

Abstract
DNA double-strand breaks (DSBs) are the most hazardous lesions arising in the genome of eukaryotic organisms, and yet occur normally during DNA replication, meiosis, and immune system development. The efficient repair of DSBs is crucial in maintaining genomic integrity, cellular viability, and the prevention of tumorigenesis. As a consequence, eukaryotic cells have evolved efficient mechanisms that sense and respond to DSBs and ultimately repair the break. The swiftness of the DNA DSB response has paved to the identification of sensors and transducers which allowed to generate a hierarchical signaling paradigm depicting the transduction of the damage signal to numerous downstream effectors (Fig. 1). The function of such effectors involve posttranslational modifications through phosphorylation, acetylation, and methylation of the substrates. This review will address the control of DSBs in damaged eukaryotic cells, the physiological processes that require the introduction of a DSB into the genome, and the maintenance of DSBs in non-damaged cells.
Keywords: DNA, DSB, signaling, recombination
The DSB is probably the most dangerous of the many types of DNA damage that exist within the cell. They arise from exogenous agents such as ionizing radiation (IR) and certain chemotherapeutic drugs, from endogenously generated reactive oxygen species and from chromosomal stress. A DNA replication fork that encounters DNA single-strand breaks or other types of lesion will also produce DSBs. They can occur at the ends of chromosomes due to defective metabolism of telomeres [Pandita, 2002a,b]. In addition, DNA DSBs can also form in a programmed manner during development. They are generated to initiate recombination between homologous chromosomes during meiosis and occur as intermediates during developmentally regulated rearrangements, such as V(D)J recombination and immunoglobulin class-switch recombination.
The inability to respond properly to, or to repair, DNA DSBs has the potential to lead to genomic instability, which in turn may either lead to cell death or increase the risk of pathological consequences such as cancer development. The repair of DNA DSBs must occur in the context of chromatin, and there is increasing evidence that the modulation of chromatin plays an integral role in the DNA DSB repair process. It is becoming evident that a defect in the signaling and repair of DSBs is instrumental in the development of a number of human cancers. There are also a number of human disorders, which are characterized by defects in proteins that function in the control of DSBs. The study of these disorders has provided enormous insight into the cellular control of DSBs.
DSB DISORDERS
There are a number of human genetic disorders which are characterized by a defective DSB response [O’Driscoll and Jeggo, 2006] (Table I). These disorders also have a number of typical characteristics, such as developmental defects, immunodeficiency, neurological degeneration, and cancer predisposition. The use of cell-lines and mouse models of these DSB disorders has greatly enhanced our understanding of the cellular process that regulates the maintenance and processing of DSBs. Mutations in the evolutionarily conserved Mre11/Nbs1/Rad50 complex results in Ataxia-telangiectasia like disorder (ATLD) and Nijmegen breakage syndrome (NBS), respectively. As yet no human condition has been identified for mutated Rad50. Knockout mouse models of all three genes are embryonic lethal, highlighting the significance of this complex. The major role of this complex is thought to be in the sensing of the DSB, which is discussed in more detail in the next section. Mutations in a group of proteins which belong to the phosphatidylinositol 3-kinase related kinases (PIKKs)—ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia and RAD3 related (ATR), and DNA protein kinase catalytic subunit (DNA-PKcs) result in ataxia-telangiectasia and Seckel syndrome, respectively. The PIKKs are essential in the efficient signal transduction pathways which are activated in response to DSBs. Lastly, there are several syndromes associated with proteins that are involved in the DSB repair process. Defective LIG4 and Artemis result in LIG4 syndrome and severe combined immunodeficiency (SCID) syndrome. Both proteins function in the non-homologous end-joining (NHEJ) pathway. BRCA2 mutations give rise to Fanconi anemia (specifically FANCD1) which results in a defective homologous recombination (HR) pathway. The broad spectrum of clinical features of these DSB disorders clearly highlights the importance in regulating the repair of this type of lesion.
TABLE I.
DSB Disorders
| Syndrome | Gene | Phenotype |
|---|---|---|
| AT | ATM | Immunodeficiency, neurodegeneration, cancer predisposition |
| ATLD | Mre11 | Milder form of AT, no reported cancer phenotype |
| NBS | Nbs1 | Immunodeficiency, developmental defects, cancer predisposition |
| Seckel syndrome | ATR | Developmental defects, no reported cancer phenotype |
| Fanconi anemia | BRCA2 (FANCD1) | Immunodeficiency, developmental defects, cancer predisposition |
| RS-SCID | Artemis | Immunodeficiency, no reported cancer phenotype |
| LIG4 syndrome | LIG4 | Immunodeficiency, developmental defects, cancer predisposition |
SENSING DSBs
Clearly, sensing the lesion is the first essential step in the cellular response to DSBs. Recent studies implicate the Mre11 complex is having an early role in the detection of the DSB. Its role in sensing the break and initial end processing will be discussed below. We will also discuss the growing importance of the modifications in chromatin structure and its potential role as a sensor of DSBs.
Early Response and Processing of DSBs by Mre11 Complex
Significant amount of data suggests that Mre11, Rad50, and Xrs2/Nbs1 (Xrs2 in S. cerevisiae; Nbs1 in mammalian cells) comprise a nuclease complex (also termed the Mre11 complex) with multiple roles in signaling and repair in meiotic and mitotic DSB response networks, as well as telomere maintenance [Grenon et al., 2001]. The complex is involved in early steps of DSB end processing prior to repair by multiple pathways and may maintain sister chromatids or broken ends in close proximity. These proteins bind DNA quickly after DSB induction, and influence the rate of 5′ to 3′ resection of DSB ends, along with Sae2/Com1 [Rattray et al., 2001]. While all evidence indicates that the heteroduplex is required for the initial processing of broken DNA ends, the nature of the processing is unclear. The nuclease activity of Mre11 degrades DNA in the 3′ to 5′ direction, leading one group to hypothesize that the complex recruits an unidentified nuclease of the correct polarity or that the helicase activity of the complex provides a substrate for the endonuclease activity of Mre11 [Nairz and Klein, 1997; Usui et al., 1998; Moreau et al., 1999]. Exo1 can partially compensate for loss of Mre11 activity in mitotic cells but not meiotic cells suggesting that Mre11 acts differently in the context of Spo11-mediated DSBs [Moreau et al., 1999, 2001]. Initial studies showed a requirement for the Mre11 complex after irradiation that produces multiple types of DNA ends, raising the possibility that processing is likely limited to an initial “cleaning” or removal of the ends rather than extensive nuclease activity. In a related way, the proposed activity of Mre11 to assist in removal of Spo11 from DNA after cleavage during meiosis may be analogous to removal of a bulky adduct from a DNA end in somatic cells.
The crystal structure of the Mre11 complex revealed strong similarities to the Smc proteins that tether replicated homologs together until appropriate anaphase signaling and separation. When complexed as a trimer, the N and C termini of Rad50 associate together separated by a long flexible arm with a zinc hook at the tip. Mre11 and Xrs2 sit together with the N and C termini [Hopfner et al., 2001, 2002; de Jager et al., 2001]. This structure along with initial genetic evidence with MRE11S and RAD50S yeast mutants [Nairz and Klein, 1997] led to the suggestion that heterotrimers associate at the zinc hooks during S phase to tether two chromatids together. Electron microscopy studies in mammalian cells suggested that multiple complexes may associate to hold together the two ends of a broken DNA duplex [de Jager et al., 2001; Wyman and Kanaar, 2002].
Although null mutants of each of the three proteins lead to embryonic lethality in mice, several hypomorphic strains have been engineered and are viable [Bender et al., 2002; Williams et al., 2002]. As expected, these mice exhibit meiotic defects, as well as radiation sensitivity and predisposition to cancer similar to the cognate human syndromes. Interestingly the Rad50S mutant mice display a distinct hematopoietic cell defect and bone marrow failure not observed in mutants of the other complex proteins [Bender et al., 2002]. The reason for the defect remains unclear; possibilities include a specific role for Rad50 in tissue-specific stem cell proliferation or differentiation, or, alternatively, a role in anti-apoptotic signaling. Since hematopoietic cells are particularly susceptible to apoptosis after DNA damage, partial loss of Rad50 function may be sufficient to demonstrate this phenotype in somatic cells. Taken together with elevated levels of apoptosis in mammalian spo11−/− spermatocytes and oocytes, ATM−/− human neural cells, and MEI304 Drosophila mutants, unrepaired DSBs in both meiotic and mitotic systems seem sufficient to lead to apoptosis [Pandita et al., 1999; Scherthan et al., 2000].
Histone H2AX
One of the immediate targets of the ataxia-telangiectasia mutated (ATM) kinase in response to DNA damage is the histone H2A variant H2AX [Redon et al., 2002]. Histone H2AX, the major isoform in yeast and a minor H2A species in mammals, is phosphorylated at the carboxy-terminal serine 139 immediately following Spo11-induced DSB formation [Hunter et al., 2001; Mahadevaiah et al., 2001]. Similarly phosphorylation occurs in somatic cells in response to damage-induced DSBs [Rogakou et al., 1998], and in response to Rag-mediated cleavage [Chen et al., 2000]. Phosphorylated H2AX (termed γ-H2AX) appears within minutes of damage over large chromatin regions extending tens of kb in yeast and up to 2 Mb in mammalian cells [Rogakou et al., 1999]. The analysis of H2AX-deficient mice has demonstrated a role for H2AX in a variety of responses to DSBs, including DNA repair, checkpoint signaling, and immunoglobulin gene class switching [Petersen et al., 2001; Bassing et al., 2002; Celeste et al., 2002; Fernandez-Capetillo et al., 2003; Reina-San-Martin et al., 2003]. H2AX−/− mice exhibit male-specific sterility, which is likely due to defects in chromatin remodeling during meiosis [Fernandez-Capetillo et al., 2003]. γ-H2AX immunostaining of mouse spermatocytes is detected in leptotene and zygotene, and co-localizes with Rad51-Dmc1 foci. By pachytene, staining is limited to unsynapsed regions such as non-homologous arms of the sex chromosomes. By contrast, staining is nearly absent at all times in SPO11−/− mutants that do not form DSBs. It is tantalizing to suggest that coating of large chromatin regions by γ-H2AX may act as a structural signal to recruit recombination and repair proteins, as well as structural proteins involved in chromatid pairing and SC formation in pachytene. The existing literature argues a strong correlation between defective DSB repair, genomic instability, and telomere dysfunction [Sharma et al., 2003a,b,c]. The most recent studies suggest that the repair of DNA DSBs must occur in the context of chromatin, and there is increasing evidence that the modulation of chromatin plays an integral role in the DNA DSB repair process (Fig. 2). Several chromatin-modifying factors have been identified that play a role in DNA damage response [Pandita, 2003; Sharma et al., 2003c; Kusch et al., 2004; Gupta et al., 2005].
Fig. 2.

Recruitment of factors involved in the repair of DNA DSBs. Multiple proteins with different activity for posttranslational modifications of histones present at the DNA DSB allow to open the chromatin in order to make the DNA accessible to DNA repair machinery. Some of the histone modifications are necessary to remodel the nucleosomes during the repair process.
hMOF Influences ATM Activation
hMOF, the human ortholog of the Drosophila MOF gene (males absent on the first), encoding a protein with histone acetyltransferase (HAT) activity, has been shown to interact with the ATM [Gupta et al., 2005]. In response to DSBs, hMOF-dependent acetylation of its target substrate, lysine 16 (K16) of histone H4 is enhanced, independent of ATM function. Blocking the IR-induced increase in acetylation of histone H4 at K16, either by the expression of a dominant negative mutant hMOF or by RNA interference-mediated hMOF knockdown, resulted in decreased ATM autophosphorylation, ATM kinase activity, and the phosphorylation of downstream effectors of ATM and DNA repair while increasing cell killing (Fig. 3). In addition, decreased hMOF activity was associated with loss of the cell-cycle checkpoint response to DSBs. These results suggest that hMOF functions upstream of ATM and its modification of histone H4 at lysine 16 may play some role in sensing the DSB.
Fig. 3.
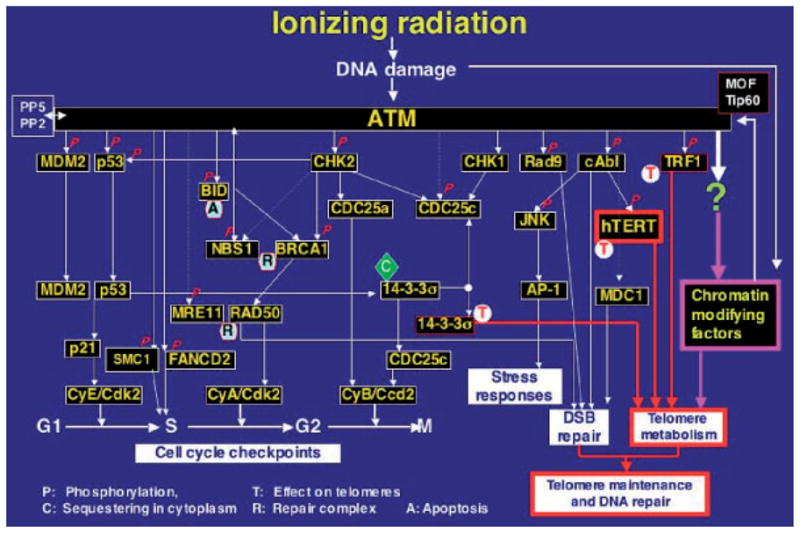
Regulatory role of ATM in cellular responses to DNA DSB repair. ATM kinase activity increases immediately after double-strand breaks (DSBs) occur in DNA following exposure to IR. ATM mediates the early stages of the rapid induction of several signaling pathways, which include activation of the DNA-DSB pathway, regulation of the cell-cycle checkpoint controls, activation of stress responses, and maintenance of telomeres. ‘P’ with solid arrows indicates reported phosphorylation events; dashed arrows represent possible signaling steps and do not imply direct interaction between proteins; ‘C’ indicates sequestering in cytoplasm; ‘R’ indicates repair complexes; and ‘T’ indicates a role for the protein in telomere metabolism. AP-1, apetala1 transcription factor; BRCA1, breast cancer susceptibility gene product 1; c-Abl, abelson protein tyrosine kinase; CDK, cyclin-dependent kinase; CHK, checkpoint kinase; FANCD2, fanconi anemia protein; JNK, Jun N-terminal protein kinase; MRE11, meiotic recombination 11 gene product; MDM2, mouse double minute 2 (p53-binding protein); NBS1, Nijmegen breakage syndrome 1 protein (p95); SMC1, structural maintenance of chromosome 1; RAD50, a radiation-damage-repair-associated protein; TRF1, telomere-repeat-finding factor 1; hTERT, human catalytic unit of telomerase; hMOF, the human ortholog of the Drosophila MOF gene (males absent on the first).
TIP60 as Sensor of DSBs
TIP60, a HAT has been shown to acetylate core histones H2A, H3, and H4 [Yamamoto and Horikoshi, 1997; Kimura and Horikoshi, 1998]. TIP60 appears to play an important role in DNA damage repair, as cells expressing catalytically inactive TIP60 accumulate DSBs [Ikura et al., 2000]. Sun et al. [2005] showed that TIP60 forms a distinct stable complex with ATM and in response to damage activates ATM by direct acetylation. The catalytic activity of TIP60 is stimulated in response to DNA damage, but does not appear to be regulated by ATM. As is the case with hMOF, it has been suggested that TIP60 functions upstream of ATM, possibly sensing DNA damage-caused chromatin changes and signaling them to ATM.
In addition to the above-mentioned chromatin modifying factors, there are several other chromatin-modifying factors with chromodomains, for example, isoforms of heterochromatin protein 1 (HP1) which have been linked with the repair of IR-induced DNA damage [Sharma et al., 2003c]. The isoforms of HP1 have potential sites that can be phosphorylated by ATM. It remains to be determined whether isoforms of HP1 interact with ATM or any other DNA damage response elements.
DSB SIGNALING AND CHECKPOINT ACTIVATION
In response to DNA DSBs a complex network of cell-cycle checkpoint proteins is activated, resulting in cell-cycle arrest in all three DNA damage cell-cycle checkpoints (G1-S, intra-S, and G2-M). During this time the DSB is repaired by either HR or NHEJ. The signaling molecules that orchestrate the DNA damage cell-cycle checkpoints are the PIKKs class of protein kinases; ATM, ATR, and DNA-PK. The survival of cells with impaired ATM, ATR function after DNA damage is compromised and they are defective in initiating DNA damage-induced cell-cycle arrest.
ATM
The ATM protein kinase is primarily activated in response to DNA DSBs caused by IR or radiomimetic drugs (Fig. 3). ATM is observed at the sites of DNA damage, where it is autophosphorylated and is dissociated from its non-active dimeric form to the active monomeric form [Bakkenist and Kastan, 2003]. The ATM protein appears to be a part of the sensory machinery that detects DSBs during meiosis or mitosis, or breaks consequent to the damage by free radicals. After the recruitment to the sites of DNA damage, ATM phosphorylates a number of substrates, including Chk1 and Chk2, which in turn target other proteins to induce cell-cycle arrest and facilitate DNA repair (Fig. 3). Cells deficient in ATM have been shown to have a high frequency of spontaneous chromosomal aberrations, high rates of intra-chromosomal recombination, and error-prone recombination [Pandita, 2002a, 2003]. Such cells have a higher initial and residual chromosomal aberrations in G1 and G2 phases after IR exposure as determined by premature chromosome condensation technology [Pandita and Hittelman, 1992a,b; Morgan et al., 1997]. Cells defective in ATM function also display higher frequency of chromosomal aberrations after IR exposure as determined by karyotypic examination of metaphase chromosome spreads [Morgan et al., 1997]. Mice mutated in the homolog of the Atm gene (ATM) display similar pleiotropic defects [Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996]. Atm-deficient mice show aberrant synapsis with unpaired axial cores, non-homologous synapsis, and fragmented synaptonemal complexes [Pandita et al., 1999; Scherthan et al., 2000]. Consistent with a role for Atm in meiosis, individuals with ataxia telangiectasia (A-T) display gonadal atrophy and spermatogenetic failure, a phenotype which is mirrored in Atm-deficient mice [Pandita, 2003].
The ATM protein belongs to a growing family of phosphatidylinositol-3 kinase-related kinases and seems to play a role as an intrinsic part of the cell-cycle machinery that surveys genomic integrity, cell-cycle progression, and processing of DNA damage [Pandita, 2002a, 2003; Kurz and Lees-Miller, 2004]. It shows similarity to several yeast and mammalian proteins involved in meiotic recombination and cell-cycle progression, namely, the products of MEC1 in the budding yeast S. cerevisiae and RAD3 of the fission yeast S. pombe [Bentley et al., 1996; Lydall et al., 1996], and the Tor proteins of yeasts and mammals [Keith and Schreiber, 1995; Savitsky et al., 1995]. The Atm protein and its relative Atr (Atm and Rad3 related) have been regarded as important components in the machinery monitoring progression of meiotic recombination, DSB repair, and homolog pairing [Moens et al., 1999], which is in agreement with the location of murine Atm throughout meiotic chromatin [Flaggs et al., 1997; Barlow et al., 1998; Schertham et al., 2000]. Detection and signaling of DNA damage are possibly mediated through downstream targets of Atm like p53, Mdm2, Brca1, Chk2, and Smc1 proteins (Fig. 3) [Pandita, 2003]. Furthermore, the yeast Atr ortholog Mec1, is known to exert checkpoint function in the mitotic and meiotic cell cycle, and its absence mediates a defect in synapsis [Lydall et al., 1996; Grushcow et al., 1999]. Mec1 is required for phosphorylation of Rpa as a response to IR-induced DNA damage [Brush et al., 1996] and in turn Rpa has been shown to interact with Rad51, which plays an important role in meiotic recombination [Shinohara et al., 1992, 1997] and localizes to meiotic recombination complexes [Terasawa et al., 1995; Anderson et al., 1997; Tarsounas et al., 1999].
DNA damage checkpoint pathways in S. cerevisiae are governed by the Atm homolog Tel1 and the Mre11 complex [Usui et al., 2001]. In mitotic cells, the Tel1–Mre11 complex pathway triggers Rad53 activation and its interaction with Rad9, whereas in meiosis it acts via Rad9 and the Rad53 paralog Mre4/Mek1. Activation of the Tel1–Mre11 complex pathway checkpoint functions appears to depend upon the Mre11 complex as a damage sensor and, at least in meiotic cells, to depend on unprocessed DNA DSBs. The DSB repair functions of the Mre11 complex are enhanced by the pathway, suggesting that the complex both initiates and is regulated by the Tel1-dependent DSB signal. These findings suggest Mre11 complex has a role in the meiotic recombination as well [Usui et al., 2001].
As a protein kinase, ATM directly phosphorylates p53 and interacts with many other molecules involved in homologous and non-homologous DSB repair, as well as in cell signaling. Some of these molecular targets include Atr, c-Abl, Chk-1, Chk-2, Rpa, Brca1, Brca2, NF-κB/IκB alpha, beta-adaptin, and autophosphorylation of ATM itself. Thus, ATM is a “hierarchical kinase,” (Figs. 1, 3) capable of initiating many pathways simultaneously [Pandita, 2003].
Fig. 1.

Major regulatory steps in the process of DNA DSBs repair. DNA damage repair is accompanied by various cellular functions, which involve the recognition of damage, modification of chromatin at the site of DNA damage, recruitment of repair factors accompanied with the processes involved in cell-cycle check points. Several proteins have been reported to have multiple functions that are involved in the regulation of the DNA DSB repair, whether the damage requires be repaired by NHEJ or HR.
ATR
The ATR (ATM and Rad3 related) protein kinase primarily responds to replication stress and other forms of DNA damage, such as UV. ATR is recruited to the sites of DNA damage by ATR-interacting protein (ATR-IP) to RPA-coated single-stranded DNA (ssDNA) that accumulates at stalled DNA replication forks or is generated by the processing of the initial DNA damage. Once at the break, like ATM, ATR phosphorylates Chk1, and Chk2 thus inducing cell-cycle arrest.
It was previously thought that although both ATM and ATR had overlapping but distinct roles in the response to DNA damage, new studies suggest that in fact they are working in a coordinated manner. Trenz et al. [2006] demonstrate that both ATM and ATR promote Mre11-dependent restart of collapsed replication forks and prevent accumulation of DNA DSBs. A study by Myers and Cortez [2006], shows that ATR is activated rapidly by IR and both ATM and Mre11 enhance ATR signaling. They postulate that ATM and Mre11 may stimulate the ATR signaling pathway by converting DNA damage generated by IR into structures that recruit and activate ATR.
DNA-PK
Components of the DSB repair DNA-dependent protein kinase DNA-PK complex, including Ku70, Ku86, and DNA-PK catalytic subunit (DNA-PKcs), are found in the radiosensitive spermatogonia [Hamer et al., 2003]. Although p53 induction is unaffected, spermatogonial apoptosis occurs faster in the irradiated DNA-PKcs-deficient SCID testis. These results suggest that spermatogonial DNA-PK functions in DNA damage repair rather than p53 accumulation. Despite the fact that early spermatocytes lack the Ku proteins, spontaneous apoptosis of these cells occurs in the SCID testis. The majority of these apoptotic spermatocytes are found at stage IV of the seminiferous epithelium where a meiotic checkpoint has been suggested to exist. DSBs are less accurately repaired in SCID spermatocytes that then fail to pass the meiotic checkpoint. Thus the role for DNA-PKcs during the meiotic prophase differs from that in mitotic cells since it is not influenced by IR and is independent of the Ku heterodimer.
REPAIR OF DSBs
Once the cell has sensed the DSB and the appropriate signaling to the cell-cycle checkpoints achieved, the DNA repair machinery is then recruited to the break. The two major pathways for the repair of DSBs are homologous recombination (HR) and NHEJ. The precise mechanisms the cell used to decide which repair pathway to use is an important question that remains to be answered.
Homologous Recombination
Homologous recombination rejoins DSBs using a sister homolog as a template. As a consequence, this method of repair provides very high fidelity. An early step in HR involves the generation of a single-stranded region of DNA, followed by invasion of the template strand, which creates a Holliday junction. DNA synthesis using the sister strand as a template is followed by branch migration and subsequent resolution of the heteroduplex [West, 2003]. RAD51, a central player in HR, is loaded onto ssDNA and promotes strand invasion, with BRCA2 having a role in delivering RAD51 to the DNA [Pellegrini et al., 2002]. BRCA1 is also required for HR, possibly in a regulatory capacity. Other proteins that are involved in HR are RAD52, XRCC2, and XRCC3. Recently, hRad9 was found to influence HR as the cells with hRad9 knockdown had about three- to fourfold reduced levels DNA DSB HR repair [Pandita et al., 2006].
HR has a key role in rejoining DSBs that arise as a result of replication-fork stalling. Most proteins that are required for HR are essential, probably because stalling of replication forks occurs during most cycles of replication. Cells that lack HR are only mildly sensitive to ionizing radiation sensitive, but are highly sensitive to DNA-crosslinking agents [Thompson and Schild, 2001]. This phenotype is consistent with the idea that the primary function of HR is to repair DSBs at the replication fork, whereas NHEJ primarily repairs DSBs that have been generated elsewhere in the DNA.
Non-Homologous End-Joining
DNA NHEJ is a predominant pathway of DNA double-strand break repair in mammalian cells. The proteins that are required for NHEJ include the heterodimer of Ku70 and Ku80, and the catalytic subunit of the DNA-dependent protein kinase, DNA-PKcs. Heterodimers of Ku bind to the ends of DNA DSBs, which in turn recruit DNA-PKcs to the site of damage. Activated DNA–PKcs complex recruits artemis, XRCC4, LIG4, and DNA polymerase μ. Artemis has endonuclease activity necessary to provide the appropriate DNA ends for ligation and gap filling by LIG4 and DNA polymerase μ. Ahnesorg et al. [2006] have recently identified a previously uncharacterized XRCC4-like factor (XLF, also named Cernunnos), that has weak sequence homology with XRCC4 and is predicted to display structural similarity to XRCC4. They show that XLF directly interacts with the XRCC4-Ligase IV complex and that downregulation of XLF in human cell lines leads to radiosensitivity and impaired NHEJ.
NHEJ functions throughout the cell cycle and is the predominant DSB-rejoining mechanism in G1 phase. NHEJ also rejoins DSBs that are introduced during V(D)J recombination, which is discussed in more detail below. Consequently, viable mice that have defective NHEJ pathway function suffer from SCID. KU and DNA-PKcs also have roles in telomere-length maintenance. Premature senescence is seen in KU-deficient mice, whereas DNA-PKcs-deficient mice have progressively shortened telomeres over successive generations. The roles of DNA DSB repair proteins at the telomeres are distinct from the roles in NHEJ.
REGULATION OF DSBs IN NON-DAMAGED CELLS
In addition to the random introduction of DSBs that occur as a result of cellular exposure to DNA damaging agents, DNA DSBs can also form in a programmed manner during development. They are generated to initiate recombination between homologous chromosomes during meiosis [Richardson et al., 2004] and occur as intermediates during developmentally regulated rearrangements, such as V(D)J recombination and immunoglobulin class-switch recombination.
Initiation of Meiotic Recombination by DSBs
At the beginning of meiosis, each chromosome must recognize its homolog, then the two become intimately aligned along their entire lengths, which allows the exchange of DNA strands between homologous sequences to generate genetic diversity. DNA double-strand breaks initiate meiotic recombination in a variety of organisms. Numerous studies have identified both the genomic loci of the initiating DSBs and the proteins involved in their formation. Meiotic recombination initiates with DSBs formed by Spo11, a topoisomerase II-like protein (Spo11 in S. cerevisiae and vertebrates, Rec12 in S. pombe, Mei-W68 in Drosophila) [Keeney et al., 1997, 1999; McKim and Hayashi-Hagihara, 1998; McKim et al., 1998; Romanienko and Camerini-Otero, 1999; Lichten, 2001]. DSB formation also requires the products of at least nine other genes that act by stabilization or recruitment mechanisms. These include meiotic-specific Mei4 [Menees et al., 1992], Mer2 [Rockmill et al., 1995], Rec102, Rec104, Rec114 (Rec7 in S. pombe) [Molnar et al., 2001a,b], Rec103 (Ski8; Rec14 in S. pombe) [Gardiner et al., 1997] as well as ubiquitously expressed Mre11, Rad50, and Xrs2 (Nbs1 in mammals) genes [Ivanov et al., 1992; Ajimura et al., 1993; Johzuka and Ogawa, 1995]. The additional accessory factor Sae2/Com1 is required for DSB processing.
It is well established that DSBs are potent inducers of HR in both meiotic and somatic cell systems [Jasin, 1996; Keeney et al., 1997; Liang et al., 1998; Richardson et al., 2000]. The localized Spo11-mediated DSBs appear before the formation of joint molecules, and their frequency correlates with the frequency of gene conversion and crossing-over. Meiotic DSBs also initiate DSB response checkpoints that ensure the completion of recombination before the exit from pachytene.
As expected, the lack of DSBs in yeast SPO11−/− mutants blocks recombination initiation, synapsis, and sporulation. Similarly, although viable, SPO11−/− mice are infertile and display multiple meiotic pairing, synapsis, and recombination defects [Baudat et al., 2000; Romanienko and Camerini-Otero, 2000]. Mammalian SPO11−/− spermatocytes and oocytes undergo elevated levels of apoptosis. Consistent with the interdependence of meiotic recombination and synapsis, DSBs may serve as the initial regulatory or structural signal required for progression through meiosis without which apoptosis occurs [Hunter et al., 2001]. Conversely, it is possible that the lack of DSBs in SPO11−/− mice induces cell cycle arrest and produces a pro-apoptotic signal [Baudat et al., 2000]. Spo11 could also serve a structural role secondary to DSB formation [Romanienko and Camerini-Otero, 2000] leading to the prediction that separation of functional mutants will demonstrate independent catalytic and structural roles as well as an understanding of apoptotic signaling in the absence of Spo11 function. Although highest in testis and ovary, the expression of mammalian Spo11 and alternative transcripts are also detected in several somatic tissues including lymphocytes [Baudat et al., 2000; Romanienko and Camerini-Otero, 2000] suggesting a possible role of these gene products in other DSB-mediated developmental programs such as somatic hypermutation or class switching [Tokuyama and Tokuyama, 2000, 2001]. However, analysis of SPO11−/− mice to date has not revealed any role of Spo11 outside of meiosis [Klein et al., 2002].
Meiotic DSB Response and Recombination: The Rad52 Epistasis Group
The Rad52 epistasis group members; Rad50, Rad51, Rad52, Rad54, Rdh54/Tid1 (Rad54B), Rad55, Rad57, (Xrcc2, Xrcc3), Mre11, and Xrs2 (Nbs1) are central to both HR in meiotic cells and DSB repair by homology-directed mechanisms in mitotic cells (Fig. 1). Consistent with evolutionary conservation of proper chromosome segregation and genetic variation during sexual reproduction, homologs of many of these genes have been identified in multiple species based on sequence similarity, and studies have determined which are functional homologs or related protein family members. Species differences in function have further defined their biochemical function. In mammals, many of the Rad52 epistasis group homologs are expressed in multiple tissue types, but with highest levels of expression seen in the testis and proliferating cells, indicating their involvement in both meiotic and mitotic recombination. The protein–protein interactions among various members of the Rad52 epistasis group suggest that two different complexes are involved in DSB-induced recombination; the first is involved in presynaptic functions including the processing of the DSB ends, and the second involved is in synaptic functions for invasion, creation of repair intermediates, and resolution [Petukhova et al., 2003]. This second complex has been termed a recombinosome but has not yet been isolated.
V(D)J Recombination
During early B and T cell development, the exons that encode immunoglobulin (Ig) and T cell receptor (TCR) variable regions are assembled from germline variable (V), diversity (D), and joining (J) segments via V(D)J recombination. The reaction is initiated when the recombinase-activating gene-1 and -2 protein complex (RAG) introduces DNA DSBs at specific sites within the border of two gene segments and their flanking recombination signals. DNA cleavage only occurs after the two recombination signals are brought together in a synaptic complex and leads to the formation of a pair of blunt phosphorylated signal ends and a pair of hairpin-sealed coding ends. The signal and coding end pairs are processed by NHEJ to generate a signal join and coding join, respectively. A recent study by Bredemeyer et al. [2006] have elegantly demonstrated that ATM function directly in the repair of chromosomal DNA DSBs by maintaining DNA ends in their repair complexes generated during lymphocyte antigen receptor gene assembly. This study explains the increase in lymphoid tumors with translocations involving antigen receptor loci associated with ataxia-telangiectasia.
FUTURE STUDIES AND DIRECTIONS
Considerable advances have been made in recent years in elucidating the mechanisms and pathways by which cells regulate the repair of DSBs. However, there are several key issues, which are the subject of intense research in the field at the moment. The first of these is the mechanism by which cells initially detect the presence of DNA DSBs. It is becoming increasingly evident that the higher order nuclear organization of chromatin (Fig. 2) plays a key role in the cells ability to sense the presence of DSBs. The identification of new chromatin associated proteins and elucidation of new roles for known chromatin associated proteins will greatly enhance our understanding of DNA DSB repair processes. The second issue relates to the mechanism by which cells regulate which DNA repair process, either HR or NHEJ, is activated at DSBs. It is clear that during certain phases (G1) of the cell cycle that NHEJ is the predominant mode of repair. However, during late S and G2 both HR and NHEJ can equally contribute to DSB repair. In this case, how does the cell decide which repair system will utilize to repair the DSBs. It is interesting to speculate that the nature of the broken ends of the DNA and their initial end processing may ultimately determine whether the DSB is repaired by HR or NHEJ. The last issue concerns the global regulation of DSBs. We currently have a reasonably good understanding of how cells activate the DSB response but little is known about how cells downregulate the DSB response once the damage is repaired. It is becoming increasingly evident that just as proteins are activated by phosphorylation in response the DSBs, they are also subjected to other modifications, such as dephosphorylation and ubiquitination, to return them to a ready but inactive state. Greater insight into the cellular control of DNA double-stand breaks will be important not only for increased understanding of cellular responses to stress and cancer in general, but also for the development of new therapeutic treatments for individuals with dysfunctional DSB repair pathways.
Acknowledgments
Grant sponsor: NIH; Grant numbers: NS34736, CA98666; Grant sponsor: Department of Army, USA; Grant sponsor: Department of Radiation Oncology, Washington University School of Medicine, St. Louis.
References
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Ajimura M, Leem SH, Ogawa H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 1993;133:51–66. doi: 10.1093/genetics/133.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LK, Offenberg HH, Verkuijlen WM, Heyting C. RecA-like proteins are components of early meiotic nodules in lily. Proc Natl Acad Sci USA. 1997;94:6868–6873. doi: 10.1073/pnas.94.13.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, Wynshaw-Boris A. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development. 1998;125:4007–4017. doi: 10.1242/dev.125.20.4007. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell. 2000;6:989–998. doi: 10.1016/s1097-2765(00)00098-8. [DOI] [PubMed] [Google Scholar]
- Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, Mirzoeva OK, Oltz EM, Petrini JH. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002;16:2237–2251. doi: 10.1101/gad.1007902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006 doi: 10.1038/nature04866. (on line) [DOI] [PubMed] [Google Scholar]
- Brush GS, Morrow DM, Hieter P, Kelly TJ. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc Natl Acad Sci USA. 1996;93:15075–15080. doi: 10.1073/pnas.93.26.15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, Nussenzweig A. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Flaggs G, Plug AW, Dunks KM, Mundt KE, Ford JC, Quiggle MR, Taylor EM, Westphal CH, Ashley T, Hoekstra MF, Carr AM. Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr Biol. 1997;7:977–986. doi: 10.1016/s0960-9822(06)00417-9. [DOI] [PubMed] [Google Scholar]
- Gardiner JM, Bullard SA, Chrome C, Malone RE. Molecular and genetic analysis of REC103, an early meiotic recombination gene in yeast. Genetics. 1997;146:1265–1274. doi: 10.1093/genetics/146.4.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenon M, Gilbert C, Lowndes NF. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat Cell Biol. 2001;3:844–847. doi: 10.1038/ncb0901-844. [DOI] [PubMed] [Google Scholar]
- Grushcow JM, Holzen TM, Park KJ, Weinert T, Lichten M, Bishop DK. Saccharomyces cerevisiae checkpoint genes MEC1, RAD17 and RAD24 are required for normal meiotic recombination partner choice. Genetics. 1999;153:607–620. doi: 10.1093/genetics/153.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Sharma GG, Young CSH, Agarwal M, Smith ER, Paull TT, Lucchesi JC, Khanna KK, Ludwig T, Pandita TK. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25:5292–5305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer G, Roepers-Gajadien HL, van Duyn-Goedhart A, Gademan IS, Kal HB, van Buul PP, de Rooij DG. DNA double-strand breaks and gamma-H2AX signaling in the testis. Biol Reprod. 2003;68:628–634. doi: 10.1095/biolreprod.102.008672. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, Carney JP, Petrini JH, Tainer JA. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Hunter N, Boerner V, Lichten M, Kleckner N. Recombinational DNA double strand breaks in mice precede synapsis. Nat Genet. 2001;27:236–238. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Ivanov EL, Korolev VG, Fabre F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics. 1992;132:651–664. doi: 10.1093/genetics/132.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Johzuka K, Ogawa H. Interaction of Mre11 and Rad50: Two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics. 1995;139:1521–1532. doi: 10.1093/genetics/139.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- Keeney S, Baudat F, Angeles M, Zhou ZH, Copeland NG, Jenkins NA, Manova K, Jasin M. A mouse homolog of the Saccharomyces cerevisiae meiotic recombination DNA transesterase Spo11p. Genomics. 1999;61:170–182. doi: 10.1006/geno.1999.5956. [DOI] [PubMed] [Google Scholar]
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998;3:789–800. doi: 10.1046/j.1365-2443.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- Klein U, Esposito G, Baudat F, Keeney S, Jasin M. Mice deficient for the type II topoisomerase-like DNA transesterase Spo11 show normal immunoglobulin somatic hypermutation and class switching. Eur J Immunol. 2002;32:316–321. doi: 10.1002/1521-4141(200202)32:2<316::AID-IMMU316>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichten M. Meiotic recombination: Breaking the genome to save it. Curr Biol. 2001;11:R253–R256. doi: 10.1016/s0960-9822(01)00131-2. [DOI] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- McKim KS, Hayashi-Hagihara A. mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: Evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev. 1998;12:2932–2942. doi: 10.1101/gad.12.18.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim KS, Green-Marroquin BL, Sekelsky JJ, Chin G, Steinberg C, Khodosh R, Hawley RS. Meiotic synapsis in the absence of recombination. Science. 1998;279:876–878. doi: 10.1126/science.279.5352.876. [DOI] [PubMed] [Google Scholar]
- Menees TM, Ross-MacDonald PB, Roeder GS. MEI4, a meiosis-specific yeast gene required for chromosome synapsis. Mol Cell Biol. 1992;12:1340–1351. doi: 10.1128/mcb.12.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens PB, Tarsounas M, Morita T, Habu T, Rottinghaus ST, Freire R, Jackson SP, Barlow C, Wynshaw-Boris A. The association of ATR protein with mouse meiotic chromosome cores. Chromosoma. 1999;108:95–102. doi: 10.1007/s004120050356. [DOI] [PubMed] [Google Scholar]
- Molnar M, Bahler J, Kohli J, Hiraoka Y. Live observation of fission yeast meiosis in recombination-deficient mutants: A study on achiasmate chromosome segregation. J Cell Sci. 2001a;114:2843–2853. doi: 10.1242/jcs.114.15.2843. [DOI] [PubMed] [Google Scholar]
- Molnar M, Parisi S, Kakihara Y, Nojima H, Yamamoto A, Hiraoka Y, Bozsik A, Sipiczki M, Kohli J. Characterization of rec7, an early meiotic recombination gene in Schizosaccharomyces pombe. Genetics. 2001b;157:519–532. doi: 10.1093/genetics/157.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Morgan EA, Symington LS. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics. 2001;159:1423–1433. doi: 10.1093/genetics/159.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SE, Lovly C, Pandita TK, Shiloh Y, Kastan MB. Fragments of ATM which have dominant-negative or complementing activity. Mol Cell Biol. 1997;17:2020–2029. doi: 10.1128/mcb.17.4.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz K, Klein F. mre11S—a yeast mutation that blocks double-strand-break processing and permits non-homologous synapsis in meiosis. Genes Dev. 1997;11:2272–2290. doi: 10.1101/gad.11.17.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M, Jeggo PA. The role of double-strand break repair-insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Pandita TK. ATM function and telomere stability. Oncogene. 2002a;21:611–618. doi: 10.1038/sj.onc.1205060. [DOI] [PubMed] [Google Scholar]
- Pandita TK. Telomeres and telomerase. Encyclopedia of Cancer. 2002b;4:355–362. [Google Scholar]
- Pandita TK. A multifaceted role for ATM in genome maintenance. Exp Rev Mol Med. 2003;5:1–21. doi: 10.1017/S1462399403006318. [DOI] [PubMed] [Google Scholar]
- Pandita TK, Hittelman WN. The contribution of DNA and chromosome repair deficiencies to the radio-sensitivity of ataxia-telangiectasia. Radiat Res. 1992a;131:214–223. [PubMed] [Google Scholar]
- Pandita TK, Hittelman WN. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat Res. 1992b;130:94–103. [PubMed] [Google Scholar]
- Pandita TK, Westphal CH, Anger M, Sawant SG, Geard CR, Pandita RK, Scherthan H. Atm inactivation results in aberrant telomere clustering during meiotic prophase. Mol Cell Biol. 1999;19:5096–5105. doi: 10.1128/mcb.19.7.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, Gupta A, Wellinger RJ, Zhang J, Powell SN, Roti Roti JL, Lieberman HB, Pandita TK. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–1864. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova GV, Romanienko PJ, Camerini-Otero RD. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell. 2003;5:927–936. doi: 10.1016/s1534-5807(03)00369-1. [DOI] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: A role for SAE2/COM1. Genetics. 2001;158:109–122. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Horikoshi N, Pandita TK. The role of the DNA double-strand break response network in meiosis. DNA Repair (Amst) 2004;3:1149–1164. doi: 10.1016/j.dnarep.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Rockmill B, Engebrecht JA, Scherthan H, Loidl J, Roeder GS. The yeast MER2 gene is required for chromosome synapsis and the initiation of meiotic recombination. Genetics. 1995;141:49–59. doi: 10.1093/genetics/141.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanienko PJ, Camerini-Otero RD. Cloning, characterization, and localization of mouse and human SPO11. Genomics. 1999;61:156–169. doi: 10.1006/geno.1999.5955. [DOI] [PubMed] [Google Scholar]
- Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell. 2000;6:975–987. doi: 10.1016/s1097-2765(00)00097-6. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Scherthan H, Jerratsch M, Dhar S, Wang YA, Goff SP, Pandita TK. Meiotic telomere distribution and Sertoli cell nuclear architecture are altered in Atm- and Atm-p53-deficient mice. Mol Cell Biol. 2000;20:7773–7783. doi: 10.1128/mcb.20.20.7773-7783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, Pandita TK. hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene. 2003a;22:131–146. doi: 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- Sharma GG, Hall EJ, Dhar S, Gupta A, Rao PH, Pandita TK. Telomere stability correlates with longevity of human beings exposed to ionizing radiations. Oncol Rep. 2003b;10:1733–1736. [PubMed] [Google Scholar]
- Sharma GG, Hwang KK, Pandita RK, Gupta A, Dhar S, Parenteau J, Agarwal M, Worman HJ, Wellinger RJ, Pandita TK. Human heterochromatin protein 1 isoforms HP1(Hsalpha) and HP1(Hsbeta) interfere with hTERT-telomere interactions and correlate with changes in cell growth and response to ionizing radiation. Mol Cell Biol. 2003c;23:8363–8376. doi: 10.1128/MCB.23.22.8363-8376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- Shinohara A, Gasior S, Ogawa T, Kleckner N, Bishop DK. Saccharomyces cerevisiae recA homologues RAD51 and DMC1 have both distinct and overlapping roles in meiotic recombination. Genes Cells. 1997;2:615–629. doi: 10.1046/j.1365-2443.1997.1480347.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Morita T, Pearlman RE, Moens PB. RAD51 and DMC1 form mixed complexes associated with mouse meiotic chromosome cores and synaptonemal complexes. J Cell Biol. 1999;147:207–220. doi: 10.1083/jcb.147.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa M, Shinohara A, Hotta Y, Ogawa H, Ogawa T. Localization of RecA-like recombination proteins on chromosomes of the lily at various meiotic stages. Genes Dev. 1995;9:925–934. doi: 10.1101/gad.9.8.925. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res. 2001;477:131–153. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- Tokuyama H, Tokuyama Y. Mouse homolog of Saccharomyces cerevisiae spo11 is induced in normal mu(+)B-cells by stimuli that cause germline C(H) transcription and subsequent class switch recombination. Cell Immunol. 2000;202:1–5. doi: 10.1006/cimm.2000.1647. [DOI] [PubMed] [Google Scholar]
- Tokuyama H, Tokuyama Y. Class switch recombination signals induce lymphocyte-derived Spo11 expression and Spo11 antisense oligonucleotide inhibits class switching. Cell Immunol. 2001;211:123–130. doi: 10.1006/cimm.2001.1830. [DOI] [PubMed] [Google Scholar]
- Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell. 1998;95:705–716. doi: 10.1016/s0092-8674(00)81640-2. [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol. 2002;12:648–653. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- Wyman C, Kanaar R. Chromosome organization: Reaching out to Embrace New Models. Curr Biol. 2002;12:R446–R448. doi: 10.1016/s0960-9822(02)00941-7. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Horikoshi M. Novel substrate specificity of the histone acetyltransferase activity of HIV-1-Tat interactive protein Tip60. J Biol Chem. 1997;272:30595–30598. doi: 10.1074/jbc.272.49.30595. [DOI] [PubMed] [Google Scholar]