturation of ribbon synapses in hair cells is driven by thyroid hormone. J Neurosci. 2007;27:3163–3173. doi: 10.1523/JNEUROSCI.3974-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T. Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Synaptotagmins: why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Bergles DE. Developmental regulation of spontaneous activity in the mammalian cochlea. J Neurosci. 2010;30:1539–1550. doi: 10.1523/JNEUROSCI.3875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- von Poser C, Ichtchenko K, Shao X, Rizo J, Südhof TC. The evolutionary pressure to inactivate. A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem. 1997;272:14314–14319. doi: 10.1074/jbc.272.22.14314. [DOI] [PubMed] [Google Scholar]
- Washington NL, Ward S. FER-1 regulates Ca2+-mediated membrane fusion during C. elegans spermatogenesis. J Cell Sci. 2006;119:2552–2562. doi: 10.1242/jcs.02980. [DOI] [PubMed] [Google Scholar]
- Wölfel M, Schneggenburger R. Presynaptic capacitance measurements and Ca2+ uncaging reveal submillisecond exocytosis kinetics and characterize the Ca2+ sensitivity of vesicle pool depletion at a fast CNS synapse. J Neurosci. 2003;23:7059–7068. doi: 10.1523/JNEUROSCI.23-18-07059.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Lawrence ND, Münkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W. Elementary properties of CaV1.3 Ca2+ channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588:187–199. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]