

Abstract
The calcium- and integrin-binding protein 1 (CIB1) is a ubiquitous Ca2+-binding protein and a specific binding partner for the platelet integrin αIIb cytoplasmic domain, which confers the key role of CIB1 in hemostasis. CIB1 is also known to be involved in apoptosis, embryogenesis, and the DNA damage response. In this study, the solution structures of both Ca2+-CIB1 and Mg2+-CIB1 were determined using solution-state NMR spectroscopy. The methyl groups of Ile, Leu, and Val were selectively protonated to compensate for the loss of protons due to deuteration. The solution structure of Ca2+-CIB1 possesses smaller opened EF-hands in its C-domain compared with available crystal structures. Ca2+-CIB1 and Mg2+-CIB1 have similar structures, but the N-lobe of Mg2+-CIB1 is slightly more opened than that of Ca2+-CIB1. Additional NMR experiments, such as chemical shift perturbation and methyl group solvent accessibility as measured by a nitroxide surface probe, were carried out to further characterize the structures of Ca2+-CIB1 and Mg2+-CIB1 as well as their interactions with the integrin αIIb cytoplasmic domain. NMR measurements of backbone amide proton slow motion (microsecond to millisecond) dynamics confirmed that the C-terminal helix of Ca2+-CIB1 is displaced upon αIIb binding. The EF-hand III of both Ca2+-CIB1 and Mg2+-CIB1 was identified to be directly involved in the interaction of CIB1 with αIIb. Together, these data illustrate that CIB1 behaves quite differently from related EF-hand regulatory calcium-binding proteins, such as calmodulin or neuronal calcium sensor proteins.
Keywords: Calcium-binding Proteins, Integrin, NMR, Peptide Interactions, Protein Structure, Calcium and Integrin-binding Protein, Magnesium, Methyl Labeling
Introduction
The calcium- and integrin-binding protein 1 (CIB1) is a member of the regulatory Ca2+-binding helix-loop-helix or EF-hand protein family (1). CIB1 is a ubiquitous 191-residue (22 kDa) protein with several functions in cell signaling (2, 3). CIB1 has been reported to interact with a number of protein targets, including the platelet integrin αIIb subunit (4), sphingosine kinase 1 (5), p21-activated kinase (6), apoptosis signal-regulating kinase (7), polo-like protein kinases (8), and a recently reported new target in the regulation of cardiac hypertrophy, calcineurin B (9). The interaction of CIB1 with the integrin αIIb subunit has been studied extensively (10–13). The integrin αIIb subunit usually associates with the integrin β3 subunit, and the heterodimeric αIIbβ3 protein is involved in both so-called “inside-out” and “outside-in” signaling pathways (3). CIB1 is believed to be capable of specifically binding to the αIIb cytoplasmic domain and dissociating the αIIbβ3 dimer, which in turn triggers integrin activation (11, 12). The interactions between Ca2+-CIB1 and a fragment of the integrin αIIb subunit (residues 983–1008, hereafter referred to as the αIIb peptide) have been suggested to mainly involve a hydrophobic pocket in the C-domain of Ca2+-CIB1 with a dissociation constant in the submicromolar range (10).
The calcium-bound CIB1 (Ca2+-CIB1) structure consists of four helix-loop-helix (EF-hand) Ca2+-binding motifs (EF-I to EF-IV) (14, 15), in which only EF-III and EF-IV have the capacity to bind Ca2+. In addition, CIB1 contains a myristoylated N-terminal extension in vivo (16), and a short C-terminal extension that was postulated to be involved in a displacement mechanism to increase its target-binding specificity (12). So far, two x-ray crystal structures of Ca2+-CIB1 have been deposited in the Protein Data Bank, in which 1XO5 (15) is a monomer and 1Y1A (14) is a head-to-tail homodimer. These two crystal structures have a similar arrangement of the secondary structural elements, but large differences exist in their relative orientations of the N- and C-domains as well as the secondary structure of their N- and C-terminal extensions. Our initial residual dipolar coupling (RDC)3 NMR study (12) suggested that the monomeric crystal structure of CIB1 (1XO5) more closely resembles the overall conformation of the protein in solution, except for its N-terminal extension.
Because the magnesium ion is constantly present in the cytosol in millimolar concentrations, the magnesium-bound form of CIB1 (Mg2+-CIB1) is considered to be the physiologically relevant form of CIB1 in the resting state of the cell (17). CIB1 has been found to bind Mg2+ only at its EF-III site, but it still possesses an overall well folded tertiary structure (17). It is further suggested that Mg2+-CIB1 and Ca2+-CIB1 have similar tertiary structures with subtle differences (18). In addition, as both Ca2+-CIB1 and Mg2+-CIB1 can bind to αIIb with similar binding affinities, the role of CIB1 for being a specific Ca2+ sensor has been brought into question (10). Therefore the high resolution structure and dynamics of Mg2+-CIB1 are of interest to better understand the mechanism of activation of CIB1 and the regulation of the interactions of CIB1 with its binding targets.
Here, we present the solution structures of both Ca2+-CIB1 and Mg2+-CIB1, as well as a biophysical and NMR dynamics characterization of the interaction of CIB1 with αIIb. The construct used for CIB1 had a His tag and a linker, with a total molecular mass of ∼24 kDa. Thus, the acquisition of high quality NMR data required the use of transverse relaxation optimized spectroscopy-type experiments (19) and perdeuteration of the protein (20). The solution structures of Ca2+-CIB1 and Mg2+-CIB1 NMR were subsequently determined by using chemical shift information, two sets of backbone RDCs (1DC′N and 1DNH) in combination with the NOEs from backbone amide protons (NH) as well as selectively labeled methyl groups (Ile/Leu/Val) in an otherwise perdeuterated protein sample. The Carr-Purcell-Meiboom-Gill (CPMG) NMR method for measuring slow motion (microsecond to millisecond scale) dynamics (21) was employed to study the CIB1 backbone dynamic behavior in both the free state and the αIIb peptide-bound state. Additionally, the TEMPOL paramagnetic surface probe (22, 23) was used to assess the solvent accessibility of the methyl groups in the hydrophobic pocket in different states of the protein.
EXPERIMENTAL PROCEDURES
Sample Preparation
The CIB1 protein was prepared as described previously (17). The 13C,2H,15N-uniformly labeled sample was expressed and purified according to an established protocol (12), and this sample was used to acquire the backbone 1DNH and 1DC′N RDCs. The 13C,2H,15N-uniformly and Ile-δ1-13CH3,Leu,Val-13CH3,12CD3-labeled sample was prepared by expression in M9 minimal media in D2O supplemented with 15NH4Cl, 13C-labeled glucose, and the methyl labeling precursors 2-keto-3-d2-1,2,3,4-13C-butyrate (Cambridge Isotope Laboratories) and 2-keto-3-methyl-3-d1-3–1,2,3,4-13C-butyrate (Cambridge Isotope Laboratories) (24); this sample was used for the acquisition of methyl assignment spectra. The 12C,2H,15N-uniformly and Ile-δ1-13CH3,Leu,Val-13CH3,12CD3-labeled CIB1 was prepared using M9 minimal media in D2O supplemented with 15NH4Cl, 13C-labeled glucose, and the methyl-labeling precursors 2-keto-3-methyl-d3-3-d1-4-13C-butyrate (Cambridge Isotope Laboratories) and 2-keto-3-d2-4-13C,d1-butyrate (24); this sample was used to acquire the 15N-edited NOESY and 13C-edited NOESY spectra. Finally, the 2H,15N-uniformly labeled sample was prepared with M9 minimal media in D2O supplemented with 15NH4Cl and d7-glucose; this sample was used to record the CIB1 backbone CPMG-relaxation dispersion spectra. The synthetic 26-residue αIIb peptide (Ac-LVLAMWKVGFFKRNRPPLEEDDEEGQ-OH) is the same as used in earlier studies (12), and it corresponds to amino acids 983–1008 of the platelet integrin αIIb subunit, with Gln-1008 as the C-terminal residue. The synthetic peptide was purchased from GenScript Corp. and was over 95% pure as determined by mass spectrometry and high performance liquid chromatography. Protein and peptide concentrations were either determined by using the Bio-Rad protein assay kit or UV absorbance measurements based on the extinction coefficients ϵ280 = 3105 for CIB1 and ϵ280 = 5500 for the αIIb peptide.
NMR Experiments
NMR spectra were recorded at 37 °C on a Bruker AVANCE 500 MHz or a Bruker AVANCE 700 MHz NMR spectrometer each equipped with a triple resonance cryoprobe. The CIB samples for the 13C-edited NOESY experiments contained 50 mm HEPES in 99.9% D2O. For other NMR CIB1 samples with or without αIIb, they all contain 0.6–0.7 mm CIB1 in 50 mm HEPES, 100 mm KCl, 0.1 mm 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS), 10 mm DTT, 10% D2O, pH 7.5 ± 0.05. For Mg2+-CIB1 samples, 0.5 mm EGTA and 6 mm MgCl2 were added; for Ca2+-CIB1, 4 mm CaCl2 was added. The 13C,2H,15N-uniformly labeled samples were used to acquire two sets of backbone RDCs (1DC′N and 1DNH). In this sample, 14 mg/ml pf1 phage (Asla-Biotech) was used to achieve partial alignment of the protein molecules. 1DNH RDCs were measured using a two-dimensional IPAP-HSQC experiment (25) with complex points of 1024 × 1024. After linear prediction and zero filling, the digital resolution was 0.83 Hz/pt in the 15N dimension. 1DC′N RDCs were measured using the three-dimensional IPAP-J-HNCO (CA) experiment (26), with 1024 × 128 × 40 complex points. The digital resolution was 2.06 Hz/pt in F2 (13C) after linear prediction and zero filling. A scale factor of 4 was used in the measurement of the 1DC′N RDC. The obtained 1DNH RDC of Mg2+-CIB1 was analyzed against the solution structure of Ca2+-CIB1 with the program PALES (27) to evaluate the correlation between the Mg2+-CIB1 and Ca2+-CIB1 solution structures. In the PALES analysis, a linear correlation indicates the degree to which the experimentally obtained RDCs correspond to an existing protein structure, with a high correlation factor R and a low quality factor Q indicating good agreement.
A recently published three-dimensional multiple quantum (MQ) (H)CCmHm-TOCSY experiment was employed to assign the methyl groups of CIB1 (28). This methyl assignment approach requires preliminary knowledge of the chemical shifts of Cα and Cβ, which have been previously obtained from the backbone assignment work (12, 18, 29). Stereospecific assignments for Val and Leu methyl groups were obtained using the 10% [13C]glucose labeling strategy (30) for Ca2+-CIB1 and Mg2+-CIB1 and Ca2+-CIB1 and Mg2+-CIB1 in complex with the αIIb peptide. The methyl chemical shift perturbation (CSP) was calculated using Equation 1, which is revised based on a previously published study (31), in which the δH and δC indicate the proton and carbon chemical shift changes, respectively.
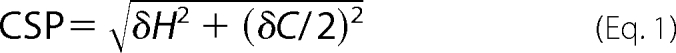 |
Titrations with 4-hydroxyl-2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPOL) (Sigma) were performed using 600 μm samples of 2H,15N-uniformly and Ile-δ1-13CH3,Leu,Val-13CH3,12CD3- labeled CIB1, being titrated with 6 eq of a freshly prepared concentrated stock solution of TEMPOL in 100 mm KCl, 99% D2O, pH 7.5 (not corrected for isotope effects).
The backbone amide 15N relaxation dispersion measurements were carried out using a CPMG relaxation dispersion experiment (21) implemented with an inter-scan delay of 2 s and a total constant CPMG length (TCP) of 61.08 ms. Two sets of CT-CPMG experiments were acquired at CPMG field strengths (νCPMG) of 50 and 500 Hz, respectively. For each CPMG field strength, two replicate data sets were collected each with 32 dummy scans and 64 scans per t1 point. Duplicates were co-added using NMRPipe prior to processing. Duplicates at each frequency were acquired and analyzed for standard error. Peaks affected by partial or total overlap were excluded from analysis. The NMR relaxation dispersion (ΔR2,eff) was quantified as (32) as shown in Equations 2–4,
Structure Calculations
A two-stage simulated annealing approach (33) using the program XPLOR-NIH 2.18 (34) was implemented for the structure determination of Ca2+-CIB1 and Mg2+-CIB1. The experimental restraints include two sets of backbone RDCs (1DC′N and 1DNH) (26), the dihedral angle restraints predicted from NMR chemical shifts using TALOS (35), hydrogen bonding restraints from the Chemical Shift Index (36), NOEs (NH-NH, NH-methyl, and methyl-methyl), and metal ion binding coordinates (15). For the NH-NH and NH-methyl NOEs from the 15N-edited HSQC-NOESY experiment (mixing time 120 ms), two categories (1.8–5.0 and 1.8–6.0 Å) were created based on the peak intensities; for the methyl-methyl NOEs from 13C-HMQC-NOESY (mixing time 200 ms) acquired with a sample in D2O solvent, two slightly larger categories (1.8–6.0 and 1.8–8.0 Å) were implemented. Because the N-terminal region of both Ca2+-CIB1 and Mg2+-CIB1, including the His tag, and the first six residues could not be observed by NMR, we excluded this region from structure calculation. The details of structure calculation were elaborated in the supplemental material. Procheck 3.5.4 (37) was used to check the calculated structures for stereochemical quality. The inter-helical angles of the EF-hands were measured using an in-house script. The structural differences between Ca2+-CIB1 and Mg2+-CIB1 were assessed with difference distance matrix plot (DDMP) (38).
Data Deposition
Backbone and methyl chemical shift assignments for Ca2+-CIB1 and Mg2+-CIB1 have been deposited in the BioMagResBank with the accession numbers 17329 and 17328, respectively. The coordinates of the calculated structure ensembles and corresponding experimental restraints used in the structure calculation have been deposited in the Protein Data Bank with the accession codes 2L4H for Ca2+-CIB1 and 2L4I for Mg2+-CIB1, respectively.
Supplemental Material
This section contains the details of the structure calculation procedure of using a two-stage low temperature simulated annealing. It also contains the complete assignments of the methyl resonances for Ca2+-CIB1, Mg2+-CIB1, and Ca2+-CIB1 bound to the αIIb peptide and Mg2+-CIB1 bound to the αIIb peptide. In addition, the HSQC spectra of TEMPOL titration in Ca2+-CIB1/αIIb and Mg2+-CIB1/αIIb are also included.
RESULTS
Secondary Structure of Ca2+-CIB1 and Mg2+-CIB
Two crystal structures of Ca2+-CIB1 have been reported, in which one is a monomer (15) and the other one is a head-to-tail homodimer (14) with a glutathione (GSH) molecule noncovalently bound to the N-domain of chain B. These two crystal structures have significant differences in the relative orientation of the N- and C-domains (12, 39). Even though the amide NH-RDC analysis results suggested that the monomer crystal structure of CIB1 (Protein Data Bank 1XO5) more closely resembles the conformation of the protein in solution (12), the secondary structure of 1XO5 differs from the solution state conformation in the N- and C-terminal extensions (Fig. 1). To some extent, the structural differences for these extensions can be explained by the presence of weak Ca2+-binding sites and the extremely high calcium concentration used to induce crystallization (39). In contrast, 1Y1A has good agreement with the NMR study in terms of secondary structure, including the N- and C-terminal extensions (Fig. 1). In the solution state, Mg2+-CIB1 has an overall similar secondary structure arrangement as Ca2+-CIB1 (Fig. 1), but the Mg2+ can only occupy EF-III of CIB1 (17), leaving site EF-IV in intermediate conformational exchange between the Mg2+-bound and -unbound states. This is similar to other calcium-binding proteins, e.g. calmodulin (40) and soybean calmodulin isoform 4 (41). Unfortunately, this means that most resonances in the C-terminal portion (residues 158–191) are missing for Mg2+-CIB1 (18). In addition, backbone heteronuclear {1H}-15N NOE dynamics for both Ca2+-CIB1 (12) and Mg2+-CIB1 (18) support the notion that the N-terminal end (residues 8–20) of CIB1 should adopt relatively rigid secondary structure rather than being an unstructured flexible tail.
FIGURE 1.

Secondary structure arrangements of the two available x-ray crystal structures of Ca2+-CIB1 (1XO5 and 1Y1A) and the NMR solution structures of Ca2+-CIB1 and Mg2+-CIB1. The positions of the four EF-hand helix-loop-helix structures are indicated. Boxes indicate α-helices, and arrows indicate β-strands.
Methyl Group Assignments and Their Utilization in the Structure Determination
To compensate for the loss of proton signals caused by deuteration of the protein, the methyl groups of the hydrophobic residues Ile-δ1-13CH3,Leu,Val-13CH3,12CD3 were selectively labeled with a published protocol (24). The methyl groups of Ile, Leu, and Val in Ca2+-CIB1 and Mg2+-CIB1 could be assigned using a three-dimensional multiple quantum (MQ) (H)CCmHm-TOCSY (28) experiment. The stereospecific assignment of the methyl groups of the Ca2+-CIB1 (supplemental Fig. 1A), Mg2+-CIB1 (supplemental Fig. 1B), Ca2+-CIB1 in complex with the αIIb peptide (supplemental Fig. 2A), and Mg2+-CIB1 in complex with the αIIb peptide (supplemental Fig. 2B), obtained by using the 10% 13C-glucose approach (30), are displayed in the supplemental material. For Mg2+-CIB1, the backbone assignment for the C-terminal region (residues 158–191) is largely missing, but the methyls on this fragment could still be assigned by referring to the assignment of Ca2+-CIB1 (supplemental Fig. 1A) as well as the knowledge about the numbers of Ile/Leu/Val residues in CIB1. The methyl-labeled CIB1 samples have been used for the acquisition of 15N-, and 13C-edited NOESY spectra, and the NOEs obtained in this manner were used in the structure determination of Ca2+-CIB1 and Mg2+-CIB1.
Solution Structures of Ca2+-CIB1 and Mg2+-CIB1
To understand the molecular details of the regulatory mechanism for the interactions of CIB1 with its various targets, we determined the solution structures of Ca2+-CIB1 and Mg2+-CIB1. In our calculation, a two-stage low temperature simulated annealing approach (33) was employed to refine the crystal structure of Ca2+-CIB1 because of high similarity in secondary structure between 1XO5 and the results predicted by the NMR chemical shift index. Moreover, 1XO5 is a monomeric protein, and our previous NMR data are consistent with the protein being in a monomeric form in solution (12, 42). The same protocol has been successfully employed in the structure determination of several other proteins (33, 41, 43, 44). The 13C- and 15N-edited NOESY provided distance restraints for our structure calculation in addition to the dihedral angle, hydrogen bonds, and the RDC restraints for the protein backbone. The calculation results are summarized in Table 1. The refined solution structure of Ca2+-CIB1 is overall similar to the 1XO5 crystal structure with a backbone r.m.s.d. of 2.71 Å for the well defined regions (residues 24–136 and 146–191). In the newly determined Ca2+-CIB1 solution structure, the secondary structures of the N- and C-terminal extensions (H0 and H10) have been corrected to be consistent with the solution state studies (12) and the other crystal structure 1Y1A.
TABLE 1.
Experimental restraints and structural statistics for the 20 lowest total energy structures of CIB1
| Ca2+-CIB1 (residues 8–191) | Mg2+-CIB1 (residues 8–157) | |
|---|---|---|
| PDB structure deposition | 2L4H | 2L4I |
| No. of experimental restraints | 1069 | 1007 |
| Metal-binding coordination restraints | 14 | 6 |
| Hydrogen bond distance restraints | 128 | 104 |
| Dihedral angle restraints | 349 | 293 |
| H-N RDCs | 86 | 94 |
| C′N RDCs | 56 | 77 |
| NOE restraints | 436 | 433 |
| Intra (|i − j| = 0) | 80 | 63 |
| Sequential (|i − j| = 1) | 167 | 164 |
| Medium (1<|i − j|≤4) | 71 | 109 |
| Long (|i − j|>4) | 118 | 97 |
| r.m.s.d. from experimental restraints | ||
| Average distance restraints violation | 0.086 ± 0.005 Å | 0.080 ± 0.005 Å |
| Average dihedral angle restraints violation | 2.40 ± 0.10° | 1.58 ± 0.05° |
| Average H-N RDCs violation | 0.342 ± 0.015 Hz | 0.230 ± 0.015 Hz |
| Average C′N RDC violation | 0.82 ± 0.05 Hz | 0.68 ± 0.05 Hz |
| r.m.s.d. from idealized covalent geometry | ||
| Bond | 0.003 ± 0.001 Å | 0.003 ± 0.001 Å |
| Angle | 0.47 ± 0.03° | 0.47 ± 0.02° |
| Ramachandran analysis (%) | Residues 8–136 and 146–191 | Residues 8–136 and 146–157 |
| Residues in favored regions | 81.3 | 76.9 |
| Residues in additional allowed regions | 15.8 | 20.5 |
| Residues in generously allowed regions | 2.9 | 2.6 |
| Residues in disallowed regions | 0 | 0 |
Interestingly, we found that the solution structure of Ca2+-CIB1 has a more closed conformation than 1X05. For EF-hand calcium-binding proteins, the interhelical angles have been widely employed to characterize the opening of EF-hands (41, 45–47). We used an in-house script to analyze the interhelical angles of each EF-hand in Ca2+-CIB1 structures (1XO5, 1Y1A, and the solution structure) (Table 2) with smaller angles indicating larger openings. Site EF-I was excluded from this analysis because it is atypical as it contains several short α-helices (Fig. 1). In the Ca2+-CIB1 solution structure, site EF-II in the N-domain has a similar opening of 119.9 ± 2.2° compared with 1XO5 (116.4°); however, the EF-III (122.8 ± 1.7°) and EF-IV (122.2 ± 0.7°) sites in the C-domain have smaller openings compared with 1XO5 (EF-III 113.3° and EF-IV 109.0) (Table 2). Therefore, sites EF-III and EF-IV in the solution structure of Ca2+-CIB1 are about 10° less open compared with 1XO5 (Fig. 3C), which is similar to the case of calmodulin (CaM), where the solution structure is also found to be more closed than the crystal structure (46).
TABLE 2.
Interhelical angles of the EF-hands in two crystal structures of Ca2+-CIB1 (1XO5 and 1Y1A) and the Ca2+-CIB1 and Mg2+-CIB1 solution structures
H4 consists of residues 71–75/88–97 that show the two fragments used for the measurement of inter-helical angles; H5 consists of residues 104–115/125–136, and H6 consists of residues 148–160/171–178.
| Helix pair | 1XO5 (x-ray) | 1Y1A (x-ray) | Ca2+-CIB1 (NMR) | Mg2+-CIB1 (NMR) |
|---|---|---|---|---|
| H4/H5 (EF-II) | 116.4 | 128.0 | 119.9 ± 2.2 | 113.2 ± 1.1 |
| H6/H7 (EF-III) | 113.3 | 116.6 | 122.8 ± 1.7 | 121.2 ± 1.2 |
| H8/H9 (EF-IV) | 109.0 | 112.2 | 122.2 ± 0.7 | NDa |
a ND means not determined.
FIGURE 3.

Solution structures of Ca2+-CIB1 and Mg2+-CIB1. A, ensemble of the 20 best solution structures of Ca2+-CIB1. B, superimposed solution structure (red) and crystal structure (1XO5, blue) of Ca2+-CIB1 with an r.m.s.d. of 2.71 Å for the well defined areas (residues 24–136 and 146–191). C, superimposed EF-III (H6 and H7) and EF-IV (H8 and H9) region (residues 102–180) of the Ca2+-CIB1 crystal (1XO5, blue) and solution structures (red) demonstrate that the solution structure has a more closed conformation than the crystal structure. The two structures were superimposed based on H7 and H8; thus, the opening was highlighted by the orientations of H6 and H9, respectively. D, ensemble of the calculated 20 best structures of Mg2+-CIB1. E, superimposed solution structure of Ca2+-CIB1 (red) and Mg2+-CIB1 (green) with an r.m.s.d. of 3.27 Å for the well defined areas (residues 24–136 and 146–157). F, superimposed EF-II (H4 and H5) of the solution structures of Ca2+-CIB1 (red) and Mg2+-CIB1 (green) demonstrate that Mg2+-CIB1 has a more opened conformation than Ca2+-CIB1. The two structures were superimposed based on H4; thus, the opening was highlighted by the orientations of H5. The letter N in italics indicates the N terminus of the protein.
It has been suggested that the solution structure of Mg2+-CIB1 is quite similar to that of Ca2+-CIB1 on the basis of NMR-HSQC and fluorescence spectroscopy data combined with mutagenesis results (18); however, it has also been suggested that the relatively small structural differences between Mg2+-CIB1 and Ca2+-CIB1 may contribute to their different thermodynamic behavior in their interactions with the αIIb peptide (10). Therefore, the structure determination for Mg2+-CIB1 was also conducted. The 1DNH RDC values for Mg2+-CIB1 show that there is a good correlation between the Mg2+-CIB1 and Ca2+-CIB1 solution structures (Fig. 2A) with a relatively high correlation factor of R = 0.93 and a quality factor of Q = 36%. In fact, the differences between these two structures are distributed throughout the whole protein sequence with the outliers Leu-13, Tyr-16, Glu-26, Phe-55, Asp-93, Ala-111, and Cys-134 (Fig. 2B). Because the backbone resonances of residues 158–191 are largely missing, we chose to only present the solution structure for residues 8–157 for Mg2+-CIB1. The solution structure of Mg2+-CIB1 is overall similar to the Ca2+-CIB1 solution structure with an r.m.s.d. 3.28 Å for the well defined area (residues 24–136 and 146–157) (Fig. 3E). The interhelical angles were also measured to evaluate the opening of the EF-hands in Mg2+-CIB1 (Table 2). Site EF-III in the C-domain of Mg2+-CIB1 has a similar degree of opening compared with Ca2+-CIB1. However, the interhelical angle (113.2 ± 1.1°) of site EF-II in the N-domain of Mg2+-CIB1 is slightly more open than Ca2+-CIB1 (119.9 ± 2.2°) (Fig. 3F and Table 2); this result was unexpected compared with several other calcium-binding proteins, e.g. CaBP1 (45) and CaM (33).
FIGURE 2.

Correlation between the Ca2+-CIB1 and Mg2+-CIB1 solution structures revealed by RDC analysis. The backbone NH-RDC of Mg2+-CIB1 shows a good correlation with the solution structure of Ca2+-CIB1 (A), in which the outliers are labeled. The absolute values of the difference between the observed RDC and calculated RDC are plotted as a function of the primary sequence (B), and a horizontal line highlights the outliers.
Because the structural differences between Ca2+-CIB1 and Mg2+-CIB1 are relatively small, a DDMP (Fig. 4) was used to illustrate the subtle differences. Overall, the differences are distributed all over the examined fragments (residues 8–157) with the most significant differences in helix 3b and 5 (Fig. 4). Because we have observed that the EF-II (containing H4 and H5) of Mg2+-CIB1 is more open than that of Ca2+-CIB1 (Fig. 3), the H3b is another new site showing difference between these two structures. Because site EF-I (containing H2, H3a and H3b) has several short α-helices (Fig. 1), it was not examined for an inter-helical angle.
FIGURE 4.

Difference distance matrix plot to compare the structural differences between Ca2+-CIB1 and Mg2+-CIB1 (residues 8–157). The secondary structure was labeled with filled boxes indicating helices and arrows indicating β-strands. The program DDMP gives real values of differences concerning Cα atoms in two structures. For example, the distance between the Cα atoms of i and j residues in the Ca2+-CIB1 structure is 10 Å, and the corresponding distance in structure Mg2+-CIB1 is 15 Å, and hence [(Ca2+-CIB1)-(Mg2+-CIB1)]ij = 10–15 = −5 Å. The red and blue color shading is indicated above the figure.
TEMPOL Reveals the Presence of Solvent-accessible Methyl Groups
To assess the solvent exposure of the methyl groups in the hydrophobic pocket of CIB1, we titrated the paramagnetic surface probe TEMPOL into methyl 13C,1H-labeled but otherwise deuterated Ca2+-CIB1 and Mg2+-CIB1. HSQC NMR spectra were used to monitor the effects of this soluble spin label on Ile/Leu/Val methyl resonances. The paramagnetic nitroxide group of TEMPOL is known to increase the relaxation rate of solvent-exposed residues in protein hydrophobic pockets (22, 23) thereby broadening their peaks in NMR spectra, although buried residues remain unaffected. However, if hydrophobic residues are located in a polar surface or on a flexible terminus of a protein, TEMPOL usually will not have specific interaction with such residues, and thus the intensities of these residues in NMR spectra will not be affected. In our study, 6 eq of TEMPOL were added into samples of Ca2+-CIB1 and Mg2+-CIB1. The addition of TEMPOL induces essentially no peak shifting in any of the spectra, indicating that weakly bound TEMPOL does not alter the conformation of Ca2+-CIB1 or Mg2+-CIB1 (Fig. 5). For Ca2+-CIB1, the methyl resonances of three Ile (Ile-114, Ile-73, and Ile-153), four Leu (Leu-95, Leu-131, Leu-135, and Leu-152), and three Val (Val-97, Val-132, and Val-176) residues experienced a complete loss of intensity in the presence of 6 eq of TEMPOL (Fig. 5A). We conclude that these methyl groups are located in the hydrophobic pocket of CIB1 and are exposed to the solvent. However, in the solution structure of Ca2+-CIB1, these methyl groups are normally buried in the hydrophobic pocket, and the accessibility to solvent would be shielded by the C-terminal extension of the protein, which folds back over this region in the crystal structure (readers are referred to Fig. 6B for the visualization of the C-terminal extension). A possible mechanism is that these buried hydrophobic methyl groups become exposed to TEMPOL because of the flexibility of the C-terminal extension. It seems that this hydrophobic pocket experiences an open/close switching, which allows a transient interaction of TEMPOL with certain methyl groups.
FIGURE 5.

Superimposed 13C-HSQC spectra of the Ile, Leu, and Val methyl groups of CIB1 and CIB1 bound to αIIb in the absence (black) and in the presence of 6 eq of TEMPOL (red). The same amount of TEMPOL (6 eq) was added into 12C,2H,15N-uniformly and Ile-δ1-13CH3,Leu,Val-13CH3,12CD3-labeled Ca2+-CIB1 (A) and Mg2+-CIB1 (B) samples at the same concentration (0.3 mm). The peaks marked with an asterisk are most likely from the 13C isotopic natural abundance of TEMPOL as those peaks do not appear for the protein alone.
FIGURE 6.

Using Ile-δ1,Leu-δ1,δ2,Val-γ1,γ2-methyl groups to probe the interaction between CIB1 and αIIb. The methyl CSP of CIB1 upon binding of αIIb is plotted as a function of methyl residues for Ca2+-CIB1 (A) and Mg2+-CIB1 (D). Methyl groups were mapped on the solution structure of Ca2+-CIB1 (B) and Mg2+-CIB1 (E) (residues 8–157). All methyl groups were labeled as round spheres, with nonaffected or marginally affected (CSP <0.2) methyl groups in gray, slightly affected (0.2 ≤ CSP <0.5) methyl groups in green, and significantly affected (CSP >0.5) methyl groups in red. The C-terminal extension (residues 179–191) of the Ca2+-CIB1 solution structure is colored golden to demonstrate the C-terminal displacement mechanism. C and F show the significantly affected methyl groups for Ca2+- and Mg2+-CIB1 upon binding of the αIIb peptide, respectively. The letter N in italics indicates the N terminus of the protein.
Unexpectedly, Mg2+-CIB1 appears to have more methyl groups exposed to the solvent than Ca2+-CIB1 as follows: five Ile (Ile-73, Ile-114, Ile-153, Ile-177, and Ile-189), seven Leu (Leu-19, Leu-64, Leu-95, Leu-131, Leu-135, Leu-144, and Leu-191), and three Val (Val-97, Val-132, and Val-176) (Fig. 5B). When 5 mm Ca2+ was subsequently added into the Mg2+-CIB1/TEMPOL sample to replace the Mg2+ and occupy the metal binding loops of EF-III and EF-IV of CIB1 (17), an identical HSQC spectrum was obtained compared with the Ca2+-CIB1/TEMPOL (result not shown). We conclude that the difference of the methyl group exposure is essentially caused by the intrinsic structural and dynamic differences between Mg2+- and Ca2+-CIB1.
The experiments were also done with saturating amounts of the αIIb peptide added into the TEMPOL saturated Ca2+- (supplemental Fig. 3A) and Mg2+-CIB1 (supplemental Fig. 3B) samples. Similar to the results obtained with peptide-bound CaM (22), addition of the αIIb peptide results in a regular binding mode of CIB1 in the presence of TEMPOL. In both cases (supplemental Fig. 3, A and B), Ca2+-CIB1/αIIb/TEMPOL and Mg2+-CIB1/αIIb/TEMPOL show HSQC spectra identical to the protein complex samples without TEMPOL, except for the weakened signal of the δ1 and δ2 methyl groups on Leu-95 and the γ1 methyl group on Val-132. Given that almost all methyl groups (except for Leu-95 and Val-132) have recovered their intensity compared with samples with no TEMPOL added, it seems likely that binding of the αIIb peptide sterically prevents the access of TEMPOL to the hydrophobic pockets in both Ca2+-CIB1 and Mg2+-CIB1. The solvent exposure of residues Leu-95 and Val-132 in the complexes may be caused by the remaining structural plasticity of the complex. Moreover, these data suggest that the complex structures of Ca2+-CIB1/αIIb and Mg2+-CIB1/αIIb will be quite similar.
Methyl Groups Probe the Interaction between CIB1 and αIIb
The methyl groups of the αIIb peptide-bound forms of Ca2+- and Mg2+-CIB1 have been assigned as well (supplemental Fig. 2). The binding of the αIIb peptide induces significant CSP, which are shown in Fig. 6, A and D. For Ca2+-CIB1 (Fig. 6, A–C), the methyl-containing residues Ile-114, Leu-123, Leu-131, Leu-135, Ile-168, and Ile-177 have the largest CSP values, and they are likely in the interface between CIB1 and αIIb. As expected, these residues are located on the C-domain of CIB1, which is consistent with previous suggestions that the hydrophobic pocket in the C-domain provides the primary binding site (13, 48). In addition, several methyl groups in EF-I surrounding Leu-64 have noticeable chemical shift changes upon αIIb binding, suggesting that this area in the N-domain of Ca2+-CIB1 could also be involved in the interaction with αIIb.
Mg2+-CIB1 shows a similar pattern as Ca2+-CIB1 when interacting with the αIIb peptide (Fig. 6, D–F), with the Ile-114, Leu-123, Leu-128, Leu-131, Leu-135, Ile-168, and Ile-177 having the largest CSP values. All the above residues in the C-domain of CIB1, and again some residues around Leu-64 in the N-domain are also slightly affected by the αIIb binding.
To further map the αIIb interactions with CIB1, the methyl groups were classified based on the CSP and mapped onto the solution structures of Ca2+- (Fig. 6B) and Mg2+-CIB1 (Fig. 6E). In Fig. 6B, the C-terminal extension (residues 179–191) of Ca2+-CIB1 solution structure is colored golden to demonstrate the C-terminal displacement mechanism. Overall, it seems that the major driving force for the interactions between Ca2+- and Mg2+-CIB1 is the hydrophobic interaction that occurs in the C-domain, whereas the area in the N-domain around Leu-64 is also involved in this interaction. Specifically, the 7th helix (H7) is clearly extensively involved in the interaction of CIB1 with the αIIb peptide in both the Ca2+-bound (Fig. 6C) and Mg2+-bound (Fig. 6F) states, with the majority of the methyl group CSP located on H7.
Microsecond Dynamics Reveals the Plasticity of CIB1 and the CIB1-αIIb Complex
The heteronuclear {1H}-15N NOE dynamic approach has been employed previously to study the fast motion dynamics (nanosecond to picosecond time scale) of Ca2+-CIB1, Ca2+-CIB1 in complex with the αIIb peptide (12), and Mg2+-CIB1 (29) to analyze the backbone flexibility of the CIB1 protein under different conditions. It has been widely accepted that slow motion dynamics (microsecond to millisecond time scale) is directly relevant to the biological function of many proteins (20, 49, 50). In particular, solution dynamics CPMG relaxation dispersion measurements have been used to characterize protein conformational exchange (21), ligand-binding site chemical exchange (51), and allostery mapping (32). In our studies, the CPMG slow motion dynamics experiments for the backbone residues of Ca2+-CIB1, Mg2+-CIB1, as well as Ca2+-CIB1 and Mg2+-CIB1 in complex with the αIIb peptide were carried out. The change in effective relaxation rates ΔR2,eff = R2,eff (500 Hz) − R2,eff (50 Hz) can be used to characterize the intrinsic slow motion (microsecond to millisecond time scale) of the protein as well as its contributions to exchange upon interaction with αIIb. The N- and C-domains of Ca2+-CIB1 have been observed to have different types of slow motion dynamics (Fig. 7B). The ΔR2,eff values for the N-domain of Ca2+-CIB1 are between 0 and 7 s−1 with an evenly distributed fluctuation throughout the entire N-domain (Fig. 7B), indicating the plasticity of the N-domain of Ca2+-CIB1. In contrast, the ΔR2,eff values for the C-domain are relatively more convergent, with the exceptions of the loop area between EF-III and EF-IV (residues 138–143) and the C-terminal extension (residues 184–191). Once Ca2+-CIB1 binds the αIIb peptide, Ca2+-CIB1 displays an overall increased conformational and/or chemical exchange behavior, with most residues falling in the ΔR2,eff value range of 0–15 s−1. Helices 6, 7, 9, and 10 in the C-domain of the CIB1·αIIb complex demonstrate significantly increased exchange rates upon binding the αIIb peptide, suggesting their involvement in the interaction with the αIIb peptide. In the Ca2+-CIB1·αIIb complex, the loop area in the C-domain of Ca2+-CIB1 retained slow motion flexibility, and the C-terminal end has acquired increased slow motion with residues 174–191 having a significantly higher ΔR2,eff (Fig. 7C) than the nearby C-domain (residues 160–173). These observations provide additional support for the proposed C-terminal displacement mechanism (12). The slow motional on/off switching of the C-terminal extension not only allows for the interactions between Ca2+-CIB1 and the αIIb peptide, it also prevents nonspecific interactions between other nonpolar substances and the hydrophobic pocket of the C-domain of Ca2+-CIB1 (12).
FIGURE 7.

Slow motion dynamics (microsecond to millisecond) backbone CPMG experiments of both CIB1 and CIB1 complexed with αIIb. A, secondary structure of Ca2+-CIB1, and microsecond dynamics of Ca2+-CIB1 (B) and Ca2+-CIB1 (C) in complex with αIIb; D, secondary structure of Mg2+-CIB1, and microsecond time scale dynamics of Mg2+-CIB1 (E) and Mg2+-CIB1 (F) in complex with αIIb. These experiments were acquired on a 2H,15N-uniformly labeled CIB1 at 37 °C, pH 7.5, at a field strength of 500 MHz. The errors were obtained based on the analysis of duplicate experiments.
Similarly, the Mg2+-CIB1·αIIb complex is more dynamic than Mg2+-CIB1 alone (Fig. 7, E and F). Although the C-terminal end (residues 158–191) of Mg2+-CIB1 could only be partially assigned (18), the corresponding part of Mg2+-CIB1, once complexed with the αIIb peptide, can be almost fully assigned. Apparently, the presence of the αIIb peptide increases the binding affinity of the EF-IV loop for Mg2+ resulting in the stabilization of the structure in the EF-IV region (29). Comparison of the ΔR2,eff of Mg2+-CIB1 and the αIIb-bound form of the protein reveals that H7 and H9 could be directly involved in the interaction with the αIIb peptide, which is consistent with the CSP results (Fig. 6F). In addition, for both Ca2+- and Mg2+-CIB1, H0 and partially H1 in the N-terminal domain have enhanced exchange upon binding αIIb, which might imply the involvement of the N-terminal myristoyl group in these interactions, and this has been observed in the case of sphingosine kinase 1 (5).
DISCUSSION
In this study, the solution structures of Ca2+-CIB1 and Mg2+-CIB1 were determined by using several solution NMR restraints, including two sets of backbone RDCs and NOEs (NH-NH, NH-methyl, and methyl-methyl). The Ca2+-CIB1 structure that was obtained shows a relatively small deviation from the 1XO5 crystal structure, with an r.m.s.d. of 2.71 Å. However, the opening of sites EF-III and EF-IV in Ca2+-CIB1 solution structure are smaller than in 1XO5 by ∼10°, which is actually similar to what has been observed for CaM (46). In the case of 1XO5, 300 mm Ca2+ was necessary to induce crystallization, thus over 99.99% CIB1 molecules are Ca2+-saturated given the Ca2+/CIB1 dissociation constant Kd ∼10−6 m (10). For our solution NMR experiments, only 4 mm Ca2+ was used to mimic physiological conditions; thus, it is possible that our NMR results represent an ensemble of Ca2+-occupied CIB1 and apo-CIB1. In addition, in 1XO5, the C-terminal extension is helical, folds back, and has a tight contact with the hydrophobic pocket of the C-domain, whereas in solution the C-terminal extension experiences opening/closing motions as suggested by the relaxation dispersion results described herein. It has also been reported that a weak calcium-binding site exists in this region of CIB1, which affects the local folding (39). It appears that the large degree of opening of EF-III and EF-IV in 1XO5 is stabilized by the C-terminal extension being bound to the hydrophobic pocket. Furthermore, the opening of sites EF-III (116.6°) and EF-IV (112.2°) in the other available crystal structure 1Y1A is more closed than 1XO5 (EF-III 113.3° and EF-IV 109.0°) but slightly larger than the solution structure by about 6–10° (Table 2). For the EF-II in the N-domain of Ca2+-CIB1, 1XO5 has the same degree of opening as the solution structure, and both are 116–119°.
When comparing the solution structures of Ca2+- and Mg2+-CIB1, site EF-III in the C-domain of the protein has essentially the same degree of opening (Table 2), but Mg2+-CIB1 has a more open conformation for site EF-II in the N-domain than Ca2+-CIB1. Other EF-hand proteins, such as CaM (46, 52) and CaBP1 (45), possess a more open conformation when bound to Ca2+; hence, the influx of Ca2+ into the cytoplasm of the cell can regulate the interactions of these proteins with their specific binding targets in a calcium-dependent manner. Interestingly, in the case of CIB1, Mg2+-CIB1 has a larger opening of the EF-II site, whereas its EF-III site is the same as Ca2+-CIB1, causing Mg2+-CIB1 to have a larger solvent-exposed hydrophobic area. Consistent with this, more methyl resonances of Mg2+-CIB1 are affected by the addition of the surface probe TEMPOL. The discovery that the Mg2+-CIB1 solution structure has a larger exposed hydrophobic surface area than the Ca2+-CIB1 solution structure is also consistent with the results of fluorescence experiments using the probe 1-anilino-8-naphthalene sulfonate (ANS) to detect its nonpolar interaction with apo-, Mg2+-, and Ca2+-CIB1, in which the Mg2+-CIB1 has a more substantial interaction with ANS than apo- and Ca2+-CIB1 (17).
Another interesting observation is that the structural differences between Ca2+-CIB1 and Mg2+-CIB1 seem to be mainly in the N-domain (EF-I and EF-II) as revealed by DDMP (Fig. 4), although the metal ion-binding sites are exclusively in the C-domain. The allosteric effects of these divalent metal ions on CIB1 make CIB1 quite different compared with other calcium-binding proteins, e.g. CaM and CaBP1.
For the interaction of CIB1 and the αIIb peptide, even though both the backbone (12) and methyl CSP suggest that the hydrophobic interactions between the N-terminal end of the αIIb peptide and the C-domain of the CIB provide the major driving force, the N-domain of CIB1 is still likely involved in this interaction, e.g. with Arg-33 (12), forming electrostatic salt bridges. Ca2+-CIB1 has been predicted to take an orientation with the α-helical N-terminal end of the αIIb peptide buried into the hydrophobic pocket of Ca2+-CIB1 and the negatively charged and extended C-terminal end of the αIIb peptide interacting with the N-domain of Ca2+-CIB1 (53). We suggest that the side chain of Lys-65 also forms electrostatic interactions with αIIb based on the relatively large chemical shift changes observed around Leu-64 (Fig. 6). The regulation of integrin activity is intriguing, and a role for CIB1 in the regulation of αIIbβ3 activation has been suggested by several groups (3, 11). Salt bridges (e.g. αIIb (Arg-995)/β3 (Asp-723) and αIIb (Arg-995)/β3 (Glu-726)) and aromatic ring stacking (e.g. αIIb (Phe-992 to Phe-993)/β3 (Trp-715)) between αIIb and β3 of integrin are important in maintaining the heterodimeric structure of the αIIb·β3 complex (54, 55). We suggest that CIB1 could interrupt these interactions between αIIb and β3 and further dissociate the αIIb and β3, resulting in the activation of integrin.
It has been discussed that there are three typical mechanisms for the regulation of neuronal calcium censor proteins (56), which is a protein family homologous to CIB1 as follows: (i) the calcium-independent type, e.g. frequenin; (ii) the calcium-myristoyl switch, e.g. recoverin; and (iii) a relatively small structural but substantial change in dynamics, e.g. GCAP1. Like the neuronal calcium censor proteins, CIB1 possesses two lobes that interact with each other, unlike CaM, where the two lobes rotate almost independently (57). However, based on what we observed in these studies, it seems that CIB1 has a distinct regulatory mechanism, which resembles the aforementioned type (iii) mechanism with relatively small structural differences between Ca2+-CIB1 and Mg2+-CIB1 and slightly different degree opening of site EF-II. The CPMG experiments also indicate the flexible loop between EF-III and EF-IV (residues 138–145) of Mg2+-CIB1, and Ca2+-CIB1 might be involved in the regulation of the activities of CIB1 by working as a spring-like apparatus to adjust the size of the hydrophobic pocket in the C-domain upon binding various targets. Taken together, the comparison of the solution structures of Ca2+-CIB1 and Mg2+-CIB1 leads to the conclusion that Mg2+-CIB1 in the cytoplasm has a more open and more dynamic conformation compared with Ca2+-CIB1, which is likely to contribute to its physiological functions.
Supplementary Material
Acknowledgments
The Bio-NMR Center at the University of Calgary was maintained through funds provided by the CFI-IOF program. We thank Dr. Deane McIntyre for maintaining the NMR instrumentation. Differences distance matrix plots were generated using the DDMP program from the Center for Structural Biology at Yale University.
This work was supported in part by the Canadian Institutes of Health Research.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3 and additional references.
- RDC
- residual dipolar coupling
- HSQC
- heteronuclear single quantum coherence
- CSP
- chemical shift perturbation
- CaM
- calmodulin
- TEMPOL
- tempol 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl
- DDMP
- difference distance matrix plot
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Gifford J. L., Walsh M. P., Vogel H. J. (2007) Biochem. J. 405, 199–221 [DOI] [PubMed] [Google Scholar]
- 2. Yamniuk A. P., Vogel H. J. (2006) Calcium Bind. Proteins 1, 150–155 [Google Scholar]
- 3. Leisner T. M., Yuan W., DeNofrio J. C., Liu J., Parise L. V. (2007) Curr. Opin. Hematol. 14, 255–261 [DOI] [PubMed] [Google Scholar]
- 4. Naik U. P., Patel P. M., Parise L. V. (1997) J. Biol. Chem. 272, 4651–4654 [DOI] [PubMed] [Google Scholar]
- 5. Jarman K. E., Moretti P. A., Zebol J. R., Pitson S. M. (2010) J. Biol. Chem. 285, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leisner T. M., Liu M., Jaffer Z. M., Chernoff J., Parise L. V. (2005) J. Cell Biol. 170, 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon K. W., Cho J. H., Lee J. K., Kang Y. H., Chae J. S., Kim Y. M., Kim J., Kim E. K., Kim S. E., Baik J. H., Naik U. P., Cho S. G., Choi E. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 17389–17394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kauselmann G., Weiler M., Wulff P., Jessberger S., Konietzko U., Scafidi J., Staubli U., Bereiter-Hahn J., Strebhardt K., Kuhl D. (1999) EMBO J. 18, 5528–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heineke J., Auger-Messier M., Correll R. N., Xu J., Benard M. J., Yuan W., Drexler H., Parise L. V., Molkentin J. D. (2010) Nat. Med. 16, 872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamniuk A. P., Vogel H. J. (2005) Protein Sci. 14, 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsuboi S. (2002) J. Biol. Chem. 277, 1919–1923 [DOI] [PubMed] [Google Scholar]
- 12. Yamniuk A. P., Ishida H., Vogel H. J. (2006) J. Biol. Chem. 281, 26455–26464 [DOI] [PubMed] [Google Scholar]
- 13. Barry W. T., Boudignon-Proudhon C., Shock D. D., McFadden A., Weiss J. M., Sondek J., Parise L. V. (2002) J. Biol. Chem. 277, 28877–28883 [DOI] [PubMed] [Google Scholar]
- 14. Blamey C. J., Ceccarelli C., Naik U. P., Bahnson B. J. (2005) Protein Sci. 14, 1214–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gentry H. R., Singer A. U., Betts L., Yang C., Ferrara J. D., Sondek J., Parise L. V. (2005) J. Biol. Chem. 280, 8407–8415 [DOI] [PubMed] [Google Scholar]
- 16. Stabler S. M., Ostrowski L. L., Janicki S. M., Monteiro M. J. (1999) J. Cell Biol. 145, 1277–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamniuk A. P., Nguyen L. T., Hoang T. T., Vogel H. J. (2004) Biochemistry 43, 2558–2568 [DOI] [PubMed] [Google Scholar]
- 18. Yamniuk A. P., Silver D. M., Anderson K. L., Martin S. R., Vogel H. J. (2007) Biochemistry 46, 7088–7098 [DOI] [PubMed] [Google Scholar]
- 19. Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kay L. E. (2005) J. Magn. Reson. 173, 193–207 [DOI] [PubMed] [Google Scholar]
- 21. Loria J. P., Rance M., Palmer A. G. (1999) J. Am. Chem. Soc. 121, 2331–2332 [Google Scholar]
- 22. Yuan T., Ouyang H., Vogel H. J. (1999) J. Biol. Chem. 274, 8411–8420 [DOI] [PubMed] [Google Scholar]
- 23. Howarth J. W., Krudy G. A., Lin X., Putkey J. A., Rosevear P. R. (1995) Protein Sci. 4, 671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tugarinov V., Kanelis V., Kay L. E. (2006) Nat. Protoc. 1, 749–754 [DOI] [PubMed] [Google Scholar]
- 25. Ottiger M., Bax A. (1998) J. Am. Chem. Soc. 120, 12334–12341 [Google Scholar]
- 26. Yang D. W., Venters R. A., Mueller G. A., Choy W. Y., Kay L. E. (1999) J. Biomol. NMR 14, 333–343 [Google Scholar]
- 27. Zweckstetter M., Bax A. (2000) J. Am. Chem. Soc. 122, 3791–3792 [Google Scholar]
- 28. Yang D., Zheng Y., Liu D., Wyss D. F. (2004) J. Am. Chem. Soc. 126, 3710–3711 [DOI] [PubMed] [Google Scholar]
- 29. Yamniuk A. P., Gifford J. L., Linse S., Vogel H. J. (2008) Biochemistry 47, 1696–1707 [DOI] [PubMed] [Google Scholar]
- 30. Neri D., Szyperski T., Otting G., Senn H., Wüthrich K. (1989) Biochemistry 28, 7510–7516 [DOI] [PubMed] [Google Scholar]
- 31. Grzesiek S., Bax A., Clore G. M., Gronenborn A. M., Hu J. S., Kaufman J., Palmer I., Stahl S. J., Wingfield P. T. (1996) Nat. Struct. Biol. 3, 340–345 [DOI] [PubMed] [Google Scholar]
- 32. Das R., Abu-Abed M., Melacini G. (2006) J. Am. Chem. Soc. 128, 8406–8407 [DOI] [PubMed] [Google Scholar]
- 33. Chou J. J., Li S., Bax A. (2000) J. Biomol. NMR 18, 217–227 [DOI] [PubMed] [Google Scholar]
- 34. Schwieters C. D., Kuszewski J. J., Clore G. M. (2006) Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 36. Wishart D. S., Sykes B. D., Richards F. M. (1992) Biochemistry 31, 1647–1651 [DOI] [PubMed] [Google Scholar]
- 37. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 38. Richards F. M., Kundrot C. E. (1988) Proteins 3, 71–84 [DOI] [PubMed] [Google Scholar]
- 39. Yamniuk A. P., Anderson K. L., Fraser M. E., Vogel H. J. (2009) Protein Sci. 18, 1128–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohki S., Ikura M., Zhang M. (1997) Biochemistry 36, 4309–4316 [DOI] [PubMed] [Google Scholar]
- 41. Huang H., Ishida H., Vogel H. J. (2010) Protein Sci. 19, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weljie A. M., Yamniuk A. P., Yoshino H., Izumi Y., Vogel H. J. (2003) Protein Sci. 12, 228–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pennestri M., Melino S., Contessa G. M., Casavola E. C., Paci M., Ragnini-Wilson A., Cicero D. O. (2007) J. Biol. Chem. 282, 667–679 [DOI] [PubMed] [Google Scholar]
- 44. Park C. J., Lee J. H., Choi B. S. (2005) Nucleic Acids Res. 33, 4172–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li C., Chan J., Haeseleer F., Mikoshiba K., Palczewski K., Ikura M., Ames J. B. (2009) J. Biol. Chem. 284, 2472–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chou J. J., Li S., Klee C. B., Bax A. (2001) Nat. Struct. Biol. 8, 990–997 [DOI] [PubMed] [Google Scholar]
- 47. Ishida H., Huang H., Yamniuk A. P., Takaya Y., Vogel H. J. (2008) J. Biol. Chem. 283, 14619–14628 [DOI] [PubMed] [Google Scholar]
- 48. Denofrio J. C., Yuan W., Temple B. R., Gentry H. R., Parise L. V. (2008) Thromb. Haemost. 100, 847–856 [PMC free article] [PubMed] [Google Scholar]
- 49. Boehr D. D., Dyson H. J., Wright P. E. (2006) Chem. Rev. 106, 3055–3079 [DOI] [PubMed] [Google Scholar]
- 50. Palmer A. G., 3rd, Kroenke C. D., Loria J. P. (2001) Methods Enzymol. 339, 204–238 [DOI] [PubMed] [Google Scholar]
- 51. Mulder F. A., Hon B., Muhandiram D. R., Dahlquist F. W., Kay L. E. (2000) Biochemistry 39, 12614–12622 [DOI] [PubMed] [Google Scholar]
- 52. Zhang M., Tanaka T., Ikura M. (1995) Nat. Struct. Biol. 2, 758–767 [DOI] [PubMed] [Google Scholar]
- 53. Hwang P. M., Vogel H. J. (2000) J. Mol. Recognit. 13, 83–92 [DOI] [PubMed] [Google Scholar]
- 54. Lau T. L., Kim C., Ginsberg M. H., Ulmer T. S. (2009) EMBO J. 28, 1351–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T., Plow E., Qin J. (2002) Cell 110, 587–597 [DOI] [PubMed] [Google Scholar]
- 56. Orban T., Bereta G., Miyagi M., Wang B., Chance M. R., Sousa M. C., Palczewski K. (2010) Structure 18, 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barbato G., Ikura M., Kay L. E., Pastor R. W., Bax A. (1992) Biochemistry 31, 5269–5278 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.