

Abstract
Vibrio cholerae is the causative agent of the disease cholera, characterized by profuse watery diarrhoea. Two of the main virulence factors associated with the disease are cholera toxin (CT) and toxin-coregulated pilus (TCP). Expression of CT and TCP is regulated via a complex cascade of factors that respond to environmental signals, but ultimately ToxT is the direct transcriptional activator of the genes encoding CT and TCP. Recent studies have begun to unveil the mechanisms behind ToxT-dependent transcription. We review current knowledge of transcriptional activation by ToxT and the environmental stimuli that allow ToxT to regulate virulence gene expression, resulting in cholera pathogenesis.
Keywords: AraC family, cholera, pathogenicity island, transcription activation, virulence
Introduction
Vibrio cholerae is the aquatic bacterium responsible for the disease cholera. In addition to being well-equipped to survive various environmental conditions, V. cholerae is also capable of infecting humans, resulting in substantial loss of fluid in the form of diarrhoea and frequently leading to death due to dehydration, if left untreated. The human intestine provides the ideal conditions for the expression of virulence factors that allow the organism to colonize, cause disease, and prepare for re-entry into the environment. The two main virulence factors are cholera toxin (CT) and toxin-coregulated pilus (TCP). CT is an ADP-ribosylating toxin composed of two subunits that causes an increase in cAMP in intestinal cells, leading to diarrhoea due to the osmotic imbalance1. The genes encoding CT, ctxAB, are encoded on a lysogenic filamentous bacteriophage, CTXΦ2 and are under the direct control of the regulatory factor ToxT.
TCP is a type IV bundle-forming pilus that is critical for intestinal colonization. The tcp genes are located on the Vibrio pathogenicity island (VPI), and are under the direct control of ToxT. The toxT gene is also encoded within the VPI, as are additional ToxT-dependent factors, such as the accessory colonization factor (ACF) genes (acfA, acfB, acfC, acfD, Fig. 1). Disruption of any of these four genes results in a colonization defect within the infant mouse3. The function of the various ACF proteins is not well characterized, with the exception of AcfB. AcfB, like another ToxT-dependent VPI protein TcpI, encodes a methyl-accepting chemotaxis protein (MCP)4. Deletion of both acfB and tcpI leads to a decrease in colonization5, suggesting that these ToxT-dependent MCPs facilitate chemotaxis to the correct intestinal location for productive colonization. Reviews have been published earlier on the ToxR regulon6 discussing as to how environmental signals induce toxT transcription. This review will focus on ToxT-dependent transcription, highlighting the complexity of virulence gene expression in V. cholerae.
Fig. 1.
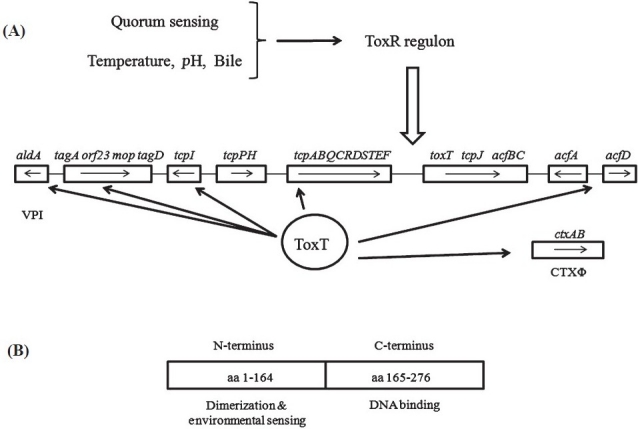
(A). The ToxR regulon. The ToxR regulon is responsible for activating transcription of toxT, which facilitates virulence gene expression. Environmental factors such as temperature, bile and pH modulate the ToxR regulon. ToxT is the direct transcriptional activator of the ctxAB (located in the CTXΦ), tcp, acfA, acfD, tcpI, aldA and tag genes (all located in the VPI), and it also responds to environmental signals to modulate expression of these genes. (B). ToxT is an AraC-like protein composed of two domains. The N-terminal domain, comprised of amino acids (aa) 1 to 164, is involved in dimerization and environmental sensing, while the C-terminal domain (aa 165 to 276) is responsible for DNA binding.
ToxT domain structure and function
A large amount of research from a number of laboratories has elucidated much of the signaling cascade that results in the expression of CT and TCP. This cascade is frequently referred to as the ToxR regulon. In this regulon, various environmental factors influence a number of regulatory proteins (including ToxR) and this ultimately culminates in the transcription of toxT.
ToxT, an AraC-like activator protein, directly activates transcription of the ctxAB, tcpABQCRDSTEF, tcpI, tagA orf23 mop tagD, aldA, acfD and acfA operons7 (Fig. 1A). Because the toxTtcpJacfBCtagE operon is located downstream of the tcpA operon, ToxT also autoregulates its own expression as well as that of the other genes in this operon, which are also under the control of the ToxR-TcpP-dependent promoter immediately upstream of the toxT gene8. Thus, with the exception of the tcpPH genes, the entire VPI is under the regulatory control of ToxT, emphasizing the importance of this virulence regulator to V. cholerae pathogenesis.
ToxT is composed of two domains: the N-terminus is involved in dimerization and environmental sensing, while the C-terminus contains two helix-turn-helix (HTH) motifs that facilitate binding to the promoter regions of the ctx, tcp, and acf genes9–11 (Fig. 1B). Comprehensive scanning alanine mutagenesis of ToxT helped to identify specific residues involved in dimerization, DNA binding and transcriptional activation10. Residue F151, which lies in a putative alpha helical region of the N-terminus, is critical for dimerization, while residues within HTH1 and HTH2 within the C-terminus failed to activate virulence gene expression highlighting their potential in DNA binding. A computer threading model of the ToxT C-terminus with the AraC family protein MarA predicts that residue T253 in HTH2 makes base-specific contact with the ToxT binding site, which is supported by our unpublished data. Interestingly, various alanine substitution mutants also exhibited differential activation of the ctx, tcp, and acf genes, indicating that modulation of ToxT may allow for such differential activation in vivo. This is supported by previous findings that tcp gene transcription precedes ctx gene transcription in vivo, despite both being activated by ToxT12.
DNA binding by ToxT
Recent work has begun to clarify how ToxT binds to the promoter regions of ToxT-activated genes. ToxT binds to a 13-bp DNA sequence that has been labeled a “toxbox” (yrTTTTwwTwAww)13. Two “toxboxes” are positioned as direct repeats in the tcpA, ctxAB and tcpI-2 promoters, and as inverted repeats in the acfD/acfA, tagA and tcpI-1 promoters (two separate binding sites have been identified in the tcpI promoter region). Interestingly, the aldA promoter appears to only contain one toxbox13. Mutational analysis of the two toxboxes within the tcpA promoter revealed 10 nucleotides within these sequences that are critical for ToxT-dependent transcription, and notably the “T” triplet found in each toxbox was important (Fig. 2A). These triplets are spaced 17 bp apart, which is similar to the spacing between triplets found in the ctx (Fig. 2B) promoter that lie within the region bound by ToxT, suggesting that specific protein-DNA contacts may be made in these triplets. The ctxA promoter region from -76 to +1 is sufficient for ToxT-dependent transcription. Although similar analysis of the specific basepairs required for ToxT-dependent transcription has not been performed at the ctx promoter, we have noted the similarity in “T” triplet spacing (Fig. 2), which would require ToxT to be bound in the opposite orientation with respect to RNA polymerase (due to the opposite orientation of toxboxes) in comparison with the tcpA promoter.
Fig. 2.
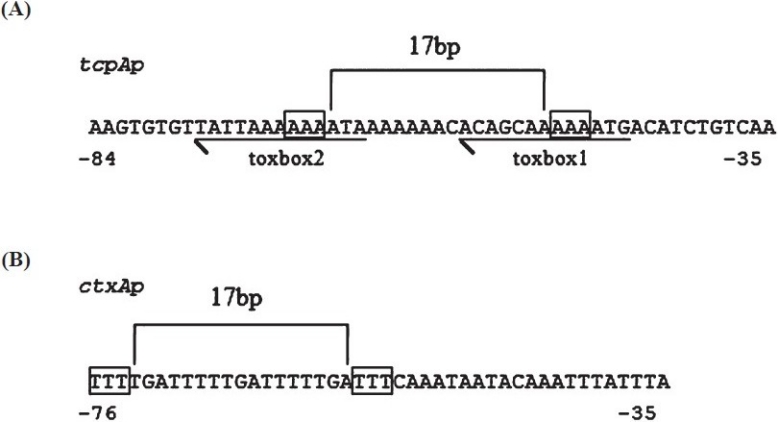
ToxT–activated promoters. A schematic representation of the tcpA and ctxA promoter regions to which ToxT binds to activate transcription. (A). The tcpA promoter contains two “toxboxes” (underlined). The “A/T” triplets within the boxes are critical nucleotides for ToxT-dependent transcription. (B). The ctxA promoter represented in this figure is sufficient for ToxT-dependent transcriptional activation and therefore should contain two “toxboxes”. The boxes outline “A/T” triplets that are spaced identically to the triplets in the tcpA promoter, which suggests that ToxT is oriented in the opposite direction with respect to RNA polymerase in ctxA promoter as compared to the tcpA promoter
Separating the toxboxes in the tcpA promoter by 5bp or 10bp disrupted ToxT-dependent transcription, despite evidence that ToxT could still protect these sites in footprinting assays performed in vitro13. Likewise, insertions between the toxboxes of the acfA/acfD, as well as the tagA promoters disrupted transcriptional activation of these genes14. These data indicate that interaction between the ToxT proteins bound at each site is critical for transcriptional activation. Our own data have supported the idea that ToxT requires dimerization in order to bind to the tcpA, ctxA, and acfA promoter sites10,11, which is consistent with the findings of other laboratories9. We suggest that the apparent binding of ToxT to the tcpA toxboxes separated by 5 and 10 bp is due to the high concentration of ToxT in in vitro footprinting reactions, and that in vivo ToxT must be dimerized to bind to these sites. However, other sites that ToxT binds may be bound by its monomeric forms.
The spacing between the ToxT binding sites and the -35 region bound by RNA polymerase (RNAP) is also critical for transcriptional activation14. The tcpA, acfA, acfD, tagA and aldA promoters were only able to tolerate slight changes in spacing (± 1 to 2 bp) between the proximal toxbox and the -35 region of RNAP. Since the “toxboxes” are all located upstream of the -35 promoter region, this places all ToxT-dependent promoters under the class I promoter category of AraC family activated promoters. This suggests that ToxT directly interacts with the α subunit C-terminal domain (α-CTD) of RNAP to activate transcription, which was supported by transcription experiments performed with ToxT and a α-CTD deletion mutant of RNAP15.
Factors that affect ToxT-dependent transcription
Nucleoid-associated proteins (H-NS) bind to DNA and have the capability of altering its structure, which in turn can affect transcription. In V. cholerae, a deletion of hns (vicH) resulted in high level expression of the tcpA and ctx genes, even under non-inducing conditions16. High levels of ctx and tcpA expression were also observed in an hns mutant lacking ToxT. These data indicate that H-NS binds these promoters to repress virulence gene expression in the absence of ToxT. Under inducing conditions, H-NS repression at the ctx and tcpA promoters can be overcome by ToxT displacement of H-NS16.
Integration host factor (IHF) has been shown to positively regulate expression of tcpA and ctx17. IHF is a heterodimeric protein encoded by ihfA and ihfB, which causes sharp bends upon binding to DNA. The deletion of these two genes resulted in approximately five-fold reduction in ctx and tcpA expression. It has also been demonstrated that IHF binds at the -162 region of the tcp Apromoter and bends the DNA. This region also overlaps with the binding region of H-NS, allowing IHF to displace H-NS, which in turn permits virulence gene expression. However, IHF has not been shown to bind the ctx promoter. Presumably, positive ToxT-dependent regulation at the tcpA promoter leads to increased levels of ToxT, which in turn enhances ctx expression17.
cAMP-CRP similar to H-NS, also negatively regulates the expression of CT and TCP under certain environmental conditions. However, these are due to effects on the ToxR regulon leading to ToxT expression, rather than effects on ToxT-dependent transcription18,19. Cyclic diguanylate (c-diGMP) levels influence transcription of ctxA, but surprisingly do not seem to affect tcpAA expression. Because low levels of c-diGMP favour ToxT transcription as well as ctx transcription, it is not clear whether c-diGMP specifically affects ToxT-dependent transcription at the ctx but not tcpAA promoter or whether this phenomenon only involves ToxT expression but not ToxT activity20.
Environmental factors that directly affect ToxT
Initial studies showed that environmental signals are able to modulate ToxT activity. Both variations in temperature and/or the presence of bile could reduce or eliminate ToxT-dependent transcription of ctxA, tcpAA and several other genes21,22. Further studies on the effects of bile on V. cholerae virulence factor transcription revealed that unsaturated fatty acids (arachidonic, linoleic, and oleic acids) within bile repressed ctxAB and tcpAA expression23, suggesting that these components may be directly interacting with ToxT to repress its activity. The presence of high levels of bile within the intestinal lumen may prevent premature ToxT activity prior to the organisms’ arrival within intestinal crypts. Anaerobiosis was also found to specifically inhibit ctxA transcription, but this effect is likely to be mediated by the effect on H-NS binding, rather than the effect on ToxT activity24.
A synthetic small molecule, virstatin, was identified for its ability to inhibit ToxT-dependent transcription25. Virstatin inhibits the dimerization of the N-terminus of ToxT, and thus prevents ToxT-dependent transcription activation of the ctxAB, tcpA, acfD and tagA promoters9,26. It is tempting to speculate that virstatin is a synthetic mimic of the effect of bile components on ToxT.
Bicarbonate has the opposite effect of bile/virstatin on ToxT activity. Low levels of bicarbonate result in low ToxT dependent transcription of tcpA and ctxA, whereas elevated levels of bicarbonate lead to maximal transcription of ctx and tcp. It is suggested that bicarbonate is found at high concentrations in the intestinal epithelium and, thus, may act as a relevant in vivo signal modulating the activity of ToxT27. From these collective data, it is evident that ToxT activity is modulated by various environmental signals, and that the protein receives signals either indirectly or directly through binding of effector molecules.
ToxT as a repressor of gene expression
The type IV mannose-sensitive haemagglutinin (MSHA) contributes to V. cholerae biofilm formation and persistence in the aquatic environment, but it is repressed during in vivo growth by ToxT. Repression of MSHA allows V. cholerae to evade binding of sIgA and establish residence within the host28. ToxT inhibits mshA transcription by binding to three binding sites within the msh operon29. Interestingly, ToxT dimerization is not required for repression of mshA transcription; ToxT either containing a dimerization point mutation, or lacking the entire N-terminus was able to repress msh transcription30. As mentioned earlier, dimerization mutants fail to activate the ctx and tcp genes, suggesting that ToxT binding sites can be differentiated between those that require dimerization for binding (ToxT-activated promoters) and those that can bind monomeric ToxT (ToxT-repressed promoters). One notable exception appears to be the ToxT-activated aldA promoter, which appears to only bind a monomer of ToxT13,14,31.
Conclusion
ToxT-dependent transcription is a critical component of V. cholerae pathogenesis. ToxT is able to differentially regulate virulence gene expression by directly binding to degenerate promoter DNA sequences. Available data imply that ToxT needs to dimerize in order to activate genes (ctxA, tcpA, acfA, acfD, tcpI), but it can bind as a monomer to repress (msh) and, possibly, even activate (aldA) other genes. ToxT binds to “toxboxes” that is oriented differently at the various promoters. This suggests a model whereby ToxT shows flexibility in its DNA binding capabilities and interactions with RNAP. ToxT activity is modulated by environmental signals resulting in optimal temporal and spatial virulence factor expression within the host. Further research is required to fully characterize the complexity of ToxT-dependent virulence gene expression in V. cholerae.
Acknowledgments
Author (KEK) acknowledges financial support by an NIH grant (AI51333), USA.
References
- 1.Prouty MG, Klose KE. Vibrio cholerae: the genetics of pathogenesis and environmental persistence. In: Thompson FL, Austin B, Swings J, editors. The biology of the vibrios. Washington, D.C: ASM Press; 2006. pp. 311–39. [Google Scholar]
- 2.Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–4. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 3.Peterson KM, Mekalanos JJ. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect Immun. 1988;56:2822–9. doi: 10.1128/iai.56.11.2822-2829.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everiss KD, Hughes KJ, Kovach ME, Peterson KM. The Vibrio cholerae acfB colonization determinant encodes an inner membrane protein that is related to a family of signal-transducing proteins. Infect Immun. 1994;62:3289–98. doi: 10.1128/iai.62.8.3289-3298.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaparro AP, Ali SK, Klose KE. The ToxT-dependent methyl-accepting chemoreceptors AcfB and TcpI contribute to Vibrio cholerae intestinal colonization. FEMS Microbiol Lett. 2010;302:99–105. doi: 10.1111/j.1574-6968.2009.01835.x. [DOI] [PubMed] [Google Scholar]
- 6.Childers BM, Klose KE. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol. 2007;2:335–44. doi: 10.2217/17460913.2.3.335. [DOI] [PubMed] [Google Scholar]
- 7.Higgins DE, Nazareno E, DiRita VJ. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J Bacteriol. 1992;174:6974–80. doi: 10.1128/jb.174.21.6974-6980.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu RR, DiRita VJ. Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J Bacteriol. 1999;181:2584–92. doi: 10.1128/jb.181.8.2584-2592.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc Natl Acad Sci USA. 2007;104:2372–7. doi: 10.1073/pnas.0611643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Childers BM, Weber GG, Prouty MG, Castaneda MM, Peng F, Klose KE. Identification of residues critical for the function of the Vibrio cholerae virulence regulator ToxT by scanning alanine mutagenesis. J Mol Biol. 2007;367:1413–30. doi: 10.1016/j.jmb.2007.01.061. [DOI] [PubMed] [Google Scholar]
- 11.Prouty MG, Osorio CR, Klose KE. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol Microbiol. 2005;58:1143–56. doi: 10.1111/j.1365-2958.2005.04897.x. [DOI] [PubMed] [Google Scholar]
- 12.Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–34. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 13.Withey JH, DiRita VJ. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol Microbiol. 2006;59:1779–89. doi: 10.1111/j.1365-2958.2006.05053.x. [DOI] [PubMed] [Google Scholar]
- 14.Bellair M, Withey JH. Flexibility of Vibrio cholerae ToxT in transcription activation of genes having altered promoter spacing. J Bacteriol. 2008;190:7925–31. doi: 10.1128/JB.00512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulbert RR, Taylor RK. Mechanism of ToxT-dependent transcriptional activation at the Vibrio cholerae tcpA promoter. J Bacteriol. 2002;184:5533–44. doi: 10.1128/JB.184.20.5533-5544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nye MB, Pfau JD, Skorupski K, Taylor RK. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol. 2000;182:4295–303. doi: 10.1128/jb.182.15.4295-4303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stonehouse E, Kovacikova G, Taylor RK, Skorupski K. Integration host factor positively regulates virulence gene expression in Vibrio cholerae. J Bacteriol. 2008;190:4736–48. doi: 10.1128/JB.00089-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skorupski K, Taylor RK. Cyclic AMP and its receptor protein negatively regulate the coordinate expression of cholera toxin and toxin-coregulated pilus in Vibrio cholerae. Proc Natl Acad Sci USA. 1997;94:265–70. doi: 10.1073/pnas.94.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacikova G, Skorupski K. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol. 2001;41:393–407. doi: 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- 20.Tischler AD, Camilli A. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun. 2005;73:5873–82. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S, Chowdhury R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun. 1997;65:1131–4. doi: 10.1128/iai.65.3.1131-1134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuhmacher DA, Klose KE. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J Bacteriol. 1999;181:1508–14. doi: 10.1128/jb.181.5.1508-1514.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatterjee A, Dutta PK, Chowdhury R. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun. 2007;75:1946–53. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnan HH, Ghosh A, Paul K, Chowdhury R. Effect of anaerobiosis on expression of virulence factors in Vibrio cholerae. Infect Immun. 2004;72:3961–7. doi: 10.1128/IAI.72.7.3961-3967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science. 2005;310:670–4. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- 26.Shakhnovich EA, Sturtevant D, Mekalanos JJ. Molecular mechanisms of virstatin resistance by non-O1/non-O139 strains of Vibrio cholerae. Mol Microbiol. 2007;66:1331–41. doi: 10.1111/j.1365-2958.2007.05984.x. [DOI] [PubMed] [Google Scholar]
- 27.Abuaita BH, Withey JH. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun. 2009;77:4111–20. doi: 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsiao A, Liu Z, Joelsson A, Zhu J. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci USA. 2006;103:14542–7. doi: 10.1073/pnas.0604650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiao A, Toscano K, Zhu J. Post-transcriptional cross-talk between pro- and anti-colonization pili biosynthesis systems in Vibrio cholerae. Mol Microbiol. 2008;67:849–60. doi: 10.1111/j.1365-2958.2007.06091.x. [DOI] [PubMed] [Google Scholar]
- 30.Hsiao A, Xu X, Kan B, Kulkarni RV, Zhu J. Direct regulation by the Vibrio cholerae regulator ToxT to modulate colonization and anticolonization pilus expression. Infect Immun. 2009;77:1383–8. doi: 10.1128/IAI.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Withey JH, Dirita VJ. Vibrio cholerae ToxT independently activates the divergently transcribed aldA and tagA genes. J Bacteriol. 2005;187:7890–900. doi: 10.1128/JB.187.23.7890-7900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]