

Abstract
Ethanol may cause fetal alcohol spectrum disorders (FASD) in part by inhibiting cell adhesion mediated by the L1 neural cell adhesion molecule. Azialcohols photolabel Glu-33 and Tyr-418, two residues that are predicted by homology modeling to lie within 2.8 Å of each other at the interface between the Ig1 and Ig4 domains of L1 (Arevalo, E., Shanmugasundararaj, S., Wilkemeyer, M. F., Dou, X., Chen, S., Charness, M. E., and Miller, K. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 371–375). Using transient transfection of NIH/3T3 cells with wild type (WT-L1) and mutated L1, we found that cysteine substitution of both residues (E33C/Y418C-L1) significantly increased L1 adhesion above levels observed for WT-L1 or the single cysteine substitutions E33C-L1 or Y418C-L1. The reducing agent β-mercaptoethanol (βME) reversibly decreased the adhesion of E33C/Y418C-L1, but had no effect on WT-L1, E33C-L1, or Y418C-L1. Thus, disulfide bond formation occurs between Cys-33 and Cys-418, confirming both the close proximity of these residues and the importance of Ig1-Ig4 interactions in L1 adhesion. Maximal ethanol inhibition of cell adhesion was significantly lower in cells expressing E33C/Y418C-L1 than in those expressing WT-L1, E33C-L1, or Y418C-L1. Moreover, the effects of βME and ethanol on E33C/Y418C-L1 adhesion were non-additive. The cutoff for alcohol inhibition of WT-L1 adhesion was between 1-butanol and 1-pentanol. Increasing the size of the alcohol binding pocket by mutating Glu-33 to Ala-33, increased the alcohol cutoff from 1-butanol to 1-decanol. These findings support the hypothesis that alcohol binding within a pocket bordered by Glu-33 and Tyr-418 inhibits L1 adhesion by disrupting the Ig1-Ig4 interaction.
Keywords: Alcohol, Cell Adhesion, Cysteine-mediated Cross-linking, Development, Drug action, Fetal Alcohol Spectrum Disorders, L1 Neural Cell Adhesion Molecule
Introduction
Alcohol exposure during pregnancy is the leading cause of preventable mental retardation in the Western world (1, 2). Depending on timing, dose, and duration of exposure, alcohol causes a range of facial and brain dysmorphology, growth retardation, and cognitive, neurological, and behavioral abnormalities, referred to as fetal alcohol spectrum disorders (FASD)2 (3). Alcohol is a weak, pleiotropic drug that disrupts fetal development through a variety of mechanisms (4, 5). One potentially important target molecule for alcohol is the L1 neural cell adhesion molecule (CAM), a developmentally critical protein (6–8).
Children with mutations in the gene for L1 have brain lesions that resemble those of children with FASD, including hydrocephalus, agenesis, or hypoplasia of the corpus callosum, and cerebellar dysplasia (7, 9, 10). Concentrations of ethanol attained after one drink inhibit the adhesion of L1 expressed in fibroblasts, neural cell lines, and cerebellar granule neurons (CGN) (6, 7). Furthermore, ethanol inhibits L1-mediated neurite outgrowth in CGNs with similar potency to its inhibition of L1 adhesion (11, 12). Finally, drugs that block ethanol inhibition of L1 adhesion also prevent ethanol teratogenesis in mouse embryos (13–18).
L1 is an immunoglobulin transmembrane glycoprotein (8). The extracellular domain (ECD) includes six Ig domains and five fibronectin III repeats. The first four Ig domains (L1Ig1–4) comprise the minimal elements required for L1 adhesion (19, 20). The crystal structure of neurofascin, a member of the L1 family of CAMs, and homology modeling with related CAMs suggest that L1Ig1–4 folds into a horseshoe structure, with Ig1 in apposition to Ig4 and Ig2 in apposition to Ig3 (20–25) (Fig. 6A). Electron microscopy has captured both a horseshoe and an extended conformation of L1Ig1–4, and functional studies suggest that the horseshoe conformation is associated with higher levels of adhesion (22–24, 26). Importantly, there is a disproportionate number of disease-causing missense mutations located at Ig domain interfaces (10, 21, 22, 27), many of which reduce L1 homophilic binding (25, 28).
FIGURE 6.

Homology models of WT-L1 and mutants. A, homology model of WT-L1 Ig1-Ig4 showing the position of the mutated residues (36). The secondary structure is shown with separate colors for each Ig domain, and the Ig2-Ig3 linker is colored pink. The residues to be mutated are shown with cyan carbons and in ball and stick mode. The N-terminal domain cannot be homology modeled and is omitted. The first residue of the homology model is Glu-33. B, a close-up view of the superposition of reduced (magenta) and non-reduced (dark-green) forms of the double mutant E33C/Y418C-L1. Sulfur atoms are colored yellow, and the disulfide bridge is shown as a yellow rod. The Cα carbon of Cys-33 moves 2 Å when the disulfide bond is reduced. C, similar view of WT-L1 (colored as in panel A) superimposed on non-reduced E33C/Y418C-L1, colored as in panel B. Once again the Cα carbon of Cys-33 differs in position by 2 Å, but the angle of movement is different from that in panel B, so that when WT-L1 and E33C/Y418C-L1 are superimposed, their Cα do not overlap (supplemental Fig. S1). D, similar view of WT-L1 superimposed on E33A-L1 (yellow) showing the lack of secondary structure change.
Pharmacological data support the hypothesis that ethanol inhibits L1 adhesion by interacting with a hydrophobic site on the L1-ECD. The potency of 1-alcohol inhibition of L1 adhesion increases progressively from methanol through 1-butanol, but higher 1-alcohols are inactive (6, 7, 29). This alcohol cutoff phenomenon suggests a discrete 1-alcohol binding pocket of delimited size (7, 15, 29–32). Furthermore, some molecules that lack intrinsic capacity to inhibit L1 adhesion antagonize alcohol inhibition of L1 adhesion (15, 16, 29, 33, 34). Among these, 1-octanol and certain small peptides also prevent ethanol-induced apoptosis, growth retardation, and delayed closure of the neural tube (13–17, 29, 33–35).
We recently reported that at low concentrations azibutanol and azioctanol each photoincorporated into Glu-33 on Ig1 and Tyr-418 on Ig4 of a purified protein construct comprising the first four Ig domains of L1 (L1Ig1–4) (36). Although separated by 385 residues, homology modeling located Glu-33 and Tyr-418 within 3 Å of each other at a domain interface between Ig1 and Ig4. We postulated that Glu-33 and Tyr-418 are part of a single alcohol binding pocket formed only when L1 adopts a horseshoe conformation. However, the photolabeling of these residues might have been nonspecific, the functional significance of these residues has not been investigated, and it cannot be assumed that the various conformations that intact L1 might adopt in a cellular system are accurately depicted in our crystallography-based homology model of L1Ig1–4. Here we provide evidence in a cellular system for the proximity of Glu-33 and Tyr-418 and their involvement in L1 adhesion and its inhibition by alcohol.
EXPERIMENTAL PROCEDURES
Mutagenesis
Several L1 mutants were generated from a vector containing wild-type human L1 (hL1) cDNA gene (RSLE+) kindly provided by Dr. Patricia Maness (University of North Carolina; Chapel Hill, NC). In the L1 constructs with single site mutations, residues Glu-33 and Tyr-418 were replaced with Cys or Ala. In the L1 double site mutant, E33C/Y418C-L1, both Glu-33 and Tyr-418 were replaced with Cys, as indicated in Table 1. PCR mutagenesis was performed with Phusion Hot Start DNA Polymerase (NEB). A pair of primers, 5′-AT GAA GGA CAC CAT GTG ATG TGC CCA CCT GTC ATC-3′ and 5′-CATC ACA TGG TGT CCT TCA TAT TCC TCG GGG-3′, were used to replace Glu-33 with Cys; a pair of primers, 5′-CTC TTG CTG GCC AAT GCC TAC ATC TAC GTT GTC CAG CTG-3′ and 5′-CAG CTG GAC AAC GTA GAT GTA GGC ATT GGC CAG CAA GAG-3′, were used to replace Tyr-418 with Cys. The PCR product was digested with DpnI restriction enzyme (NEB), and the purified PCR product was transformed into Escherichia coli DH5α for plasmid amplification and DNA preparation (New England Biolabs). All mutations were confirmed by DNA sequencing. The E33A-L1 mutation was created and verified by Biopioneer, Inc.
TABLE 1.
Effect of point mutations on L1 adhesion and its inhibition by ethanol and βME
The IC50 is the concentration of ethanol that produces half-maximal inhibition of L1 adhesion. The maximal level of L1 adhesion was determined at a concentration of 100 mm ethanol. βME was used at a concentration of 30 mm in all indicated experiments. The cutoff refers to the longest 1-alcohol that inhibits L1 adhesion; e.g., C4 indicates inhibition by 1-butanol, but not 1-pentanol, and C10 indicates inhibition by 1-decanol, but not 1-undecanol.
| Mutation | L1 adhesion | Ethanol, maximal inhibition | Ethanol, IC50 | βME inhibition | Alcohol cutoff | ||||
|---|---|---|---|---|---|---|---|---|---|
| % | n | % | n | mm | n | % | n | ||
| WT | 25.6 ± 1.6 | (21) | 55.9 ± 4.8 | (19) | 5.1 ± 1.6 | (7) | 5.3 ± 13.3 | (7) | C4 |
| E33C/Y418C | 34.2 ± 2.1a | (21) | 37.8 ± 3.4a | (21) | 9.3 ± 3.3 | (7) | 46.4 ± 4.7b | (7) | C4 |
| E33C | 25.4 ± 1.9 | (14) | 56.0 ± 7.2 | (14) | 5.4 ± 1.6 | (7) | 2.8 ± 1.0 | (5) | C4 |
| Y418C | 23.7 ± 2.6 | (15) | 53.3 ± 4.6 | (10) | 7.8 ± 4.6 | (7) | −2.5 ± 4.0 | (5) | C4 |
| E33A | 24.9 ± 1.9 | (10) | 56.2 ± 10.0 | (9) | 3.8 ± 1.0 | (6) | – | C10 | |
a p < 0.01, ethanol maximal inhibition, mutant compare to WT.
b p < 0.001, L1 adhesion, mutant compared to WT; βME inhibition compared to 0 inhibition.
Transient Transfection of L1
NIH/3T3 fibroblasts were plated in 50 mm silicone-coated glass plates and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum (Invitrogen) at 37 °C in an atmosphere of 90% air and 10% CO2. At 50% confluence, cells were transiently transfected with 5 μg of plasmid DNA with either wild-type L1 (WT-L1) or mutant L1 using 10 μl of PolyFect transfection reagent from Qiagen, based on the manual. Twenty-four hours after transfection, cells were carefully washed three times with 10 ml of DMEM and then detached with PBS supplemented with 2 mm EDTA. Cells were collected by centrifugation, suspended in DMEM supplemented with 10% bovine serum, plated in T25 flasks, and cultured for an additional 24 h.
Cell Aggregation Assay
Subconfluent cells were detached in PBS supplemented with 2 mm EDTA and 0.1 mg/ml DNase and separated into a single cell suspension by trituration. Between 150,000 and 200,000 cells were added to 12-well plates in a volume of 1 ml. Plates were agitated on a rotary shaker at 60 rpm for 10 min. Cells were then visualized at 200× magnification on an inverted stage Nikon microscope. Images were captured from four orthogonal fields per well located at half the distance from the center to the edge. The percentage of adherent and single cells was scored from four images of at least three replicate wells without knowledge of experimental conditions. L1 adhesion was defined as the difference in the percentage of adherent cells between cells transfected with L1 plasmid DNA and cells treated with PolyFect alone. Inhibition of L1 adhesion was calculated as [100 (1- (the ratio of L1 adhesion in the presence and absence of inhibitors))] (7, 29). Importantly, these mutational analyses were performed using transfected NIH/3T3 cells. All of our prior findings on alcohol and L1 in NIH/3T3 cells have been replicated in neural cell lines, neurons, or embryos (7, 14–16, 33, 34, 37).
Aqueous alcohol solutions were prepared daily. For longer chain alcohols (≥ octanol), stocks were made in DMSO and diluted in assay buffer. At the concentrations used, DMSO had no effect on cell adhesion (29). Alcohols and β-mercaptoethanol (βME) (Sigma-Aldrich) were added below the surface of cell suspensions just prior to the initiation of cell aggregation and did not affect cell viability, as determined by exclusion of trypan blue (not shown).
Curve Fitting of Dose Response Curve
The ethanol concentration-response curves in Fig. 4 were analyzed using the raw adhesion data to avoid skewing the data with normalizations (and compounding errors). The two curves, each containing 70 points, were fitted simultaneously with equal slopes to a function in Equation 1,
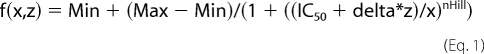 |
where the parameter delta represents the difference between the data sets, IC50 is the midpoint of the curve, nHill is the slope or Hill coefficient, Max is the control fraction of adherent cells in the absence of ethanol and Min that at high ethanol concentration. Fitting was achieved using the NLIN procedure of the SAS package.
FIGURE 4.
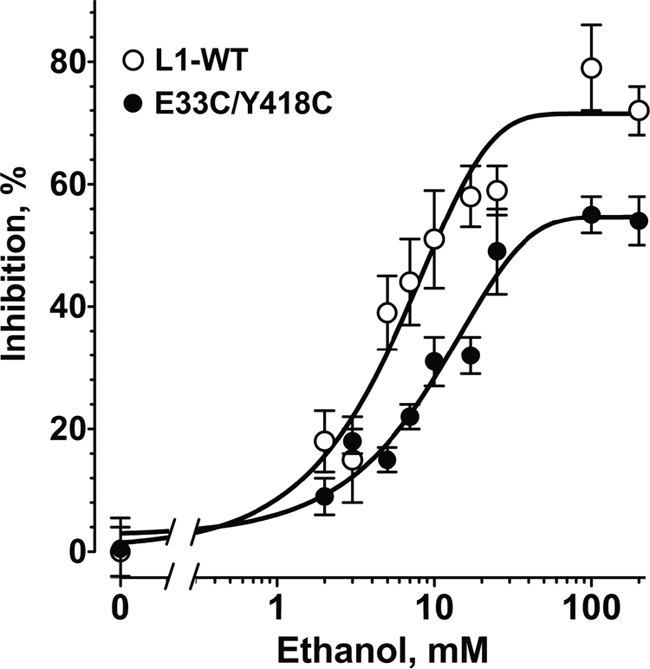
Concentration-response curves for ethanol inhibition of L1 adhesion in NIH/3T3 cells expressing WT-L1 and E33C/Y418C-L1. Values shown are the mean ± S.E. percent inhibition from seven independent experiments. Both curves use the same value of 0% inhibition for 0 mm ethanol. Curve fitting (see “Experimental Procedures”) revealed a calculated nHill of 1.1 ± 0.2 and IC50 of 9.3 ± 3.3 mm for E33C/Y418C-L1 and 5.1 ± 1.6 mm for WT-L1.
L1 Immunoblots
Cells were grown to 75–85% confluence, harvested in PBS, and pelleted by centrifugation. Cells were resuspended in Nonidet P-40 lysis buffer (NaCl 150 mm, Tris 50 mm pH 8.0, Nonidet P-40 1.0%) supplemented with 1:100 (v/v) Protease Arrest and 1:100 (v/v) Phosphatase Inhibitor Mixture (Calbiochem), incubated on ice for 5 min, and centrifuged at 4 °C at 16,000 × g for 15 min to remove the insoluble fraction. Protein was separated on 10% SDS-polyacrylamide gels and blotted onto polyvinylidene fluoride (PVDF) membranes (Millipore). Membranes were incubated with goat anti-L1 polyclonal antibody SC-1508 (Santa Cruz Biotechnology) for 2 h at room temperature. After three washes with TBS buffer (NaCl 150 mm, Tris 50 mm, pH 7.4, Tween-20 0.05%), membranes were incubated at room temperature for 1 h with an anti-goat mAb conjugated with alkaline phosphatase (Invitrogen). The PVDF membranes were washed three times again with TBS and the immunolabeled bands were visualized with 5-bromo-4-chloro-3-indoyl phosphate/nitroblue tetrazolium (BCIP/NBT) substrate (Vector Laboratories, Inc.). Protein expression levels were compared using densitometric analysis of protein bands from scanned images of PVDF membranes using NIH Image J software. Tubulin was used as a control to normalize L1 expression.
L1 Immunohistochemistry
NIH/3T3 cells were plated on 50 mm glass Petri dishes in DMEM supplemented with 10% bovine serum and grown to 75–85% confluence. Cells were fixed in 4% paraformaldehyde, blocked with PBS supplemented with 5% bovine serum (PBS/BS), and stained overnight at room temperature with mouse L1 mAb 5G3 in PBS/BS, washed three times with PBS, incubated for 3 h at room temperature with goat anti-mouse antibody conjugated with Alexa Fluor 594 (Invitrogen) in PBS/BS, and washed again with PBS. Images were captured using a Nikon Eclipse TE2000-U confocal microscope and EZ-C1 Viewer V.3.60 (Nikon). Monoclonal antibody 5G3 recognizes an epitope overlapping the Ig1 and Ig2 domains of L1 (38).
Flow Cytometry
Cells cultured in T75 flasks or 6-well plates were detached in PBS supplemented with 2 mm EDTA and separated into a single-cell suspension by trituration. Cells were incubated with anti-L1 mAb 5G3 in PBS/BS at room temperature for 1 h, washed once with PBS, and then incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) for 30 min. After three washes with PBS, cells were then fixed in 4% paraformaldehyde for flow cytometry analysis. Geometric means were calculated from 5000 gated events using a FACScalibur system (BD Biosciences). In selected experiments, surface expression of L1 was also measured using mAb UJ127 (Thermo Scientific), which recognizes an epitope on the fibronectin-like repeat-4 domain of L1 (38).
L1 Homology Modeling
The homology modeling of residues 33–424 of hL1 Ig1-Ig4 based on the structure of axonin Ig1-Ig4 has been reported previously (36). The mutated structural models were built using this model as a template. All the calculations were carried out with Discover Studio Suite 2.1 (Accelrys, CA). We used the MODELLER module to make in silico mutations at positions 33 and 418. All new models were then subjected to molecular dynamics and energy minimization over all residues using the Smart Minimizer algorithm. The temperature was increased to 3000 K and then cooled off to room temperature in 2000 steps with the time step of 0.001 ps. The spherical cutoff was used for the electrostatics parameter.
The geometry of the new hL1 Ig1-Ig4 homology models was verified using Procheck (39). The geometrical parameters (bond lengths and angles) of the molecules were within the experimental error. In the Ramachandran plot, there is no significant deviation of the π-Ψ angles compared with the WT-L1 homology model. The root mean square deviations (RMSD) for the Cα atoms were calculated using the top3d program (CCP4, 1994).
Statistic Analysis
Data are expressed as mean ± S.E., except as noted. Statistical analysis was performed using the Student's t test with Prism 5 (Graphpad Software Inc.). Statistical significance was defined as p < 0.05. Nonlinear least squares analysis was done with Igor (Wave Metrics Inc).
RESULTS
Effect of Cysteine Substitution at Glu-33 and Tyr-418 on L1 Adhesion
To determine whether Glu-33 and Tyr-418 are close enough to form a functional disulfide bond (40), we created hL1 constructs with cysteine substitutions at Glu-33 (E33C-L1), Tyr-418 (Y418C-L1), or both sites (E33C/Y418C-L1) and transiently transfected NIH/3T3 fibroblast cells with WT-L1 and mutant-L1 plasmid DNA. Forty-eight hours after transfection, ∼70% of cells showed positive L1 fluorescence staining. There were no differences in the cell surface expression of L1, measured with mAb 5G3, or in the immunolabeling of L1 extracted from whole cell lysates of WT-L1 and mutant-L1-expressing cells (Fig. 1). Likewise, flow cytometry using mAb UJ127, which recognizes a different epitope in the L1-ECD than mAb 5G3 (38), did not disclose differences in cell surface expression of L1 between WT-L1 and E33C/Y418C-L1 (not shown). Hence, differences in the adhesion of transfected cells should reflect primarily the effects of mutations on the intrinsic adhesiveness of L1. The transient expression of WT-L1 and mutant L1 increased cell adhesion above levels observed in cells treated solely with the transfection agent, PolyFect (Fig. 2A). L1 adhesion was significantly higher in cells transfected with E33C/Y418C-L1 (34.2 ± 2.1%, n = 21) than in cells transfected with WT-L1 (25.6 ± 1.6%; n = 21; p < 0.01)(Fig. 2B). In contrast, levels of adhesion did not differ between cells transfected with WT-L1 and the single cysteine mutations E33C-L1 and Y418C-L1 (Table 1).
FIGURE 1.

Expression of WT-L1 and mutant-L1. A, immunofluorescence staining of L1 in WT-L1 and mutant L1-expressing NIH/3T3 cells using mAb 5G3. Bar, 50 μm. B, Western blot analysis, using polyclonal antibody SC-1508, of L1 expression in whole cell extracts of NIH/3T3 cells transiently transfected with the indicated L1 constructs. Lanes marked Control and PolyFect are from cells treated without or with the transfection reagent, but without L1 plasmid DNA. Solid arrow, L1; open arrow, β-tubulin. C, quantitative densitometric analysis of Western blots from the experiments described in B. Data shown represent the mean ± S.E. percent of WT-L1 levels for each construct obtained from eight independent experiments. L1 expression was normalized to tubulin expression from the same samples. Expression of the mutant forms of L1 did not differ significantly from expression of WT-L1. D, flow cytometry analysis of cell surface expression of WT-L1 and L1 mutants using mAb 5G3. Geometric means of cells expressing mutant L1 were normalized to values from cells expressing WT-L1 (dotted line in C and D). Data represent the mean ± S.E. from at least four independent experiments.
FIGURE 2.

Inhibition of L1 adhesion by ethanol and βME in NIH/3T3 cells expressing WT-L1 and E33C/Y418C-L1. A, number of single and adherent cells was counted from phase contrast micrographs viewed at 100× magnification. Bar, 200 μm. B, L1 adhesion levels in NIH/3T3 cells expressing WT-L1 (open bars) or E33C/Y418C-L1 (solid bars) in the presence or absence of 100 mm ethanol or 30 mm βME. C, percent inhibition of adhesion by ethanol or βME. Data shown represent mean ± S.E., n = 7–21. *, p < 0.01; ***, p < 0.0001.
Effect of Reducing Conditions on E33C/Y418C-L1 Adhesion
The observation that the double mutant E33C/Y418C-L1 showed increased adhesion, but E33C-L1 and Y418C-L1 did not, suggests that Cys-33 and Cys-418 are close enough to form a functional disulfide bond. If cross-linking Ig1 and Ig4 increases L1 adhesion, then breaking this disulfide bond should decrease L1 adhesion. Therefore, we measured E33C/Y418C-L1 adhesion in the presence and absence of 30 mm βME, a strong reducing agent. βME inhibited E33C/Y418C-L1 adhesion by 46.4 ± 4.7% (n = 7, p < 0.001), an effect comparable to that produced by ethanol (Fig. 2). By contrast, βME had no significant effect on WT-L1, E33C-L1, or Y418C-L1 adhesion (Fig. 3). Treatment of WT-L1- or E33C/Y418C-L1-expressing cells for 10 min with ethanol or βME did not significantly change cell surface expression of L1, as determined by flow cytometry with mAb 5G3 (not shown). These data suggest that Cys-33 spontaneously forms a disulfide bond with Cys-418, rather than with other proximate cysteines.
FIGURE 3.

Effect of βME and ethanol on L1 adhesion in cells expressing WT-L1 and mutant-L1. A, βME inhibition of L1 adhesion in WT-L1 (white bars), E33C/Y418C-L1 (black bars), E33C-L1 (gray bar), or Y418C-L1 (gray bar). B, reversibility of βME inhibition on E33C/Y418C-L1 adhesion. Cells expressing WT-L1 (white bars) or E33C/Y418C-L1 (black bars) were pretreated with βME for 15 min, followed by washing with PBS supplemented with 30 mm βME (βME; left bars) or with plain PBS (PBS; right bars) to remove the βME. Assays were conducted using the same solutions as the wash. C, inhibition of E33C/Y418C-L1 adhesion by 100 mm ethanol, 30 mm βME, or both. Data are mean ± S.E. from 4–9 determinations. *, p < 0.01; ***, p < 0.0001.
To verify that the effects of βME were due to the reduction of a disulfide bond, rather than to nonspecific effects on cellular or protein integrity, we determined whether the actions of βME were reversible. L1-expressing cells were pretreated with 30 mm βME for 15 min, washed twice with PBS, and then tested for adhesion in the absence or presence of βME. Fig. 3B demonstrates that βME pretreatment does not inhibit E33C/Y418C-L1 adhesion if cells are washed prior to assay.
Combined Effects of Ethanol and βME
Ethanol inhibited WT-L1 adhesion to the same extent as βME inhibited E33C/Y418C-L1 adhesion, suggesting that ethanol and βME disrupt Ig1-Ig4 interactions at the same locus, between positions 33 and 418. Therefore, we asked whether the effects of ethanol and βME on E33C/Y418C-L1 adhesion were additive or synergistic. Cell adhesion assays were performed in the presence of 100 mm ethanol, 30 mm βME, or both. Ethanol did not potentiate βME inhibition of E33C/Y418C-L1 adhesion (Fig. 3C).
Ethanol Inhibition of WT-L1 and E33C/Y418C-L1 Adhesion
We proposed that ethanol inhibits L1 adhesion partly by reducing interactions between Ig1 and Ig4. To determine whether cross-linking Ig1 and Ig4 reduces the effects of ethanol, we measured cell adhesion in WT-L1- and E33C/Y418C-L1-expressing cells in the presence of 0–100 mm ethanol. Low concentrations of ethanol inhibited WT-L1 and E33C/Y418C-L1 adhesion in a concentration-dependent manner (Fig. 4, Table 1). The slope of the curves (nHill) was 1.1 ± 0.2, as expected for mass action or many non-interacting sites of equal affinity. Ethanol appeared less potent in inhibiting E33C/Y418C-L1 (IC50 9.3 ± 3.3; mean ± S.D.) than WT-L1 (IC50 5.1 ± 1.6 mm), although the difference was not statistically significant (p = 0.24). Maximal ethanol inhibition of L1 adhesion was determined in a series of comparisons at 100 mm ethanol, a concentration 20-fold higher than the IC50. Maximal ethanol inhibition was significantly lower in cells expressing E33C/Y418C-L1 (37.8 ± 3.4%, n = 21) than in those expressing WT-L1 (55.9 ± 4.8%, n = 19, p < 0.01)(Fig. 2C). The potency and efficacy of ethanol in these transiently transfected NIH/3T3 cells was comparable to what we reported in stably transfected NIH/3T3 cells and in NG108–15 neuroblastoma x glioma hybrid cells treated with BMP-7 to induce L1 gene expression (6, 7). Neither of the single cysteine mutations affected ethanol potency or maximal effect (Table 1). These data confirm that cross-linking Ig1 and Ig4 reduces ethanol inhibition of L1 adhesion.
Modification of the Alcohol Cutoff by the E33A Mutation
If Glu-33 and Tyr-418 surround an alcohol binding pocket, then replacing Glu-33 with Ala (E33A-L1), a residue with a less bulky side chain, might expand the binding pocket and increase the alcohol cutoff, as observed for the ρ1-GABAA and α1-glycine receptors (32). The E33A-L1 mutation had no effect on the expression or cell surface staining of L1 (Fig. 1). To characterize the 1-alcohol cutoff, we measured L1 adhesion in the absence and presence of 1-alcohols from methanol to 1-undecanol at aqueous concentrations calculated to produce equivalent membrane concentrations (29). In cells expressing WT-L1, there was a sharp cutoff in 1-alcohol inhibition of L1 adhesion between 1-butanol and 1-pentanol, and 1-pentanol antagonized ethanol inhibition of L1 adhesion (Fig. 5), as reported previously (33, 34). The E33A-L1 mutation did not alter L1 adhesion or its inhibition by ethanol (Table 1), but dramatically increased the 1-alcohol cutoff from 1-butanol to 1-decanol and converted 1-octanol from an antagonist to an agonist (Fig. 5). 1-Undecanol, the largest 1-alcohol that did not inhibit E33A-L1 adhesion, potently antagonized ethanol inhibition of E33A-L1 adhesion, as was observed for 1-pentanol in WT-L1-expressing cells.
FIGURE 5.

Effect of the E33A-L1 mutation on the cutoff for 1-alcohol inhibition of L1 adhesion. Values shown are the mean ± S.E. levels of L1 adhesion (top panels) and percent inhibition of L1 adhesion (bottom panels) for cells expressing WT-L1 (left panels, n = 4) and E33A-L1 (right panels, n = 6). The abscissa labels show the identity and concentration of each 1-alcohol as follows: (number of carbons)/(concentration, mm); e.g. C10/5 represents 5 mm 1-decanol. In WT-L1-expressing cells, the cutoff is between 1-butanol and 1-pentanol; in E33A-L1-expressing cells, the cutoff is between 1-decanol and 1-undecanol. Ethanol inhibition of L1 adhesion is antagonized by 1-pentanol in WT-L1- and by 1-undecanol in E33A-L1-expressing cells. *, p < 0.01; **, p < 0.001.
Homology Modeling of L1 Mutations
The results of the homology modeling of the first four Ig domains of L1 are shown in Fig. 6. The published WT-L1 model (36) predicts a horseshoe structure in which Ig1 and Ig4 and Ig2 and Ig3 lie opposed to each other in anti-parallel fashion (Fig. 6A). The mutated L1 models maintained the same secondary structure and the shifts in RMSD values of the α carbons (Cα) were modest (supplemental Table S1). The highest RMSD values (>1.0 Å) were obtained in comparing WT-L1 and reduced E33C/Y418C-L1. The distance between the Cα atoms of residues 33 and 418 was 8.9 ± 0.1 Å in models of WT-L1 and all of the single mutations. However, when both residues were mutated to cysteine, disulfide formation reduced the separation between the Cα of residues 33 and 418 from 9 to 6.3 Å (supplemental Table S2 and Fig. 6, B and C). Reduction of the disulfide bond increased the distance between Cys-33 and Cys-418, but the separation was still 1.6 Å less than in WT-L1 (supplemental Table S2 and Fig. S1). Reduction caused both sulfur atoms to swing away from the interface to become separated by 6.9 Å and the Cα of residue 33 moves 2.0 Å along the interface (Fig. 6B).
The most striking change in the E33A mutation was loss of the -CH2-COO moiety with a resultant increase in space between Ala-33 and Tyr-418. The E33A mutation caused a negligible change in the secondary structure (Fig. 6D and supplemental Table S1): the Cα atom separation decreased by a negligible 0.2 Å; side chain motions caused the Cβ atom separation to decrease by 0.5 Å, and the tyrosine hydroxyl moved away from Ig1 by 0.9 Å, because it no longer formed a H-bond.
DISCUSSION
Three principal findings of this study support the hypothesis that alcohol inhibits L1 adhesion partly by interacting with a binding site at the domain interface between Ig1 and Ig4 of the L1-ECD. First, Glu-33 on Ig1 and Tyr-418 on Ig4 are sufficiently close to form a disulfide bond after cysteine substitution. Second, formation and disruption of this disulfide bond modulates L1 adhesion and its inhibition by ethanol. Third, mutation of Glu-33 to Ala markedly changes the pharmacology of 1-alcohol inhibition of L1 adhesion.
Proximity of Glu-33 and Tyr-418
Our published homology model predicted that two widely separated residues that were photolabeled by azibutanol and azioctanol, Glu-33 on Ig1 and Tyr-418 on Ig4, were part of a single alcohol binding pocket within the interface between Ig1 and Ig4 (36). Because of the uncertainties inherent in homology modeling of purified peptide fragments (41), it remained possible that in cellular systems the two photolabeled residues are not closely apposed or are associated with two separate alcohol binding sites. Therefore, we sought to verify this prediction using a cross-linking strategy (40). Homology modeling predicted that after cysteine substitution, the β-carbons of Cys-33 and Cys-418 would be separated by 6.5 Å under reducing conditions (Fig. 6B). It has been suggested that disulfide formation occurs when the β-carbons of two cysteines are separated by ≤ 4.5 Å. Much greater equilibrium separations (∼15–20 Å) still lead to disulfide formation in the presence of permissive local protein dynamics (40). The observation that E33C/Y418C-L1 adhesion is greater than that of WT-L1 and is markedly decreased by reducing conditions provides strong evidence that Cys-33 and Cys-418 can approach closely enough to allow disulfide bond formation, thereby cross-linking Ig1 and Ig4. By inference, this suggests that Glu-33 and Tyr-418 can approach closely enough to allow hydrogen bonding, as predicted by our homology model.
Ig1-Ig4 Interactions and L1 Adhesion
X-ray crystallography has revealed a horseshoe structure for a number of immunoglobulin cell adhesion molecules related to L1 (21, 22, 42), and several models have been proposed in which L1 and homologous molecules alternate between the horseshoe and more extended conformations (26, 42). The fact that we can perturb adhesion by disrupting just two of the many residues that mediate interactions between Ig1 and Ig4 suggests that we are creating subtle distortions of the horseshoe conformation rather than the fully extended conformation captured by rotary-shadowed electron microscopy (23).
Adhesion studies of the double mutant E33C/Y418C-L1 under non-reducing and reducing conditions provide evidence for an equilibrium between more and less adhesive conformations of L1. The adhesion of E33C/Y418C-L1 was significantly greater than that of WT-L1, E33C-L1, or Y418C-L1, suggesting that cross-linking Ig1 and Ig4 at Cys-33-Cys-418 traps a conformation that favors adhesion. Indeed, homology modeling revealed that the α carbon of residue 33 in E33C/Y418C-L1 is closer to Ig4 than in WT-L1 or E33A-L1. Furthermore, reducing conditions reversibly decreased L1 adhesion in E33C/Y418C-L1, but had no effect in WT-L1, E33C-L1, or Y418C-L1. The most consistent interpretation of these findings is that the cysteines at Cys-33 and Cys-418 form a functional and reversible disulfide bond that stabilizes a more adhesive horseshoe conformation and potentiates cell adhesion. In effect, our adhesion data and homology modeling provide evidence for two states of the horseshoe conformation that differ significantly in their adhesive properties, but only slightly in structure.
It is noteworthy that the increase in L1 adhesion (22.2 ± 4.5%, n = 21) conferred by creating a disulfide bond by mutation of WT-L1 to E33C/Y418C-L1 was significantly less than the decrease in L1 adhesion (46.4 ± 4.7%, n = 7; p < 0.01) induced by breaking the disulfide bond in E33C/Y418C-L1 with βME. If one assumes that cross-linking Ig1 and Ig4 stabilizes the horseshoe conformation and breaking the disulfide bond shifts the equilibrium toward a less adhesive conformation, these observations imply that under our experimental conditions, the equilibrium in WT-L1 favors the more adhesive over the less adhesive conformation, consistent with observations made in purified fragments of L1 and neurofascin (21, 23). If so, then agents such as ethanol might decrease WT-L1 adhesion by shifting the equilibrium from the more adhesive to the less adhesive conformation.
Glu-33 and Tyr-418 Mediate Actions of Ethanol
Our data support the suggestion that Glu-33 and Tyr-418, the two residues in L1Ig1–4 most consistently photolabeled by azialcohols, mediate ethanol inhibition of L1 adhesion. Ethanol inhibited WT-L1 adhesion to the same extent as βME inhibited E33C/Y418C-L1 adhesion. One possible explanation for this observation is that ethanol and βME act similarly to decrease the Ig1-Ig4 interaction, ethanol, by weakening a hydrogen bond between Glu-33 and Tyr-418 in WT-L1, and βME by breaking a disulfide bond at the same location in E33C/Y418C-L1. Consistent with this hypothesis, ethanol was less effective in decreasing adhesion in E33C/Y418C-L1 than in WT-L1. The observation that ethanol retained modest inhibitory activity and unchanged potency in E33C/Y418C-L1 implies that additional actions at the Ig1-Ig4 interface or elsewhere contribute to ethanol inhibition of L1 adhesion. Of note, ethanol did not potentiate βME inhibition of L1 adhesion in the double mutant, suggesting that ethanol stabilizes a less adhesive horseshoe conformation of L1 similar to that adopted following reduction of the disulfide bond.
The mutations E33C-L1, E33A-L1, and Y418C-L1 should disrupt the hydrogen bond between Glu-33 and Tyr-418, yet none of these altered L1 adhesion or its inhibition by ethanol. Therefore, simple disruption of the interaction between residues 33 and 418 alone cannot account for the effects of ethanol. This is not surprising, given the large number of sites of interaction between Ig1 and Ig4 and Ig2 and Ig3 identified in the recent crystal structure of the Ig1-Ig4 domains of neurofascin (21). In fact, azibutanol and azioctanol did photolabel two additional residues between positions 24 and 27, a region encoded by exon2 that is important for homophilic interactions (43). Unfortunately, there is insufficient structural information about the N-terminal domain in related molecules to construct a homology model. However, if these residues are part of the same alcohol binding pocket, it is likely that they interact with Ig4 and contribute to the Ig1-Ig4 interaction.
Glu-33 Borders an Alcohol Binding Pocket
Experiments with the E33A-L1 mutation provide some of the strongest evidence that Glu-33 and Tyr-418 border an alcohol binding pocket. This mutation replaces the bulky, negatively charged side chain of glutamic acid with the small, neutral side chain of alanine. Although L1 adhesion, ethanol potency, and ethanol efficacy were unchanged in E33A-L1, there was a dramatic increase in the cutoff for 1-alcohols from 1-butanol to 1-decanol. This finding suggests that steric hindrance from the side chain of Glu-33 in WT-L1 limits occupancy within the alcohol binding pocket to alcohols of four or fewer carbons. The shift in cutoff from 1-butanol to 1-decanol in E33A-L1 is consistent with the removal of four atoms from the glutamic acid side chain and some additional small changes in the interaction between Ig1 and Ig4. A similar observation was made for amino acid substitutions within the α1-glycine and ρ1-GABAA receptors (32, 44). Mutations at critical residues increased or decreased the 1-alcohol cutoff without altering ethanol potency or the actions of glycine or GABA (32).
One of the more striking effects of the E33A-L1 mutation was the conversion of 1-octanol from an antagonist to an agonist. This transformation occurred without any alteration in baseline L1 adhesion. We showed previously that Glu-33 and Tyr-418 were each photolabeled by the agonist azibutanol and the antagonist azioctanol (36). Taken together, these findings suggest that alcohol agonists and antagonists interact at a common site at the domain interface of Ig1 and Ig4 to stabilize different horseshoe conformations of L1. It will be important to replicate these findings in a variety of neuronal and non-neuronal systems. Further characterization of this alcohol binding pocket might yield additional insights into the regulation of L1 adhesion by alcohols and the design of molecules that block the effects of ethanol.
Supplementary Material
Acknowledgments
We thank Taylor VanderWoude and Caitlin Davis for technical assistance, Dr. Hui Zheng, MA General Hospital, for statistical assistance, Dr. Patricia Maness, University of North Carolina, for providing the WT-L1 construct, and to Devon Fitzgerald and Dr. Suzhen Chen for critical review of the manuscript and helpful discussions.
This work was supported, in whole or in part, by NIAAA, National Institutes of Health Grant R37-AA12974 (to M. E. C.); NIAAA U24-AA014811, as a component of the Collaborative Initiative on Fetal Alcohol Spectrum Disorders (to M. E. C.); the Medical Research Service, Dept. of Veterans Affairs (to M. E. C.); and the Dept. of Anesthesia and Critical Care, MA General Hospital (to K. W. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and Tables S1–S2.
- FASD
- fetal alcohol spectrum disorders
- CGN
- cerebellar granule neurons
- ECD
- extracellular domain
- L1Ig1–4
- first four Ig domains of L1
- CAM
- cell adhesion molecule
- WT-L1
- wild type L1
- hL1
- human L1
- βME
- β-mercaptoethanol
- PVDF
- polyvinylidene fluoride
- Cα
- α carbon.
REFERENCES
- 1. Abel E. L., Sokol R. J. (1986) Lancet 328, 1222. [DOI] [PubMed] [Google Scholar]
- 2. May P. A., Gossage J. P., Kalberg W. O., Robinson L. K., Buckley D., Manning M., Hoyme H. E. (2009) Dev. Disabil. Res. Rev. 15, 176–192 [DOI] [PubMed] [Google Scholar]
- 3. Hoyme H. E., May P. A., Kalberg W. O., Kodituwakku P., Gossage J. P., Trujillo P. M., Buckley D. G., Miller J. H., Aragon A. S., Khaole N., Viljoen D. L., Jones K. L., Robinson L. K. (2005) Pediatrics 115, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harris R. A., Trudell J. R., Mihic S. J. (2008) Sci. Signal. 1, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodlett C. R., Horn K. H. (2001) Alcohol Res. Health. 25, 175–184 [PMC free article] [PubMed] [Google Scholar]
- 6. Charness M. E., Safran R. M., Perides G. (1994) J. Biol. Chem. 269, 9304–9309 [PubMed] [Google Scholar]
- 7. Ramanathan R., Wilkemeyer M. F., Mittal B., Perides G., Charness M. E. (1996) J. Cell. Biol. 133, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maness P. F., Schachner M. (2007) Nat. Neurosci. 10, 19–26 [DOI] [PubMed] [Google Scholar]
- 9. Fransen E., Lemmon V., Van Camp G., Vits L., Coucke P., Willems P. J. (1995) Eur. J. Hum. Genet. 3, 273–284 [DOI] [PubMed] [Google Scholar]
- 10. Kenwrick S., Watkins A., De Angelis E. (2000) Hum. Mol. Genet. 9, 879–886 [DOI] [PubMed] [Google Scholar]
- 11. Bearer C. F., Swick A. R., O'Riordan M. A., Cheng G. (1999) J. Biol. Chem. 274, 13264–13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeaney N. K., He M., Tang N., Malouf A. T., O'Riordan M. A., Lemmon V., Bearer C. F. (2009) J. Neurochem. 110, 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spong C. Y., Abebe D. T., Gozes I., Brenneman D. E., Hill J. M. (2001) J. Pharmacol. Exp. Ther. 297, 774–779 [PubMed] [Google Scholar]
- 14. Chen S. Y., Wilkemeyer M. F., Sulik K. K., Charness M. E. (2001) FASEB. J. 15, 1649–1651 [DOI] [PubMed] [Google Scholar]
- 15. Wilkemeyer M. F., Chen S. Y., Menkari C. E., Brenneman D. E., Sulik K. K., Charness M. E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8543–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilkemeyer M. F., Chen S. Y., Menkari C. E., Sulik K. K., Charness M. E. (2004) J. Pharmacol. Exp. Ther. 309, 1183–1189 [DOI] [PubMed] [Google Scholar]
- 17. Chen S. Y., Charness M. E., Wilkemeyer M. F., Sulik K. K. (2005) Dev. Neurosci. 27, 13–19 [DOI] [PubMed] [Google Scholar]
- 18. Parnell S. E., Chen S. Y., Charness M. E., Hodge C. W., Dehart D. B., Sulik K. K. (2007) Alcohol. Clin. Exp. Res. 31, 2059–2064 [DOI] [PubMed] [Google Scholar]
- 19. Haspel J., Friedlander D. R., Ivgy-May N., Chickramane S., Roonprapunt C., Chen S., Schachner M., Grumet M. (2000) J. Neurobiol. 42, 287–302 [PubMed] [Google Scholar]
- 20. Gouveia R. M., Gomes C. M., Sousa M., Alves P. M., Costa J. (2008) J. Biol. Chem. 283, 28038–28047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu H., Focia P. J., He X. (2011) J. Biol. Chem. 286, 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su X. D., Gastinel L. N., Vaughn D. E., Faye I., Poon P., Bjorkman P. J. (1998) Science 281, 991–995 [DOI] [PubMed] [Google Scholar]
- 23. Schürmann G., Haspel J., Grumet M., Erickson H. P. (2001) Mol. Biol. Cell. 12, 1765–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He Y., Jensen G. J., Bjorkman P. J. (2009) Structure 17, 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Angelis E., Watkins A., Schäfer M., Brümmendorf T., Kenwrick S. (2002) Hum. Mol. Genet. 11, 1–12 [DOI] [PubMed] [Google Scholar]
- 26. Haspel J., Grumet M. (2003) Front. Biosci. 8, 1210–1225 [DOI] [PubMed] [Google Scholar]
- 27. Bateman A., Jouet M., MacFarlane J., Du J. S., Kenwrick S., Chothia C. (1996) EMBO. J. 15, 6050–6059 [PMC free article] [PubMed] [Google Scholar]
- 28. De Angelis E., MacFarlane J., Du J. S., Yeo G., Hicks R., Rathjen F. G., Kenwrick S., Brümmendorf T. (1999) EMBO. J. 18, 4744–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilkemeyer M. F., Menkari C. E., Charness M. E. (2002) Mol. Pharmacol. 62, 1053–1060 [DOI] [PubMed] [Google Scholar]
- 30. Pringle M. J., Brown K. B., Miller K. W. (1981) Mol. Pharmacol. 19, 49–55 [PubMed] [Google Scholar]
- 31. Franks N. P., Lieb W. R. (1985) Nature 316, 349–351 [DOI] [PubMed] [Google Scholar]
- 32. Wick M. J., Mihic S. J., Ueno S., Mascia M. P., Trudell J. R., Brozowski S. J., Ye Q., Harrison N. L., Harris R. A. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6504–6509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilkemeyer M. F., Menkari C. E., Spong C. Y., Charness M. E. (2002) J. Pharmacol. Exp. Ther. 303, 110–116 [DOI] [PubMed] [Google Scholar]
- 34. Wilkemeyer M. F., Sebastian A. B., Smith S. A., Charness M. E. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3690–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou F. C., Sari Y., Powrozek T. A., Spong C. Y. (2004) J. Mol. Neurosci. 24, 189–199 [DOI] [PubMed] [Google Scholar]
- 36. Arevalo E., Shanmugasundararaj S., Wilkemeyer M. F., Dou X., Chen S., Charness M. E., Miller K. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkemeyer M. F., Pajerski M., Charness M. E. (1999) Alcohol Clin. Exp. Res. 23, 1711–1720 [PubMed] [Google Scholar]
- 38. Chen M. M., Lee C. Y., Leland H. A., Lin G. Y., Montgomery A. M., Silletti S. (2010) Mol. Biol. Cell. 21, 1671–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR. 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 40. Careaga C. L., Falke J. J. (1992) J. Mol. Biol. 226, 1219–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. (2009) Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 42. Freigang J., Proba K., Leder L., Diederichs K., Sonderegger P., Welte W. (2000) Cell 101, 425–433 [DOI] [PubMed] [Google Scholar]
- 43. Jacob J., Haspel J., Kane-Goldsmith N., Grumet M. (2002) J. Neurobiol. 51, 177–189 [DOI] [PubMed] [Google Scholar]
- 44. Ye Q., Koltchine V. V., Mihic S. J., Mascia M. P., Wick M. J., Finn S. E., Harrison N. L., Harris R. A. (1998) J. Biol. Chem. 273, 3314–3319 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.