

Abstract
Experimental data has shown that mesenteric lymph from rats subjected to trauma-hemorrhagic shock (THS) but not trauma-sham shock (TSS) induces neutrophil activation, cytotoxicity, decreased red blood cell deformability and bone marrow colony growth suppression. These data have lead to the hypothesis that gut factors produced from THS enter the systemic circulation via the mesenteric lymphatics and contribute to the progression of Multiple Organ Failure (MOF) following THS. Ongoing studies designed to identify bioactive lymph agents implicated factors associated with the heparin use in the THS procedure. We investigated if heparin itself was responsible for reported toxicity to human umbilical vein endothelial cells (HUVECs). HUVEC toxicity was not induced by lymph when alternate anti-coagulants (citrate and EDTA) were used in THS. HUVEC toxicity was induced by lymph after heparin but not saline or citrate injection into TSS and naïve animals and was dose dependent. Activities of both heparin-releasable lipases (lipoprotein (LPL) and hepatic (HL)) were detected in the plasma and lymph from THS and naïve animals receiving heparin but not citrate or saline. Lymph-induced HUVEC toxicity correlated with lymph lipase activities. Finally, incubation of HUVECs with purified LPL added to naïve lymph induced toxicity in vitro. These data show that heparin, not THS, is responsible for the reported lymph-mediated HUVEC toxicity through its release of lipases into the lymph. These findings can provide alternative explanations for several of the THS effects reported in the literature using heparin models thus necessitating a review of previous work in this field.
Keywords: Multiple Organ Failure, lipoprotein lipase, hepatic lipase, chylomicrons, endothelial injury
Introduction
Multiple organ failure (MOF) is the cause of 50–80% of deaths in the surgical intensive care units (1). Because the development of MOF is poorly understood, current treatment is mainly supportive (2). It is therefore critical to identify key events and factors that lead to the progression of MOF in order to identify new and important points of intervention.
Glenn and Lefer (3) first proposed that deleterious factors created in the hypoperfused splanchnic region due to trauma with hemorrhagic shock (THS) were transported to the systemic circulation via the lymphatics. These authors theorized that proteolytic enzymes from the ischemic pancreas created a myocardial depressant factor localized in thoracic lymph post-THS based upon the finding that pancreatic enzymes were detected in lymph (4). This concept has been expanded in recent years in the Deitch and Moore laboratories (5,6) after clinical data emerged correlating gut permeability with extent of the injury in patients (7–10). It is currently hypothesized that THS end-organ damage results from factors derived from the ischemic gut that enter the mesenteric lymphatics and are introduced to the general circulation via the subclavian vein. This theory is supported by in vivo data showing that lymph duct ligation or diversion abrogates THS-induced lung injury (11,12), neutrophil activation (PMNs, 13, 14), increased rigidity of red blood cells (RBCs,15), endothelial cell dysfunction (16–18) and bone marrow colony suppression (BM,19). Furthermore, incubation of THS but not sham-shocked (TSS) lymph with PMNs, RBCs, BM and endothelial cells in vitro could reproduce in vivo effects (12,13,15,19–21). More recently, it was shown that lung injury could be recreated in animals after injection of THS but not TSS lymph (22).
Isolation studies designed to identify the bioactive components in THS lymph are ongoing and both lipid and protein components have been implicated for neutrophil activation and cytotoxicity (8,23,24). Our current isolation methods have implicated lipoproteins in the induction of post-THS lymph-mediated human umbilical vein endothelial cell (HUVEC) toxicity (unpublished). This finding necessitated the investigation into the role of heparin used in the THS but not TSS procedure since it is well-established that heparin promotes lipoprotein metabolism through its release of lipoprotein (LPL) and hepatic (HL) lipases into the circulation (25, 26). Furthermore, LPL activity has been linked to endothelial cell damage in atherosclerosis (27) and cytotoxicity to macrophages and endothelial cells in vitro when active within hypertriglyceremic serum and triglyceride-rich lipoproteins (28,29). We therefore tested the hypothesis that HUVEC toxicity induced by mesenteric lymph is dependent on heparin and results from heparin’s release of lipases and not to THS itself.
Material and Methods
Animals
Pathogen-free, male Sprague-Dawley rats (Charles River, Troy, NY) were maintained under 12h light/dark cycles in barrier-sustained conditions, and allowed access to water and chow ad libitum (Teklad 22/5 rodent diet W-8640, Harlan Teklad, Madison, WI). For experiments using fasted animals, food was withdrawn 18h prior to surgery. Procedures were approved by the New Jersey Medical School Animal Care Committee and animals were maintained in accordance with the “Guide for the Care and Use of Laboratory Animals”.
Lymph collection
Male rats (avg wt 380±40 g) were anesthetized with pentobarbital (50 mg/kg, IP) and secured to an operating board (Fisher, Pittsburgh, PA) which was placed on a heating pad to maintain the animal’s temperature. Lymph duct cannulation and collection was as previously described (30) with minor modifications: 1) The silastic tubing (ID 0.51mm, Dow Corning, Midland, MI) used to cannulate the mesenteric lymph duct was flushed with saline rather than heparin, was decreased in length from 50cm to 8cm and was secured in place using 6-0 silk suture rather than glue; 2) lymph was collected by placing the end of the lymph catheter into a sterile 1cc syringe (BD Biosciences, Franklin Lakes, NJ) that was secured to an ice pack (Polar Tech Industries, Inc., Genoa, Il); 3) lymph volumes (avg range: 0.2–0.45ml) were determined using the syringe calibrations, then the lymph was transferred to a sterile 1.5ml eppendorf tube (VWR, Westchester, PA) and an antiprotease cocktail (Sigma, St. Louis, MO) was added (1:100 v/v) to the sample, which was then mixed by inversion; and 4) cells were removed from lymph by centrifugation in a tabletop microfuge (8,000×g, 2min, 4°C) and a 12ul aliquot was dispensed into a 0.5ml eppendorf tube and placed on ice for cell viability testing. In most cases, cell viability was performed on the same day as collection.
Naïve lymph used for in vitro experiments was collected for 4h then processed as above, stored at 4°C and used within 72h.
In experiments examining the effects of lymph duct ligation (LDL), the isolated lymph duct was ligated with 6-0 silk suture prior to abdominal closure.
Trauma-Hemorrhagic shock (THS)
In experiments using the standard fixed-pressure shock model (17), after the laparotomy to cannulate the lymphatic duct (trauma), the external jugular vein was isolated and cannulated with silastic tubing and the femoral artery was cannulated with polyethylene tubing (PE-50, ID 0.58mm, Becton Dickinson, Sparks, MD, 20cm in length). Both cannulas were filled (primed) with heparinized saline (100U/ml). The femoral catheter for THS animals was attached to a blood pressure recorder (BPA Blood Pressure Analyzer, Micro-Med, Louisville, KY) used to continuously monitor the animals’ blood pressure during the shock period. The animals were subjected to THS or TSS after a 30min stabilization period.
Hemorrhagic-shock was induced by the withdrawal of blood from the external jugular vein to a mean arterial pressure (MAP) of 30mmHg into a 10cc syringe containing heparin (heparin amounts are provided in the figure legends for specific experiments). Blood was withdrawn or reinfused during this period as needed to maintain this blood pressure for 90min. At the end of the shock period, the animals were resuscitated by reinfusion of all shed blood using a syringe pump (1ml/min, KD Scientific, Holliston, MA). The sham-shock animals were subjected to the same surgical procedures but no blood was withdrawn or reinfused nor was pressure monitored. Throughout the procedure, the animal’s temperature was maintained with the use of heating pads.
In THS procedures with alternate anti-coagulants or Lactated Ringer’s solution as a resuscitation fluid, all lines were primed with either normal saline, sodium citrate or EDTA diluted in saline (1:10 dilution from BD Vacutainers, Franklin Lakes, NJ) and sodium citrate or ethylenediaminetetraacetic acid (EDTA) replaced heparin in the syringe for blood withdrawal. For clarity, additional details are provided in the figure legends for these specific experiments.
In THS procedures without blood pressure monitoring, the femoral catheter was omitted. The jugular catheter was primed with saline. Blood was withdrawn into separate heparinized syringes in volumes and at times analogous to the standard-fixed pressure model as follows: 5ml (1min), 0.5ml (10, 15, 20, 25 and 30min), 0.3ml (35, 40, 50, 60, 70min). A total volume of 9ml was withdrawn based upon an average withdrawal of 9.9ml calculated for 30 rats. Animals were resuscitated with Lactated Ringer’s solution as described above. No anticoagulant was given to the animals at any point.
Heparin injection
In experiments in which the effects of heparin alone were investigated, the femoral catheter was omitted. Heparin, saline or citrate was injected into the external jugular vein using a 1cc syringe. The final calculated volume of heparin per weight (250, 125, 60 or 30U/kg) was increased by 50ul to account for the void volume of the syringe tip and catheter. For clarity, additional details are provided in the figure legends for these specific experiments.
Plasma collection
Plasma used in lipase measurements was obtained from blood withdrawn (100ul) at 30min intervals from the external jugular vein into a 1cc syringe with no anticoagulant. The plasma sample collected during THS, was taken from the syringe used for blood withdrawal immediately prior to reinfusion of the shed blood. After each collection, samples were immediately subjected to centrifugation (8,000×g, 2min, 4°C), transferred to a sterile eppendorf tube then stored at −80°C.
For in vitro incubation assays, naïve non-heparinized and post-THS plasmas were obtained by exsanguination from the ascending aorta into a 10cc syringe containing 100U of heparin. Cells were removed by centrifugation (8,000×g, 10min, 4°C) and plasma was aliquoted and stored at −40° or −80°C. Heparinized rat plasma was collected 30min after a bolus injection of heparin (250U/kg) in the same manner as other plasmas.
Superior Mesenteric Arterial Occlusion (SMAO)
In experiments investigating the source of lymph lipases, the superior mesenteric artery was occluded as previously described (31) except occlusion was for 60min. The lymph duct and external jugular vein were cannulated as described above. Heparin (250U/kg) was injected into the external jugular vein using a 1cc syringe immediately after occlusion. Lymph was collected for 30min prior to occlusion, 60min during occlusion-post-heparin injection, and for six 30min intervals following reperfusion of blood. Lymph was processed as described above.
Reagents and cell lines
Human umbilical vein endothelial cells (HUVECs), endothelial growth media (EGM), endothelial basal media (EBM), and Hank’s Balanced Salt Solution (HBSS) were from Lonza Walkersville (Walkersville, MD). The mitochondrial cell viability assay (MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide), antiprotease cocktail, gum arabic, isopropanol, heptane, unlabeled triolein (glyceryl trioleate), NaCl, SDS, BSA (fraction V), Orlistat and sodium oleate were purchased from Sigma (St. Louis, MO). The lipoprotein lipase monoclonal antibody (5D2) was purchased from Roar Biomedical (NY, NY). Mouse isogenic control IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Heparin (1000U/ml) was purchased from Cardinal Health (Dublin, OH). Labeled tri[9,10(n)-3H]oleate and scintillation fluid (Optiphase HiSafe 3) were from PerkinElmer (Boston, MA). Tris base was from Invitrogen (Carlsbad, CA).
Endothelial cell Viability Assay
The MTT cell viability assay was performed according to the manufacturer’s instructions. Due to their availability, HUVECs were used in viability assays since previous work has shown that THS lymph affects HUVECs to the same degree as rat pulmonary microvascular endothelial cells (32). Briefly, HUVECs (passage three or four) were grown to 100% confluency in 75cm2 tissue culture flasks (Corning, NY) then trypsinized and resuspended in EGM at 2.5×105 cells/ml. Cells were seeded (100ul/well) onto 96-well tissue culture plate (Corning, NY) and the plate was placed in a humidified incubator over night at 37°C/5%CO2. Lymph was tested at 3% or 5% v/v. Cells incubated with 3–5% fetal calf serum or 10% DMSO served as negative or positive controls for cell death, respectively. After incubation (3h or 18h) at 37°C/5%CO2 the lymph/media were removed and replaced with 90ul EBM and 10ul of a 5mg/ml MTT solution. The formazen crystals were solubilized after additional incubation at 37°C/5%CO2. The absorbance570 (SpectraMax 384 Plus, Molecular Dynamics, Sunnyvale, CA) was determined and viability was calculated as a percent of the negative control. For clarity, specific lymph volumes and incubation times are detailed in figure legends.
Lipase measurements
Measurements of LPL and HL were made using a gum Arabic emulsion as described (33) with adjustments made for a smaller sonication probe. Briefly, unlabeled (6.25mg) and labeled tri[9,10(n)-3H] oleate (8×106 dpm) were dispensed into the bottom of a disposable 16x100mm boroscilicate tube (Corning, Corning, NY) and evaporated under a stream of nitrogen. Gum Arabic and Tris-Cl (pH8.5) were added to final concentrations of 2.35% and 0.3M, respectively, in a final volume of 1.0625ml. The tube was placed in a beaker packed with ice and sonicated using a 3mm diameter probe (Vibra Cell, Sonics and Materials, Danbury, CT) using a 50% pulsed mode at medium setting until no oil droplets were visible on the surface (2 × 10min pulses). Emulsions were placed on ice and used within 3h.
In assays for the measure of HL, LPL was inactivated using high salt (1.3M NaCl) in the assay buffer. Samples (30ul naïve or pre-heparin plasma or lymph, 3ul or 10ul post-heparin plasma or lymph, respectively) were diluted in 10mM Tris-Cl (pH7.7) to a final volume of 50ul. For the measure of LPL, HL was inactivated by diluting the sample (3ul) 1:1 with 10mM Tris-Cl (pH 7.7) followed by a 1:1 dilution with 0.721% SDS and incubated at 28C for 1h followed by dilution as above. No HL activity was detected in control experiments with heparinized plasma using this method. Higher volumes of sample could not be used in the LPL assays due to interference by SDS. Duplicate sample measures were made in all assays.
Lipase assay solutions were made by mixing the emulsion with NaCl (5M in 0.2M Tris-Cl, pH8.8), BSA (20% in water, pH 8.4) and heat inactivated (HI) rat serum (56°C for 30min) to final concentrations of 2mg/ml triolein, 8% BSA, 0.15M NaCl (LPL)/1.3M NaCl (HL), 9% HI rat serum (LPL), 0.02% (w/v) heparin (LPL) and water to volume. Assay solution (150ul) was dispensed into boroscilicate tubes and incubated for 5min in a shaking water-bath (50rpm, 30°C). The assay was initiated by the addition of sample (50ul) prepared as above. Each reaction tube was individually timed. Reactions were stopped and fatty acids were extracted according to Dole (34) as described (33). Radioactive counts were determined as described (35) and activity was calculated according to Briquet-Laugier, et al (36) and expressed as nmoles of fatty acids released in 1min per 1ml (nmole FA/min/ml).
Isolation of bovine LPL
Bovine LPL was isolated from milk as described (37). Bovine milk was a generous gift from Rob Kibbe at Fosterfields Farm, Morristown, NJ.
Calcium measurements
Free calcium was measured in lymph using the Quantichrom Calcium Assay kit (BioAssay Systems, Hayword, CA) according to instructions. Interference due to the opacity of lymph was controlled for in 2 ways: 1) the kit buffer contains reagents to limit interference with lipids/chylomicrons and 2) the OD612 of corresponding time points in THS-hep & THS-cit samples were compared in a water background. Since there was no significant difference in this value, any interference from opacity was equal between like samples. Since our measurements were relative, and not absolute, any errors using a buffer-based standard were negated.
pH measurements
The pH was measured in each lymph sample immediately after each lymph collection period following centrifugation and antiprotease addition using a Symphony pH meter (VWR, Westchester, PA) with an Orion micro pH electrode (Thermo Fisher Scientific, Waltham, MA).
Lipoprotein profile analysis
Mesenteric lymph (2ul) from individual time points was applied to Titan gels (Helena Labs, Beaumont, TX) and electrophoresed and stained according to manufacturer’s instructions. Dried gels were photographed and densitometry was performed using an AlphaImager IS-3400 (AlphaInnotech, San Leandro, CA). A box of equal size was drawn in each gel lane corresponding to the area of the fast migrating band observed after heparin injection. Densities were standardized to the pre-injection lane and data are expressed as a ratio of the integrated density value for each band from each time point relative to the pre-injection standard. Background values were determined by the software.
Creation of cytotoxic lymph in vitro
Lipase activities were measured in all plasmas and LPL activity was normalized to either 40 or 115 nmoles fatty acid (FA)/min/ml in each reaction tube (final volume 20ul). Plasma was mixed with 1× PBS (pH 7.6) to a final volume of 13ul in 0.5ml eppendorf tubes and placed on ice. Working with each tube separately, 7ul of naïve lymph was added to the plasma, mixed, then dispensed in duplicate aliquots (8.6ul) to endothelial cells to achieve a final lymph concentration of 3% v/v. No more than 15min passed between the first and last sample.
For lipase inhibition studies, the LPL monoclonal antibody (5D2, 0.08–0.16mg/ml final concentration) or Orlistat (252uM final concentration) was incubated with plasma, lymph or purified LPL on ice. After 1h, PBS (pH7.7) was added to a volume of 13ul followed by addition of lymph and tested as described above. Mouse IgG and solvent (10% DMSO/90% EtOH) served as negative controls for the antibody and Orlistat, respectively.
For chylomicron (CM) depletion experiments, naïve lymph (2 tubes of 0.25ml each) was subjected to centrifugation (10,000×g, 10min, 21°C). The subnatant (125ul) from each tube was removed and combined in a fresh tube (CM depleted, CMD). The remaining CM enriched (CME) lymph (125ul) from both tubes was combined. This was repeated two more times to obtain equal volumes (250ul) of CMD and CME. An additional sample was remixed after each centrifugation and was used as the whole lymph control.
For oleic acid experiments, naïve lymph (18ul) was incubated with 2ul of oleic acid stock (in water) to yield final concentrations ranging from 0.328uM-32.8mM. Samples were analyzed on Titan gels as described above.
In studies investigating if heparin alone induced HUVEC toxicity, heparin (1U, 2U and 4U/ml, final concentration) was incubated with HUVECs for 3h. In studies investigating if heparin added to naïve lymph could induced HUVEC toxicity, the same concentrations of heparin were incubated with naïve lymph and incubated at 37°C to mimic in vivo effects. Aliquots were removed at timed intervals (0, 30, 60, 90 and 120min) and incubated with HUVECs (3% v/v) for 3h. The high concentration of 4U/ml was based upon the assumption that a 400g rat receiving 250U/kg of heparin (100U total) would have a maximum of 4U of heparin/ml in 24ml (blood volume (ml) = 0.06 * body weight (g) + 0.77) (38) of circulating blood.
Statistical analysis
Analysis of data was performed using Sigma-Plot 3.0 using a Student’s paired t-test or repeated measures analysis of variance (ANOVA) at an overall significance level of P = 0.05. The Tukey method was used for pairwise multiple comparisons within a group using at an overall significance level of P = 0.05. The correlation between lipase activities and endothelial cell viability was determined using Sigma-Plot 3.0 Spearman Rank Order Correlation Test at an overall significance level of P = 0.05.
RESULTS
Endothelial cell toxicity is the direct result of heparin injection and not of THS
In order to discern the role of heparin in mesenteric lymph-mediated HUVEC toxicity in a THS model, blood was withdrawn into a syringe containing either heparin (THS-hep, approx. 100U) or sodium citrate (THS-cit, 1.5ml of 0.105M sodium citrate) as an anticoagulant in amounts sufficient to inhibit clot formation in 10ml of whole blood. Control TSS animals received an equal volume and concentration of the respective anticoagulant at the end of the TSS period but no blood was withdrawn. Jugular and femoral cannulas were both primed with sodium citrate in both the THS and TSS groups. Lymph collected from each group was tested for induction of HUVEC toxicity. Toxicity to HUVECs was only induced by lymph from animals in which heparin was used as an anticoagulant in blood withdrawal (Fig. 1A). Although the anticoagulant properties of citrate are due to its ability to chelate calcium, no overall significance was found in calcium levels between THS rats receiving citrate and those receiving heparin (data not shown).
Fig. 1.





Trauma hemorrhagic-shock (THS) mesenteric lymph does not induce endothelial cell toxicity in the absence of heparin. Animals were subjected to THS as described in Materials and Methods using either 100U of heparin (THS-hep, in 100ul, N=4), 1.5ml of 0.105M sodium citrate (THS-cit, from three BD Vacutainer tubes, N=3) or sodium EDTA (21.6mg in 200ul of saline from two BD Vacutainer tubes) in the syringe used for blood withdrawal. Animals were resuscitated (resus) with shed blood (A,B,D) or 3volumes of Lactated Ringer’s solution (RL, C,D). Trauma sham-shocked (TSS) animals received the same amount of heparin (TSS-hep) or citrate (TSS-cit) at the same time and rate of blood reperfusion in the shocked animals. Cannulas in the surgical groups were primed with citrated-saline (10.5mM, final concentration). Lymph was collected at designated intervals: 30min prior to THS; THS: 90min during THS period when blood is withdrawn; resus: 15min period when shed blood is returned; post-THS: 30 or 60min intervals following THS. Note: 1h post-THS is 45min post-resuscitation. The numbers of animal in each group are shown in parenthesis in the legend. Lymph was tested using the MTT cell viability assay as originally described (21). Data represent cell viability expressed as mean % of control ± SD. A. Lymph was tested at 5% v/v final concentration for 18h. *THS-hep vs TSS-Cit and THS-Cit; # TSS-hep and THS-hep vs TSS-Cit and THS-Cit (One-way ANOVA with Tukey post hoc test at overall significance at P=0.05). B and C. Lymph was incubated with cells at 5% v/v for 18h. *P ≤ 0.001 and #P < 0.05, Student’s t test compared with Lactated Ringer’s with heparin. D and E. Animals were subjected to THS using a fixed volume method. Heparin, citrate and RL were in quantities described above and were administered in combination either through shed blood (D. THS-hep, THS-hep-cit) or by RL (E. THS-RL, THS-RL-hep). Lymph was incubated with cells at 3% v/v for 3h (THS-hep, THS-hep-cit) and 10% v/v for 18h (THS-RL) or 3h (THS-RL-hep). The values for THS-RL are an average of three independent measures. *P < 0.05, Student’s t test.
To test if the lack of post-THS lymph-induced HUVEC toxicity was specific to citrate, EDTA (21.6mg in 200ul of saline) was used as an alternate anticoagulant. No HUVEC toxicity was induced by post-THS lymph when EDTA was used for both blood withdrawal and priming of jugular and femoral cannulas; however, lymph-induced HUVEC toxicity was detected if heparin replaced EDTA only in priming the femoral cannula used for blood pressure monitoring (Fig. 1B). Although differences in the viabilities did not reach significance between the two EDTA groups, HUVEC toxicity was detected at one or more time points in three of the four animals in the heparin group as reflected by the high standard deviations.
Post-THS lymph-induced HUVEC toxicity has also been reported after resuscitation with Lactated Ringer’s solution (RL), rather than shed blood (39–41). We therefore investigated heparin’s influence in this model. Toxicity to HUVECs was only induced by lymph when heparin was used for blood withdrawal and priming of all cannulas. In this model, heparin is not only introduced via the femoral catheter but also by the partial reinfusion of shed blood as needed to maintain target blood pressure over the 90min period (0.2–2ml) prior to resuscitation with RL. When citrate was substituted for the heparin in the syringe and cannulas, no HUVEC toxicity was induced by lymph (Fig. 1C).
We next conducted a set of experiments to test whether the lack of post-THS lymph-induced HUVEC toxicity was due to a protective effect conferred by citrate or EDTA. To accomplish this, the jugular cannula was primed with saline and THS was induced using a methodology analogous to a fixed volume model in which a percent of total blood volume is withdrawn thus eliminating the need for the femoral catheter for pressure monitoring and the jugular cannula. Blood was withdrawn from the animals in fixed volumes at specified times as determined using our fixed pressure model, thus maintaining the same shock time frame. Animals were first resuscitated with their shed, heparinized blood to ensure the modification did not alter results. Lymph from these animals induced HUVEC toxicity (Fig. 1D). The next group of animals was resuscitated with shed blood that contained both citrate and heparin using the rationale that if citrate was protective, then the lymph should not induce HUVEC toxicity. Toxicity to HUVECs equivalent to THS-hep, was induced by lymph collected from these THS-animals (Fig. 1D). Another set of animals was resuscitated with RL, thus omitting all anti-coagulants. Lymph collected from these animals were incubated at a higher concentration (10%) and longer period (18h) to compensate for the dilution of lymph due to the high volume of RL used for resuscitation. The viabilities of HUVECs were slightly reduced by the lymph collected from these animals at 30–90min post-THS (Fig. 1E) but were not reduced to the levels when heparin was used. To further test that this lack of HUVEC toxicity was not simply due to the dilution of lymph by RL, heparin was added to the RL solution used for resuscitation. In contrast to the RL group, lymph from the RL-hep group significantly induced HUVEC toxicity after only a 3h incubation with cells (Fig. 1E) at the same concentration as RL. Because of the charge differences between heparin (negative) and citrate (positive), the pH was measured in the lymph from THS animals receiving either citrate or heparin. Although the pH of the THS-cit lymph was slightly higher at all time points and reached significance at 150min post-THS (Fig. 2), there was no correlation between pH and cell viability after incubation with the lymph (correlation coefficient = −0.231).
Fig. 2.

The pH of lymph from THS animals receiving heparin (hep) or citrate (cit). The pH was measured in each lymph sample at each time point immediately following cell removal and addition of anti-proteases (N=6 each group). *P = 0.04, Student’s t test.
Intravenous injection of heparin into naïve rats produces cytotoxic lymph
In order to separate the contribution of heparin from THS in the production of lymph that induced HUVEC toxicity, heparin alone was injected into naïve animals. The amount of heparin (250U/kg) was based on the THS model using 100U for a 400g rat. Toxicity to HUVECs was induced by mesenteric lymph collected post-heparin injection and was attributed to the supernatant and not the cellular component, consistent with original reports of THS lymph (20) (Fig 3). Lymph from animals injected with saline or citrate as a vehicle and anti-coagulant control, respectively, did not induce HUVEC toxicity (Fig. 3).
Fig. 3.

Mesenteric lymph from naïve animals post-heparin injection induces HUVEC toxicity. Cell viability after incubation with the supernatant of lymph collected from animals pre- and post heparin (250U/kg, N=5) or saline (100ul, N=3) or citrate (1.5ml of 0.105M sodium citrate from BD Vacutainer tubes, N=3) injection. The animals were surgically manipulated as in the THS model except cannulation of the femoral artery was omitted. The jugular cannula was primed with citrated-saline (10.5mM, final concentration). Injections were at the same rate as in the THS model. Lymph was collected at seven intervals: two 30min intervals post-trauma/pre-injection (pre) and six 30min intervals following injection of heparin, saline, or citrate (post). All samples were tested at 3% v/v final concentration for 3h. Data are expressed as mean ± SD. *Heparin vs saline and citrate (One-way ANOVA with Tukey post hoc test at overall significance at P=0.05).
HUVEC toxicity is lipase dependent
Because it is well established that heparin injection releases both HL and LPL into the general blood circulation (25, 26), we hypothesized that lipase activity would be present in the plasma and lymph of naïve and THS animals receiving heparin but not citrate in the surgical procedure. Consistent with this hypothesis, both HL and LPL activities were detected in both the plasma and mesenteric lymph following, but not prior to, heparin injection (naïve-hep) or THS blood reperfusion (THS-hep) (Fig. 4A–D). Maximum activity in the plasma is reached by 30min and sharply decreases for the following 60min (4A and 4B). The maximum activities measured in lymph lag the maximum plasma activities by 30–60min (compare 4C with 4D) and were sustained until 120min post-heparin injection. This time period of maximal lipase activity in lymph directly correlates with a decrease in cell viability (4E, Table 1). From 90 to 180min, lipase activities were equally distributed between the two fluids. Lipase activities in lymph following saline or citrate injection into naïve animals or post-blood reperfusion in THS-cit animals never exceeded the activity measured in the pre-injection sample (data not shown).
Fig. 4.



Hepatic (HL) and lipoprotein (LPL) lipase activities are detected in mesenteric lymph after heparin injection. Animal surgery, heparin injection (250U/kg) and lymph collection were as described in Figure Legend 2. Both HL and LPL were measured within the same plasma (A, N=5, naïve -hep; B, N=3 THS-hep) or lymph (C, N=7, naïve -hep, D, N=5, THS-hep) samples collected pre- and post heparin injection/THS (250U/kg) and are expressed as nmoles of fatty acids released in 1min per 1ml (nmole FA/min/ml). E. Cell viability after incubation with lymph from naïve -hep and THS-hep samples shown in C and D. Lymph was tested at 3% v/v final concentration for 3h. F. Plasma lipase measurements pre-and post heparin injection into an animal in which the lymph duct had been ligated (LDL). Plasma collected from both a heparinized and non-heparinized naïve animal was aliquoted and served as an internal positive and negative control, respectively, for lipase assays and an indicator of daily assay variability. The heparinized control varied by 12% (HL) and 30% (LPL) over all measurements (N=6). No more than 1nmol of fatty acid (FA)/min/ml was ever measured for either lipase in the non-heparinized control. No loss in cell viability was detected after cells were incubated with lymph collected from naïve animals receiving saline (N=3) or THS animals receiving citrate (N=3) injections and no lipase activity was detected at any time point in this lymph (N=2 each group, data not shown). Data are expressed as mean ± SD.
Table 1.
Correlation Coefficients of cell viability vs. lipase activity in animals receiving heparin
| LPL | HL | Total | N | |
|---|---|---|---|---|
| Naïve -hep | −0.75 | −0.877 | −0.869 | 53 |
| THS-hep | −0.685 | −0.759 | −0.711 | 36 |
A Spearman Rank Order Correlation Test at an overall significance level of P=0.05 was used to analyze lipase activity and corresponding cell viability shown in Figure 3D and E, resp. The number of time points (N) reflects 6–8 viability –lipase time course measurements in n=7 (naïve-hep) and n=5 (THS-hep) animals.
Since mesenteric lymph is reintroduced to general circulation and mesenteric lymph duct ligation (LDL) has been shown to abrogate THS-end organ injury (11,12), we hypothesized that LDL should reduce the amount of lipase activity measured within the plasma. Only the LPL, and not HL, was affected (Fig. 4F).
Fasted animals were next used to substantiate a direct in vivo role for LPL in heparin-induced lymph-mediated HUVEC toxicity since fasting has been shown to decrease the amount of lipase in animals (42–45). The lymph from the fasted animals was less toxic to cells when compared to fed animals in both naïve-hep and THS-hep groups (Table 2).
Table 2.
Effects of heparin concentration and fasting on lymph-induced HUVEC toxicity in naïve and shocked (THS) animals
| −60 | −30 | 30 | 60 | 90 | 120 | 150 | 180 | n | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Heparin | |||||||||||
| Naïve | Fed | 250 U/kg | 83 ± 19 | 95 ± 19 | 69 ± 40 | 29 ± 11 | 34 ± 19 | 46 ± 30 | 63 ± 35 | 76 ± 33 | 12 |
| 125 U/kg | 95 ± 5 | 91 ± 13 | 75 ± 32 | 19 ± 8 | 29 ± 11 | 54 ± 10 | 81 ± 2 | 89 ± 17 | 3 | ||
| 60 U/kg | 75 ± 6 | 76 ± 14 | 75 ± 15 | 57 ± 22* | 66 ± 26* | 78 ± 6 | 87 ± 9 | 85 ± 13 | 4 | ||
| 30 U/kg | 98 + 3 | 103 ± 7 | 92 ± 19 | 86 ± 14# | 104 ± 7* | 96 ± 11* | 88 ± 19 | 86 ± 15 | 3 | ||
| Fasted | |||||||||||
| 250U/kg | 69 ± 9 | 88 ± 12 | 81 ± 16 | 56 ± 28** | 53 ± 18 | 64 ± 15 | 76 ± 14 | NC | 5 | ||
| Time (min) relative to shock | −30 | 90 (during) | 30 | 60 | 90 | 120 | 150 | 180 | n | ||
| THS | Fed | 250 U/kg | 89 ± 19 | 101 ± 21 | 75 ± 25 | 66 ± 31 | 35 ± 8 | 53 ± 24 | 71 ± 17 | 93 ± 24 | 6 |
| 125 U/kg | 80 ± 9 | 73 ± 4 | 74 ± 16 | 77 ± 30 | 84 ± 20* | 74 ± 7 | 84 ± 20 | 82 ± 34 | 3 | ||
| 60 U/kg | 80 ± 13 | 82 ± 4 | 82 ± 13 | 76 ± 20 | 82 ± 13* | 76 ± 12 | 90 ± 13 | 91 ± 10 | 3 | ||
| Fasted | |||||||||||
| 250 U/kg | 82 ± 1 | 91 ± 13 | 100 ± 16 | 84 ± 9** | 99 ± 14** | 99 ± 4** | 104 ± 16 | 93 ± 3 | 3 |
HUVEC incubations with lymph were for 3h. Data are expressed as mean % of control ± SD. Dotted line indicates time of heparin administration. NC, not collected.
P ≤ 0.02 ANOVA compared with fed-250U/kg;,
P < 0.001 ANOVA compared with fed-250U/kg and fed-125U/kg;
P ≤ 0.02, Student’s t test compared with fed-250U/kg.
Because lipases are released into circulation due to their high affinity for heparin, we theorized that lower doses of heparin should release less lipase and therefore result in less HUVEC toxicity. Consistent with this theory, lymph-mediated HUVEC toxicity was less with lower doses of heparin in naïve-hep and to a greater degree in THS-hep (Table 2). No average cell viability below 89% was measured after incubation of endothelial cells with increasing doses of heparin alone (1, 2, and 4U/ml) or with naïve lymph that had been pre-incubated (0, 30, 60, 90, 120min) with the same doses heparin (Fig. 5).
Fig. 5.
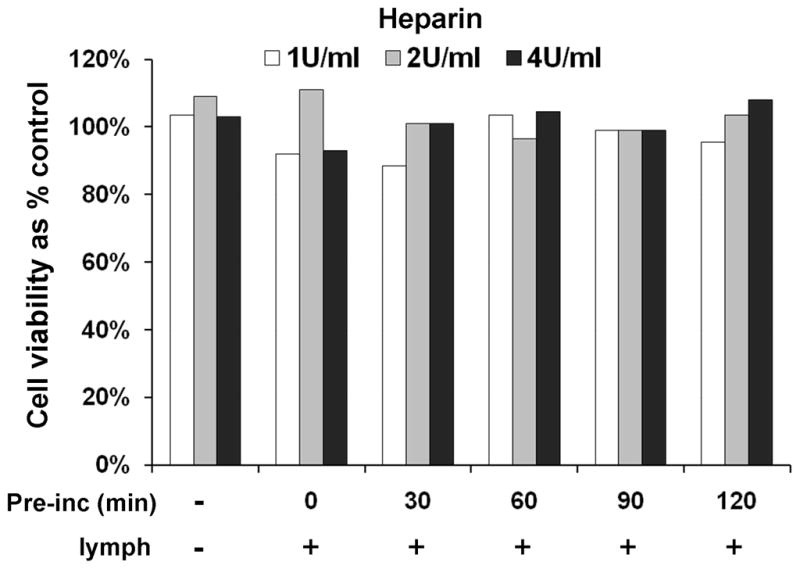
Heparin alone or pre-incubated with naïve lymph does not induce HUVEC toxicity. HUVECs were incubated (3h) with heparin (1, 2, or 4U/ml, final concentration) or with naïve lymph (3% v/v) that had been pre-incubated (pre-inc) with heparin (1, 2, or 4U/ml) for 0, 30, 60, 90, 120 min. The data represent an average of two tests in each group.
Lipases are derived from the plasma
Because lymph is a filtrate of plasma and the lymph lipase profiles mirrored those of the plasma, we hypothesized that the lipases were derived from the plasma. To test this, we reasoned that the temporary interruption of the blood supply to the area drained by the mesenteric lymphatics would delay or diminish the appearance of lipase activity and, in turn, lymph-induced HUVEC toxicity. The superior mesenteric artery was therefore occluded (SMAO) immediately prior to heparin injection and remained so for 60min before the return of blood flow. Only lymph collected immediately after the return of blood flow in the animals receiving heparin reduced cell viability (Fig. 6A). The detection of this reduced activity was delayed 30min (90min post-heparin) compared to a non-occluded naïve-hep animal (60min post-heparin, Fig. 4E and Table 2). Lipase activity was also only detected in the animals receiving heparin (Fig. 6B).
Fig. 6.
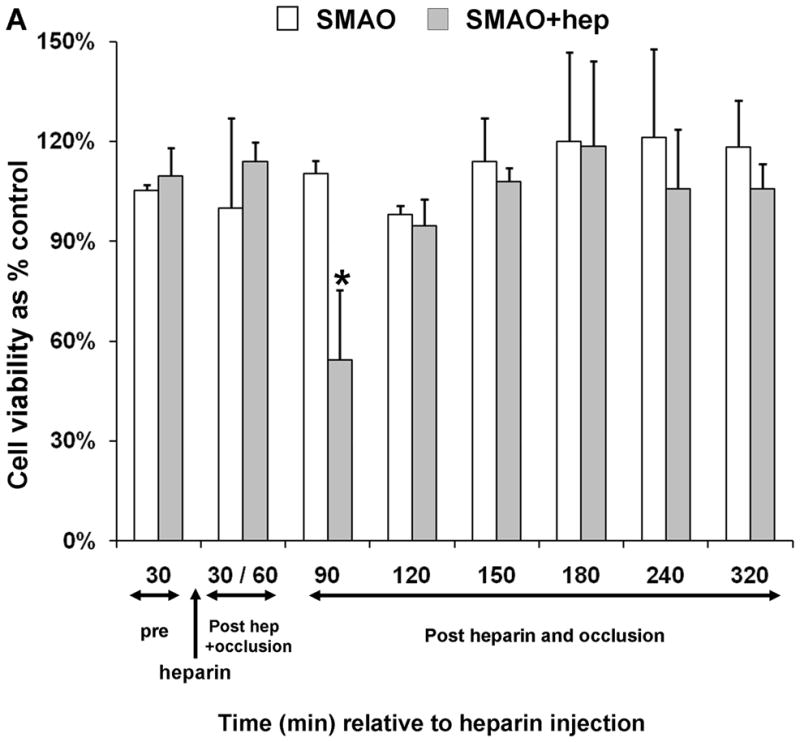
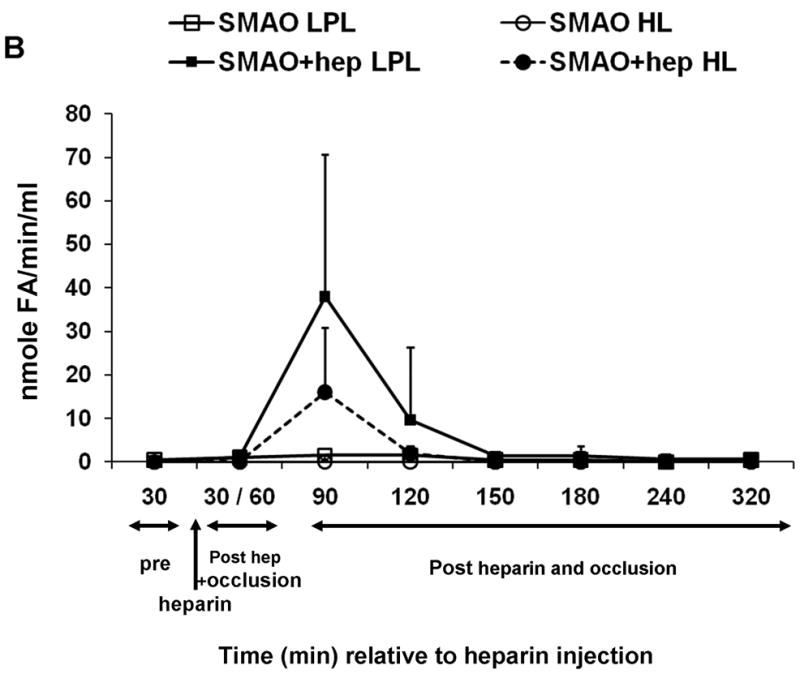
Superior mesenteric arterial occlusion (SMAO) delays appearance of lymph-induced HUVEC toxicity. (A) Lymph collected from animals pre, during and post occlusion of the superior mesenteric artery either with (N=3) or without (N=3) heparin injection (250U/kg) was incubated with endothelial cells at 3% v/v final concentration for 3h to test for induced HUVEC toxicity. (B) Lipase activities in the same samples were made as described in Fig. 4 legend. Data are expressed as mean ± SD. *P= 0.05, compared with SMAO without heparin (Student’s t test).
Bioactive lymph can be created in vitro
To begin establishing a direct role for lipase activity in the production of post-heparin lymph-mediated HUVEC toxicity and because our data indicate that post-heparin mesenteric lymph lipases enter from the plasma, we hypothesized that lymph-induced HUVEC toxicity could be recreated in vitro by combining lymph with post-heparin plasma. Lymph collected from naïve rats (no IV injections) was combined with plasma collected from naïve -hep rats 30min after receiving a bolus IV injection of 250U/kg heparin or with plasma from THS-hep rats collected 1 & 2h after resuscitation with shed blood. The amount of plasma used in each experiment was normalized to an LPL activity of either 40 or 115 nmolesFA/min/ml. These values corresponded to the lowest (THS-hep) and median (naïve -hep) LPL activity, respectively, that yielded toxicity at 60–90min post-heparin injection. In order to reflect the lymph at 60–120min post heparin injection, samples were mixed and immediately tested for the induction of HUVEC toxicity. Lymph incubated with both naïve -hep and THS-hep plasma at 115nmolesFA/min/ml induced HUVEC toxicity within 3h (Fig. 7A). Cell viability was reduced up to 40% with LPL normalized to 40 nmolesFA/min/ml after 3h and up to 90% after 18h (data not shown). The induced HUVEC toxicity was abrogated by pre-incubating the naïve -hep or THS-hep plasma with either the general lipase inhibitor, Orlistat, or the monoclonal antibody 5D2 specific for lipoprotein lipase (Fig. 7A). Depleting the lymph of chylomicrons (Fig. 7B), the substrate for LPL, prior to incubation, also reduced the amount of HUVEC toxicity (Fig. 7A). Naïve lymph mixed with plasma collected from non-heparinized rats (N=3 different plasmas) did not induce HUVEC toxicity nor did naïve plasma, chylomicron depleted or enriched lymph alone (data not shown).
Fig. 7.

Toxicity to HUVECs can be recreated in vitro. A. Plasma withdrawn 30min post-heparin injection (naïve -hep) or 1–2h post-THS with heparin use (THS-hep) was incubated with the supernatant from lymph (S) or lymph that had been depleted (CMD) or enriched (CME) with chylomicrons with and without inhibitors (Orlistat (O, 252uM), LPL monoclonal antibody (5D2, 0.08mg/ml), isogenic control (IgG, 0.08mg/ml)). The amount of plasma was normalized to an LPL activity of 115 nmoles of fatty acids released in 1min per 1ml (nmolesFA/min/ml). Data reflect duplicate measures of N=3 naïve -hep plasma and N=2 1h and N=2 2h post-THS-hep plasma. Incubations were for 3hr with 3% lymph final concentration. Viabilities are expressed as mean % of control + SD. *P < 0.05 vs. Lymph + heparinized plasma. B. An agarose lipoprotein gel depicting lymph substrates used in the chylomicron (CM) depletion assays. CMs run as a smear when present in high concentration.
We next purified bovine milk LPL to test if LPL alone mixed with naïve rat lymph could induce HUVEC toxicity. The presence of LPL was verified by western blot and by limited protein sequencing (data not shown). The purified enzyme had lipase activity that could be neutralized with Orlistat and the 5D2 antibody in the lipase assay (data not shown). Toxicity to HUVECs was induced when the purified LPL was incubated with naïve rat lymph using the same procedure and activity as described for heparinized plasma (Fig. 8A). We next tested if lipoprotein lipase was responsible for the induced HUVEC toxicity within our cell viability assays by pre-incubating lymph shown to induce HUVEC toxicity with both the 5D2 antibody and the general lipase inhibitor, Orlistat. Both inhibitors were equally protective (Fig. 8B). Neither inhibitor was protective for three lymph samples from a single naïve -hep rat (not included in the figure). It was noted that these lymph samples had very high lipase activities (both LPL and HL > 150 nmoles FA/min/ml).
Fig. 8.
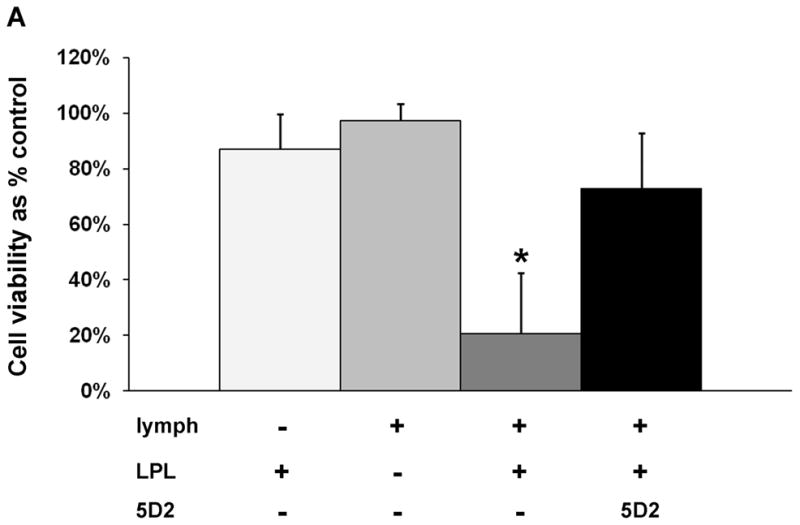

Lipoprotein lipase is responsible for lymph-induced HUVEC toxicity. A. Lipoprotein lipase purified from bovine milk was incubated with naïve lymph in the same manner as our experiments with heparinized plasma. The amount of LPL was normalized to an activity of 115 nmolesFA/min/ml. Data are expressed as mean ± SD of three measures. *P < 0.01 compared to all other groups (One-way ANOVA and Tukey post hoc test). B. Eight naïve -hep and nine THS-hep lymph samples (2–3 time point samples from three animals each group) were pre-incubated with the 5D2 monoclonal antibody/isogenic control (0.08mg/ml–0.16mg/ml, final concentration) or Orlistat/vehicle control (252uM, final concentration) for 1h at 4C then incubated with cells for 3–4 h. For proper comparison purposes, femoral artery was cannulated in the naïve -hep animals. **P = 0.002, *P < 0.001, Student’s t test.
Mesenteric lymph lipoprotein profiles are altered after heparin injection
It has been reported that electrophoretic migration of plasma lipoproteins are altered in post-heparin plasma, which the authors attributed to the action of lipase on plasma lipoprotein substrates (46). Because our data indicate that lipases are plasma derived and processing of lymph lipoproteins produces cytotoxic factors, we reasoned that similar post-heparin lipoprotein profiles should be observed in lymph. We therefore compared the electrophoretic migration patterns in the lymph collected from naïve -hep, naïve -sal, THS-hep and THS-cit animals on agarose gels. Post-heparin lymph consistently showed the appearance of fast migrating bands in a pattern identical to that previously reported for post-heparin plasma (Fig. 9, arrow). These bands did not appear in pre-heparin samples or in any time point after saline or citrate injection. Furthermore, these bands appeared in both naïve -hep and THS-hep lymph in the time points corresponding to maximal HUVEC toxicity (right lower panels, 60–120min compare with Table 2). In addition, the lipoprotein profile of lymph after in vitro incubation with heparinized plasma was analogous to those collected from naïve -hep and THS-hep animals (data not shown).
Fig. 9.

Lymph lipoprotein profiles are altered after heparin injection. The left gel is representative of lymph samples prior to heparin administration and samples post-injection of saline (naïve -sal, N=3) and in post-THS samples in which citrate was used (THS-cit, N=3). The right gel is representative of lymph samples post-heparin (naïve -hep, N=3) and in post-THS samples in which heparin was used (THS-hep, N=4). Data were analyzed by densitometry of boxes drawn around the gel migration area of the fast migrating band appearing in post-heparin samples (shown). Values of each lane were compared to the pre-injection lymph control and these ratios were graphed. The pre-injection control has an inherent value of one and is represented by the dotted line. There was no overall significance in the means in any of the band areas for either the naïve -saline or THS-cit groups. Data are expressed as mean ± SD. *P ≤ 0.05, compared with naïve -hep pre, 120, 150 180min post-heparin; **P ≤ 0.05, ANOVA compared with THS-hep pre, 30min post-THS (One-way ANOVA and Tukey post hoc test).
It has been shown that the simple association of free fatty acids (FFA’s) with lipoproteins can alter their electrophoretic migration (47). Because lipase activity creates FFA’s, we investigated if this mechanism could account for the new bands detected in post-heparin lymph by mixing oleic acid (0.328uM – 32.8mM) with naïve lymph as previously reported (47) followed by electrophoretic analysis. No bands were observed at any concentration when oleic acid alone was applied to the gels and no alterations in the banding patterns were observed in naïve lymph incubated with oleic acid (data not shown).
DISCUSSION
The study of factors created during trauma-hemorrhagic shock (THS) is paramount to understanding events leading to multiple organ failure (MOF). To this end, animal models have been developed to study the impact of THS on inflammation and end-organ injury. Within each model, the means of resuscitation may vary and include Lactated Ringer’s solution (39–41), hypertonic saline (40, 41), shed blood (11–21) or a combination of these (39). Despite these differences, these surgical procedures have a single commonality: heparin. Heparin is highly effective in preventing coagulation of shed blood and preventing clot formation in the vessel cannulas used for blood withdrawal or pressure monitoring. In controlling for this drug in our current model of THS, we found injection of heparin into control animals could produce a factor in lymph that induced HUVEC toxicity. We therefore investigated the respective roles of THS and heparin in the creation of this lymph-induced HUVEC toxicity.
Two key findings show that lymph-induced HUVEC toxicity is the direct result of heparin injection and not of THS. First, post-THS lymph did not induce HUVEC toxicity when heparin was omitted from the surgical procedure. This result was independent of the anticoagulant (citrate, EDTA, none) and the resuscitation fluid (shed blood or Lactated Ringer’s solution) since all combinations of the two yielded the same results. This lack of cytotoxicity could not be attributed to the calcium chelating abilities of citrate or the anionic charge of heparin since calcium levels between THS-cit and THS-hep lymph were not significantly different and there was no correlation between pH and cell viability. The use of citrate did not confer protection since lymph collected from THS animals receiving both citrate and heparin with shed blood could induce HUVEC toxicity to the same extent as heparin alone. Second, heparin injection alone, but not citrate or saline, into TSS and naïve animals produced lymph-induced HUVEC toxicity. Because citrate and saline were given under identical conditions, this induced HUVEC toxicity was not due to injection itself or to injection rate (Figure 2). Together, these data clearly show that heparin, not THS, is the precipitating factor in lymph-induced HUVEC toxicity.
Four key findings show that heparin-released lipases are responsible for the lymph-induced HUVEC toxicity in both naïve -hep and THS-hep animals. First, heparin-induced LPL and HL activities were detected in plasma and lymph in both heparin groups. Second, the lipase activity had similar time course profiles. Third, the lipase activity directly correlated with lymph-induced HUVEC toxicity and fasting, a well-established method of lowering LPL levels in vivo (42–45), significantly reduced the ability of lymph to induce HUVEC toxicity in both heparin groups. Lastly, HUVEC toxicity could be both created in vitro by incubating cells with purified bovine LPL with lymph from naïve animals and inhibited after pre-incubating toxic lymph with both a general lipase inhibitor and an LPL-specific neutralizing antibody. The clear heparin-lipase link to lymph-induced in both naïve -hep and THS-hep groups, are fully consistent with the notion that THS-hep does not contribute to the formation of novel cytotoxic factors.
Our data are consistent with a previous report of LPL and HL activity in dog skeletal and peripheral lymph (48), where plasma levels peaked 30min after heparin injection and 60–90 min in lymph. Contrary to their study, our absolute plasma and lymph lipase activities were lower and our ratio of lymph:plasma lipase ratios were higher. These differences could be species specific because our plasma lipase activities are fully consistent with those reported in rat (49).
In this prior study it was also suggested that heparin exiting the circulation into the interstitial space displaces LPL attached to tissues thus freeing the lipases to enter the lymph (48). Within the THS model, it is proposed that lymph factors responsible for the post-THS lymph-induced endothelial cell damage are derived from the ischemic gut (21). Our data, however, lead us to hypothesize that the lymph lipases are derived directly from the circulation. First, the similarity of plasma-lymph lipase profiles coupled with the 30min delay of lipase appearance in the lymph compared with plasma (Figure 4) support this concept. Second, incubation of post-heparin plasma with naïve lymph induced HUVEC toxicity. Third, the temporary interruption of blood (SMAO) flow prior to heparin injection delayed the appearance of both lymph-induced HUVEC toxicity and lipase activity. The lack of detectable HUVEC toxicity in the lymph collected from SMAO animals in the absence of heparin indicates that the injured gut is not a source of the lipases. Furthermore, if lipases were exiting the gut into the lymph due to ischemia, lymph-induced HUVEC toxicity would be expected to increase post-occlusion, which is inconsistent with our observed decrease. Lymph lipases were also not due to blood contamination during the surgical or collection procedure since no red blood cells were detected in the pellet after centrifugation of the lymph from either naïve -hep or THS-hep animals. Collectively, the data support the hypothesis that post-heparin mesenteric lymph lipases in THS and naïve animals, are plasma-derived.
Since the half-life of a circulating chylomicron is 5min (50), it could be argued that the detected toxicity is an artifact of collection. Although we have not rigorously tested this concept, it has not been our experience that lymph samples remaining on ice for the duration of the entire collection period become more toxic than those frozen immediately after collection. We do recognize that pooling the lymph samples most likely skews the true physiologic concentrations at any given time. However, this is not simply an in vitro phenomenon since lymph duct ligation prevents organ injury in the THS-hep model (11,12). In addition, lymph collected post-heparin injection in shorter time intervals (15min) was still able to induce HUVEC toxicity. Pooling lymph is unavoidable in order to do several difference measurements on a single sample due to low lymph volumes. Still, we have attempted to avoid any possible processing during collection by reducing the lymph cannula length to 8cm, collecting lymph directly into a syringe affixed to an ice pack and processing and freezing (−80°C) lymph at each time point immediately after each is collected.
There are two important points that have come from these data. First, the sole use of heparin in the femoral catheter used for monitoring blood pressure is sufficient to induce HUVEC toxicity in the mesenteric lymph as shown in the experiments using EDTA as an anti-coagulant (Fig. 1B). That this is a significant source of heparin is underscored in some large animal models using flow-through transducers to negate the need for heparin (51,52). Second, heparin can be used as a tool not only to visualize the reperfusion process following THS by comparing lipase activities within blood at different areas but also to investigate organ damage that could be contributed by lipoprotein metabolism during THS. Indeed, heparin (200U/kg/h) is used to amplify the effects of lipids in TLR4 signaling in a mouse model (53).
The deleterious effects of heparin-induced lipases shown in our current study can, in part, provide an explanation for several experimental findings attributed to THS when heparinized shed blood is returned in the shock model. For example, a series of experiments showed that heparinization of rats prior to THS was protective, which the authors attributed to improved microcirculation during THS due to the anticoagulation effects of heparin (54,55). In their study, shock was induced by blood withdrawal immediately after a high dose (1U/g) of heparin and then resuscitated with Lactated Ringer’s solution, not shed blood. Therefore, the protective effects of pre-heparinization could be due to the removal of heparin-released lipases from the shocked animal thus reducing lipase contributions to end-organ damage. The protective effects of hypertonic saline (1.3M NaCl) (40, 41) can be attributed to the irreversible inhibition of LPL, which is sensitive to salt concentrations as low as 0.5M NaCl (26, 33). In fact, concentrations of 1M are used routinely to inhibit LPL activity in assays of HL measurements in vitro. The protective effects of high density lipoproteins (HDL) in THS end organ-injury (56) could be due to the pivotal role of HDL particles in chylomicron remnant clearance (57), which are implicated in endothelial damage in atherosclerosis (reviewed in 58). Estrogen has been shown to decrease LPL in adipose tissue (59) and injected estradiol has been shown to preferentially bind to and inhibit HL activity (60), which could therefore account, in part, for protective effects seen in female rats subjected to THS (61,62). Furthermore, heparin-induced lymph-mediated effects may also explain the reported decrease in post-THS red blood cell deformability (15) since the same post-THS lymph fractions that we reported to induce endothelial cell toxicity (23), also affect red blood cell deformability (unpublished). Finally, myeloperoxidase (MPO), a marker used for neutrophil activation in post-THS animals (12, 17, 40), is not limited to the neutrophil but is also bound to the vascular endothelium and released by heparin into circulation (63). These examples do not negate the fact that THS clearly has a systemic impact since gut injury, inflammation, gender differences and organ injury are seen in models not using heparin (52,64,65) and slightly reduce endothelial cell viability in our RL resuscitation (Fig. 1E) model in the absence of any anticoagulants. However, viewed within the context of our study, heparin-related factors can provide alternate explanations for the results obtained from specific end-assays used to evaluate damage due to THS when heparin is used.
CONCLUSION
Our data clearly show that the use of heparin in a rat shock model causes biologic activity in mesenteric lymph that is independent of THS by demonstrating that the biologic activity in mesenteric lymph collected from naïve animals receiving an intravenous injection of heparin is indistinguishable from the biologic activity measured in lymph collected from post-shock animals using a heparin model. Our data are fully consistent with a model in which heparin injection into both naïve and THS animals releases LPL and HL into circulation and the lipases are filtered into the mesenteric lymph. Here, action of LPL on the nascent lipoproteins induces factor(s) that are toxic to HUVECs. Furthermore, the presence of the HL, a liver–specific enzyme, within post-heparin lymph indicates that the gut may not be the sole source for mesenteric lymph components and suggests factors generated by the liver during THS may also be potential cytotoxic candidates contributing to MOF via the lymphatics. Finally, these data show that use of heparin in animal models causes confounding effects as it is clear that lipoprotein metabolism is instrumental in the observed biologic effects in these models.
Acknowledgments
This study was supported by grants from the National Institutes of Health (R01-GM059841 and 1P50GM069790).
We thank Dr. Patricia Fitzgerald-Bocarsly for support throughout this project. We thank Drs. George Dikdan, Robert Donnelly and Jeffrey Wilusz for critical reading of the manuscript, Dr. Qi Lu for technical assistance and Chloe Sibona for help in manuscript preparation.
Footnotes
Conflict-of-Interest: None
References
- 1.Deitch EA. Multiple organ failure: Pathophysiology and potential future therapy. Ann Surg. 1992;216(2):117–134. doi: 10.1097/00000658-199208000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deitch EA, Goodman E. Prevention of Multiple Organ Failure. Surg Clin North Am. 1999;79(6):1471–1488. doi: 10.1016/s0039-6109(05)70088-8. [DOI] [PubMed] [Google Scholar]
- 3.Glenn TM, Lefer AM. Protective effect of thoracic lymph diversion in hemorrhagic shock. Am J Physiol. 1970;219(5):1305–1310. doi: 10.1152/ajplegacy.1970.219.5.1305. [DOI] [PubMed] [Google Scholar]
- 4.Papp M, Nemeth E, Feuer I, Fodor I. Effect of an impairment of lymph flow on experimental acute “pancreatitis”. Acta Med Acad Sci Hung. 1957;11(2):203–208. [PubMed] [Google Scholar]
- 5.Deitch EA, Xu DZ, Kaiser VL. Role of the gut in development of injury and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci. 2006;11:520–528. doi: 10.2741/1816. [DOI] [PubMed] [Google Scholar]
- 6.Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15(1):1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 7.Faries PI, Simon RJ, Martella AT. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44(6):1031–1036. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Langkamp-Henken B, Donovan TB, Pare LM. Increased intestinal permeability following blunt and penetrating trauma. Crit Care Med. 1995;23(4):660–664. doi: 10.1097/00003246-199504000-00013. [DOI] [PubMed] [Google Scholar]
- 9.Deitch EA. Intestinal permeability is increased in burn patients shortly after injury. Surgery. 1990;107(4):411–416. [PubMed] [Google Scholar]
- 10.Rush BF, Sori AJ, Murphy TF, Smith S, Flanagan JJ, Machiedo GW. Endotoxemia and bacteremia during hemorrhagic shock. Ann Surg. 1988;207(5):549–554. doi: 10.1097/00000658-198805000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deitch EA, Adams CA, Lu Q, Xu DZ. A time course study of the protective effect of mesenteric lymph duct ligation on hemorrhagic shock-induced pulmonary injury and the toxic effects of lymph from shocked rats on endothelial cell monolayer permeability. Surgery. 2001;129(1):39–47. doi: 10.1067/msy.2001.109119. [DOI] [PubMed] [Google Scholar]
- 12.Sambol JT, Xu DZ, Adams CA, Magnotti LJ, Deitch EA. Mesenteric lymph duct ligation provides long term protection against hemorrhagic shock-induced lung injury. Shock. 2000;14(3):416–420. [PubMed] [Google Scholar]
- 13.Adams CA, Hauser CJ, Adams JM, Fekete Z, Xu DZ, Sambol JT, Deitch EA. Trauma-hemorrhage-induced neutrophil priming is prevented by mesenteric lymph duct ligation. Shock. 2002;18(6):513–517. doi: 10.1097/00024382-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Zallen G, Moore EE, Johnson JL, Tamura DY, Ciesla DJ, Silliman CC. Posthemorrhagic shock mesenteric lymph primes circulating neutrophils and provokes lung injury. J Surg Res. 1999;83(2):83–88. doi: 10.1006/jsre.1999.5569. [DOI] [PubMed] [Google Scholar]
- 15.Zaets SB, Berezina TL, Caruso J, Xu DZ, Deitch EA, Machiedo GW. Mesenteric lymph duct ligation prevents shock-induced red blood cell deformability and shape changes. J Surg Res. 2003;109(1):51–56. doi: 10.1016/s0022-4804(02)00024-0. [DOI] [PubMed] [Google Scholar]
- 16.Deitch EA, Forsythe R, Anjaria D, Livingston DH, Lu Q, Xu DZ, Redl H. The role of lymph factors in lung injury, bone marrow suppression and endothelial cell dysfunction in a primate model of trauma-hemorrhagic shock. Shock. 2004;22(3):221–228. doi: 10.1097/01.shk.0000133592.55400.83. [DOI] [PubMed] [Google Scholar]
- 17.Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and potentiates lung injury following hemorrhagic shock. Ann Surg. 1998;228(4):518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu DZ, Lu Q, Adams CA, Issekutz AC, Deitch EA. Trauma-associated shock-induced upregulation of endothelial cell adhesion molecules is blunted by mesenteric lymph duct ligation. Crit Care Med. 2004;32(3):760–765. doi: 10.1097/01.ccm.0000114815.88622.9d. [DOI] [PubMed] [Google Scholar]
- 19.Anjaria DJ, Rameshwar P, Deitch EA, Xu DZ, Adams CA, Forsythe RM, Sambol JT, Hauser CJ, Livingston DH. Hematopoietic failure after hemorrhagic shock is mediated partially through mesenteric lymph. Crit Care Med. 2001;29(9):1780–1785. doi: 10.1097/00003246-200109000-00021. [DOI] [PubMed] [Google Scholar]
- 20.Adams CA, Xu DZ, Lu Q, Deitch EA. Factors larger than 100 kD in post-hemorrhagic shock mesenteric lymph are toxic for endothelial cells. Surgery. 2001;129(3):351–363. doi: 10.1067/msy.2001.111698. [DOI] [PubMed] [Google Scholar]
- 21.Upperman JS, Deitch EA, Guo W, Lu Q, Xu DZ. Post-hemorrhagic shock mesenteric lymph is cytotoxic to endothelial cells and activates neutrophils. Shock. 1998;10(6):407–414. doi: 10.1097/00024382-199812000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Senthil M, Watkins A, Barlos D, Xu DZ, Lu Q, Abungu B, Caputo FJ, Feinman R, Deitch EA. Trauma-shock mesenteric lymph causes lung injury that is dependent upon inducible nitric oxide synthase pathway. Ann Surg. 2007;246(5):822–830. doi: 10.1097/SLA.0b013e3180caa3af. [DOI] [PubMed] [Google Scholar]
- 23.Kaiser VL, Sifri ZC, Dikdan GS, Berezina T, Zaets S, Lu Q, Xu DZ, Deitch EA. Trauma-hemorrhagic shock mesenteric lymph from rat contains a modified form of albumin that is implicated in endothelial cell toxicity. Shock. 2005;23(5):417–425. doi: 10.1097/01.shk.0000160524.14235.6c. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez RJ, Moore EE, Biffl WL, Ciesla DJ, Silliman CC. The lipid fraction of post-hemorrhagic shock mesenteric lymph inhibits neutrophil apoptosis and enhances cytotoxic potential. Shock. 2000;14(3):404–408. doi: 10.1097/00024382-200014030-00028. [DOI] [PubMed] [Google Scholar]
- 25.Hahn PF. Abolishment of alimentary lipemmia following injection of heparin. Science. 1943;98(2531):19–20. doi: 10.1126/science.98.2531.19. [DOI] [PubMed] [Google Scholar]
- 26.Korn ED. Properties of clearing factor obtained from rat heart. Science. 1954;120(3114):399–400. doi: 10.1126/science.120.3114.399-a. [DOI] [PubMed] [Google Scholar]
- 27.Higgins LJ, Rutledge JC. Inflammation associated with the postprandial lipolysis of triglyceride-rich lipoproteins by lipoprotein lipase. Curr Atheroscler Rep. 2009;11(3):199–205. doi: 10.1007/s11883-009-0031-9. [DOI] [PubMed] [Google Scholar]
- 28.Chung BH, Tallis GA, Cho BH, Segrest JP, Henkin Y. Lipolysis-induced partitioning of free fatty acids to lipoproteins: effect on the biological properties of free fatty acids. J Lipid Res. 1995;36(9):1956–1970. [PubMed] [Google Scholar]
- 29.Speidel MT, Booyse FM, Abrams A, Moore MA, Chung BH. Lipolyzed hypertriglyceridemic serum and triglyceride-rich lipoprotein cause lipid accumulation in and are cytotoxic to cultured human endothelial cells. High density lipoproteins inhibit this cytotoxicity. Thromb Res. 1990;58(3):251–264. doi: 10.1016/0049-3848(90)90095-t. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser VL, Sifri ZC, Senthil M, Dikdan GS, Lu Q, Xu DZ, Deitch EA. Trauma-hemorrhagic shock mesenteric lymph from rat contains a modified form of albumin that is implicated in endothelial cell toxicity. Peptides. 2005;26(12):2491–2499. doi: 10.1097/01.shk.0000160524.14235.6c. [DOI] [PubMed] [Google Scholar]
- 31.Grotz MR, Deitch EA, Ding J, Xu D, Huang Q, Regel G. Intestinal cytokine response after gut ischemia: role of gut barrier failure. Ann Surg. 1999;229(4):478–486. doi: 10.1097/00000658-199904000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deitch EA, Adams CA, Lu Q, Xu DZ. Mesenteric lymph from rats subjected to trauma-hemorrhagic shock are injurious to rat pulmonary microvascular endothelial cells as well as human umbilical vein endothelial cells. Shock. 2001;16(4):290–293. doi: 10.1097/00024382-200116040-00010. [DOI] [PubMed] [Google Scholar]
- 33.Bengtsson-Olivecrona G, Olivecrona T. Lipoprotein Analysis: A Practical Approach. Oxford University Press; New York, New York: 1992. pp. 169–185. [Google Scholar]
- 34.Dole VP. A relation between non-esterified fatty acids in plasma and the metabolism of glucose. J Clin Invest. 1956;35(2):150–154. doi: 10.1172/JCI103259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindberg A, Olivecrona G. Lipase evolution: trout, Xenopus and chicken have lipoprotein lipase and apolipoprotein C-II-like activity but lack hepatic-like activity. Biochim et Biophys Acta. 1995;1255(2):205–211. doi: 10.1016/0005-2760(94)00233-o. [DOI] [PubMed] [Google Scholar]
- 36.Briquet-Laugier V, Ben-Zeev O, Doolittle MH. Lipase and Phospholipase Protocols. Humana Pres, Inc; Totowa, New Jersey: 1999. pp. 81–94. [Google Scholar]
- 37.Bengtsson-Olivecrona G, Olivecrona Phospholipase activity of milk lipoprotein lipase. Methods Enzymol. 1991;197:345–356. doi: 10.1016/0076-6879(91)97160-z. [DOI] [PubMed] [Google Scholar]
- 38.Lee HB, Blaufox MD. Blood volume in the rat. J Nucl Med. 1985;25(1):72–76. [PubMed] [Google Scholar]
- 39.Vega D, Badami CD, Caputo FJ, Watkins AC, Lu Q, Xu DZ, Berezina TL, Zaets SB, Feketeova E, Deitch EA. The influence of the type of resuscitation fluid on gut injury and distant organ injury in a rat model of trauma/hemorrhagic shock. J Trauma. 2008;65(2):409–414. doi: 10.1097/TA.0b013e3181719708. [DOI] [PubMed] [Google Scholar]
- 40.Deitch EA, Shi HP, Feketeova E, Hauser CJ, Xu DZ. Hypertonic saline resuscitation limits neutrophil activation after trauma-hemorrhagic shock. Shock. 2003;19(4):328–333. doi: 10.1097/00024382-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Shi HP, Deitch EA, Xu DZ, Lu Q, Hauser CJ. Hypertonic saline improves intestinal mucosa barrier function and lung injury after trauma-hemorrhagic shock. Shock. 2002;17(6):496–501. doi: 10.1097/00024382-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Doolittle MH, Ben-Zeev O, Elovson J, Martin D, Kirchgessner TG. The response of lipoprotein lipase to feeding and fasting. Evidence for posttranslational regulation. J Biol Chem. 1990;265(8):4570–4577. [PubMed] [Google Scholar]
- 43.Masuno H, Blanchette-Mackie EJ, Schultz CJ, Spaeth AE, Scow RO, Okuda H. Retention of glucose by N-linked oligosaccharide chains impedes expression of lipoprotein lipase activity: effect of castanospermine. J Lipid Res. 1992;33(9):1343–1349. [PubMed] [Google Scholar]
- 44.Park JW, Oh MS, Yang JY, Park BH, Rho HW, Lim SN, Jhee EC, Kim HR. Glycosylation, dimerization, and heparin affinity of lipoprotein lipase in 3T3-L1 adipocytes. Biochim Biophys Acta Lipids Lipid Metab. 1995;1254(1):45–50. doi: 10.1016/0005-2760(94)00161-q. [DOI] [PubMed] [Google Scholar]
- 45.Masuno H, Blanchette-Mackie EJ, Chernick SS, Scow RO. Synthesis of inactive nonsecretable high mannose-type lipoprotein lipase by cultured brown adipocytes of combined lipase-deficient cld/cld mice. J Biol Chem. 1990;265(3):1628–1638. [PubMed] [Google Scholar]
- 46.Houtsmuller AJ. Heparin-Induced post beta Lipoproteins. Lancet. 1966;7470(II):967. [Google Scholar]
- 47.Gordon RS., Jr Interaction between oleate and the lipoproteins of human serum. J Clin Invest. 1955;34(3):477–484. doi: 10.1172/JCI103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang J, Sloop CH, Roheim PS, Wong L. Lipoprotein lipase and hepatic triacylglycerol lipase activities in peripheral and skeletal muscle lymph. Arterioscler Thromb Vasc Biol. 1990;10(5):720–726. doi: 10.1161/01.atv.10.5.720. [DOI] [PubMed] [Google Scholar]
- 49.Olivecrona T, Olivecrona G. Handbook of Lipoprotein Testing. American Association for Clinical Chemistry; Washington, DC: 2000. pp. 479–498. [Google Scholar]
- 50.Park Y, Damron BD, Miles JM, Harris WS. Measurement of human chylomicron triglyceride clearance with a labeled commercial lipid emulsion. Lipids. 2001;36(2):115–120. doi: 10.1007/s11745-001-0696-6. [DOI] [PubMed] [Google Scholar]
- 51.Desselle WJ, Greenhaw JJ, Trenthem LL, Fabian TC, Proctor KG. Macrophage cyclooxygenase expression, immunosupression, and cardiopulmonary dysfunction after blunt chest trauma. J Trauma. 2001;51(2):239–251. doi: 10.1097/00005373-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 52.Ragsdale DN, Proctor KG. Acadesine and intestinal barrier function after hemorrhagic shock and resuscitation. Crit Care Med. 2000;28(12):3876–3884. doi: 10.1097/00003246-200012000-00023. [DOI] [PubMed] [Google Scholar]
- 53.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang P, Ba ZF, Chaudry IH. Endothelial cell dysfunction occurs after hemorrhage in nonheparinized but not in preheparinized models. J Surg Res. 1993;54(5):499–506. doi: 10.1006/jsre.1993.1077. [DOI] [PubMed] [Google Scholar]
- 55.Wang P, Singh G, Rana MW, Ba ZF, Chaudry IH. Preheparinization improves organ function after hemorrhage and resuscitation. Am J Physiol. 1990;259(3 Pt 2):R645–650. doi: 10.1152/ajpregu.1990.259.3.R645. [DOI] [PubMed] [Google Scholar]
- 56.Cockerill GW, McDonald MC, Mota-Filipe H, Cuzzocrea S, Miller NE, Thiemermann C. High Density Lipoproteins reduce organ injury and organ function in a rat model of hemorrhagic shock. FASEB J. 2001;15(11):1941–1952. doi: 10.1096/fj.01-0075com. [DOI] [PubMed] [Google Scholar]
- 57.Weng W, Breslow JL. Dramatically decreased high density lipoprotein cholesterol, increased remnant clearance, and insulin hypersensitivity in apolipoprotein A-II knockout mice suggest a complex role for apolipoprotein A-II in atherosclerosis susceptibility. Proc Natl Acad Sci USA. 1996;93(25):14788–14794. doi: 10.1073/pnas.93.25.14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Botham K. Oxidation of chylomicron remnants and vascular dysfunction. Atheroscler Suppl. 2008;9(2):57–61. doi: 10.1016/j.atherosclerosissup.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Hamosh M, Hamosh P. The effect of estrogen on the lipoprotein lipase activity of rat adipose tissue. J Clin Invest. 1975;55(5):1132–1135. doi: 10.1172/JCI108015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Applebaum DM, Goldberg AP, Pykälistö OJ, Brunzell JD, Hazzard WR. Effect of estrogen on post-heparin lipolytic activity. Selective decline in hepatic triglyceride lipase. J Clin Invest. 1977;59(4):601–608. doi: 10.1172/JCI108677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ananthakrishnan P, Cohen DB, Xu DZ, Lu Q, Feketeova E, Deitch EA. Sex hormones modulate distant organ injury in both a trauma/hemorrhagic shock model and a burn model. Surgery. 2005;137(1):56–65. doi: 10.1016/j.surg.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 62.Caruso JM, Deitch EA, Xu DZ, Lu Q, Dayal SD. Gut injury and gut-induced lung injury after trauma hemorrhagic shock is gender and estrus cycle specific in the rat. J Trauma. 2003;55(3):531–539. doi: 10.1097/01.TA.0000025584.46870.95. [DOI] [PubMed] [Google Scholar]
- 63.Baldus S, Rudolph V, Roiss M, Ito WD, Rudolph TK, Eiserich JP, Sydow K, Lau D, Szöcs K, Klinke A, Kubala L, Berglund L, Schrepfer S, Deuse T, Haddad M, Risius T, Klemm H, Reichenspurner HC, Meinertz T, Heitzer T. Heparins increase endothelial Nitric Oxide availability by Liberating vessel-immobolized Myeloperoxidase. Circulation. 2006;113(15):1871–1878. doi: 10.1161/CIRCULATIONAHA.105.590083. [DOI] [PubMed] [Google Scholar]
- 64.Not LG, Brocks CA, Vamhidy L, Marchase RB, Chatham JC. Increased O-linked beta-N-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 hours after trauma-hemorrhage in rats. Crit Care Med. 2010;38(2):562–571. doi: 10.1097/CCM.0b013e3181cb10b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuebler JF, Toth B, Rue LW, III, Wang P, Bland KI, Chaudry IH. Differential fluid regulation during and after soft tissue trauma and hemorrhagic shock in males and proestrus females. Shock. 2003;20(2):144–148. doi: 10.1097/01.shk.0000072127.33223.f1. [DOI] [PubMed] [Google Scholar]