

Abstract
Mutant p53 is not only deficient in tumor suppression but also acquires additional activity, called gain of function. Mutant p53 gain of function is recapitulated in knock-in mice that carry one null allele and one mutant allele of the p53 gene. These knock-in mice develop aggressive tumors compared with p53-null mice. Recently, we and others showed that tumor cells carrying a mutant p53 are addicted to the mutant for cell survival and resistance to DNA damage. To further define mutant p53 gain of function, we used the MCF-10A three-dimensional model of mammary morphogenesis. MCF-10A cells in three-dimensional culture undergo a series of morphological changes and form polarized and growth-arrested spheroids with hollow lumen, which resembles normal glandular architectures in vivo. Here, we found that endogenous wild-type p53 in MCF-10A cells was not required for acinus formation, but knockdown of endogenous wild-type p53 (p53-KD) led to partial clearance of cells in the lumen due to decreased apoptosis. Consistent with this, p53-KD altered expression patterns of the cell adhesion molecule E-cadherin, the cytoskeletal marker β-catenin, and the extracellular matrix protein laminin V. We also found that ectopic expression of the mutant G245S led to a phenotype similar to p53-KD, whereas a combination of ectopic expression of siRNA-resistant G245S with p53-KD led to a less cleared lumen. In contrast, ectopic expression of mutant R248W, R175H, and R273H disrupted normal acinus architectures with filled lumen and led to formation of irregular and multiacinus structures regardless of p53-KD. In addition, these mutants altered normal expression patterns and/or levels of E-cadherin, β-catenin, laminin V, and tight junction marker ZO-1. Furthermore, epithelial-to-mesenchymal transitions (EMT) markers, Snail, Slug, and Twist, were highly induced by mutant p53 and/or p53-KD. Together, we postulate that EMT represents a mutant p53 gain of function and mutant p53 alters cell polarity via EMT.
Keywords: Carcinogenesis, Cell Cycle, Cell Death, Mutant, p53, Cell Polarity, Mutant p53
Introduction
Mutation of p53 is the most frequent genetic alteration in human cancers. The majority of tumor-derived p53 mutations are missense point mutations and clustered within the central DNA-binding domain. Mutant p53 is defective in sequence-specific DNA binding and growth inhibition (1, 2), which defines the classical loss of function mutation for a tumor suppressor. In addition, mutant p53 with an intact domain for tetramerization is dominant negative because the mutants can form a heterotetramer with wild-type p53 (3, 4).
Mutant p53 is not only deficient in tumor suppression but also acquires additional activity, called gain of function. Mutant p53 gain of function is recapitulated in knock-in mice that carry one null allele and one mutant allele of the p53 gene (R172H or R270H). These knock-in mice develop aggressive tumors compared with p53-null mice (5, 6). Recently, we and others (7–10) showed that tumor cells carrying a mutant p53 are addicted to the mutant for cell survival and resistance to DNA damage. Mutant p53 also is found to promote invasion, loss of directionality of migration, and metastatic behavior via increased recycling of integrin (11). In addition, mutant p53 is found to facilitate somatic cell reprogramming and augments the malignant potential of reprogrammed cells (12). A recent study showed that mutant p53 can compete with wild-type p53 to promote epithelial-to-mesenchymal transition (EMT)2 and regulate the EMT-associated stem cell-like phenotype (13).
Mutant p53 gain of function is dependent on its ability to transactivate or repress specific target genes (1), such as c-Myc, Fas, and NF-κB2. In addition, recent studies showed that upon inducible knockdown of endogenous mutant p53 in SW480 and MIA PaCa-2 cells, Id2 expression is increased (9), whereas GRO1 expression is repressed (7). Interestingly, both Id2 and GRO1 are capable of maintaining the transformed phenotypes of tumor cells addicted to a mutant p53 (7, 9).
Six p53 mutations (R175H, G245S, R248W, R249S, R273H, and R282W) are described as “hot-spot” because they are among the most prevalent p53 mutations in human cancer (14). Mutant p53 is categorized into two classes: contact site mutants and conformational mutants. The contact site mutants, such as R248W and R273H, have an altered residue at a site that, in the wild-type protein, directly contacts DNA (15, 16). The conformational mutants, such as R175H and R245S, alter the structure of the protein (15). This structural alteration is detectable by an anti-p53 antibody (PAb240) that recognizes residues exposed in the mutant but cryptic in the wild-type (3). Thus, it is important to determine whether various classes of p53 mutants contain common and distinct properties for maintaining the transformed phenotypes of tumor cells. A recent study attempted to address this by generating a series of SW480 cell lines in which endogenous mutant p53 can be inducibly knocked down by shRNA, whereas an siRNA-resistant mutant p53 can be inducibly expressed (8). Using this approach, both contact site (R248W and R273H) and conformation (G245S and R249S) mutants can substitute endogenous mutant R273H/P309S in SW480 cells for maintaining the transformed phenotypes and conferring resistance to DNA damage in SW480 cells (8).
To further determine whether various classes of mutant p53 share additional common properties for their gain of function, we have utilized the MCF-10A three-dimensional model of mammary morphogenesis. MCF-10A cells in a three-dimensional culture undergo a series of morphological changes and form polarized and growth-arrested acinar structures with hollow lumen, which resembles normal glandular architectures in vivo (17–19). We found that endogenous wild-type p53 was not required for acinus formation, but p53 knockdown (p53-KD) in MCF-10A led to partial clearance of cells in the lumen due to decreased apoptosis. Interestingly, we found that ectopic expression of siRNA-resistant mutant G245S led to a phenotype similar to p53-KD, whereas a combination of ectopic expression of G245S with p53-KD only led to a less cleared lumen. In contrast, ectopic expression of R248W, R175H, and R273H disrupted normal acinus architectures with filled lumen regardless of p53-KD. Furthermore, these mutants abolished the normal expression patterns and/or levels of E-cadherin, β-catenin, laminin V, and ZO-1 and induced EMT markers, Snail, Slug, and Twist. Taken together, we postulate that MCF-10A cells undergo EMT upon ectopic expression of mutant p53 and that mutant p53 alters normal cell polarity via EMT.
EXPERIMENTAL PROCEDURES
Reagents
Growth factor-reduced Matrigel (BD Biosciences) was used for the three-dimensional culture. DMEM/F12 medium, donor horse serum, and To-Pro-3 nucleus dye were purchased from Invitrogen. Hydrocortisone, insulin, and cholera toxin were purchased from Sigma. Recombinant human EGF was purchased from Peprotech (Rocky Hill, NJ). Anti-laminin V γ2 antibody was purchased from Millipore (Temecula, CA). Normal goat serum and FITC-conjugated secondary antibody were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Antibodies against β-catenin (E-5), Snail-1, Slug, Twist, p21, and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against cleaved caspase-3 was purchased from Cell Signaling (Beverly, CA). Anti-E-cadherin and Bax were purchased from BD Transduction Laboratories. Antibody against ZO-1 was purchased from Invitrogen. Anti-actin was purchased from Sigma. 21-bp siRNA against p53, 5′- GAC UCC AGU GGU AAU CUA C dTdT-3′, and a scrambled siRNA, 5′-GCA GUG UCU CCA CGU ACU A dTdT-3′, were purchased from Dharmacon RNA Technologies.
Plasmids Constructions and Cell Line Generation
pBabe-U6-sip53 and pcDNA3 plasmids expressing siRNA-resistant tumor-derived hot-spot mutants (R248W, R273H, G245S, and R175H) were described previously (8). To generate stable p53 knockdown cell lines, pBabe-U6-siRNA was transfected into MCF-10A cells. The resulting p53-KD cell lines were selected with puromycin, and p53-KD was confirmed by Western blot analysis. To generate mutant p53-producing cell lines, pcDNA3-mutant p53 was transfected into MCF-10A cells. The resulting mutant p53-producing cell lines were selected with G418 and confirmed by Western blot analysis. To generate stable p53-KD cell lines with mutant p53 overexpression, pBabe-U6-siRNA was co-transfected with pcDNA3-mutant p53 into MCF-10A cells. The resulting cell lines were selected with puromycin and G418. Both p53-KD and mutant p53 expression were confirmed by Western blot analysis.
Cell Culture
The immortalized MCF-10A cell line was obtained from American Type Culture Collection (Manassas, VA). The medium for MCF-10A cells is composed of DMEM/F12 supplemented with 5% donor horse serum, 20 ng/ml EGF, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, and 100 ng/ml cholera toxin. The overlay three-dimensional culture was carried out based on the method described previously with some modifications (17). Briefly, four-well chamber slides (Millipore) were precoated evenly with 80 μl overnight-thawed Matrigel and placed in a 37 °C incubator for 15 min for Matrigel solidification. Single cell suspensions were plated onto Matrigel-coated chamber slides at 5000 cells/well in complete growth medium with 2% Matrigel and allowed to grow for 1–22 days. Overlay medium containing 2% Matrigel was renewed every 4 days.
Confocal Microscopy
The three-dimensional structures in Matrigel were fixed in 4% paraformaldehyde at room temperature for 20 min and permeabilized with 0.5% Triton X-100 in PBS for 30 min at 4 °C. After quenching with 100 mm glycine in PBS, the three-dimensional structures were preblocked with a primary blocking buffer A (130 mm NaCl, 7 mm Na2HPO4, 3.5 mm NaH2PO4, 0.1% BSA, 0.2% Triton X-100, and 0.05% Tween 20) containing 10% normal goat serum for 2 h and further blocked with a secondary blocking buffer B (buffer A plus 10% normal goat serum and 20 mg/ml goat anti-mouse F(ab′)2 fragments) for 1 h. Then, the three-dimensional structures were incubated overnight with primary antibodies at 4 °C, washed thoroughly with buffer A, and stained with FITC-conjugated secondary antibody (diluted 1:200 in buffer A) for 1 h. The nuclei of three-dimensional structures were stained with 5 μg/ml of To-Pro-3 in PBS for 15 min at room temperature. The three-dimensional structures were mounted under glass coverslips with 0.1% para-phenylenediamine D (PPD) and 90% glycerol in PBS. Confocal microscopic images of the acinus structures were captured by the Z-stacking function for serial confocal sectioning at 2-μm intervals (LSM-510 Carl Zeiss laser scanning microscope) and then analyzed by Carl Zeiss software. The experiments were conducted in triplicate.
RESULTS
p53-KD in MCF-10A Cells Leads to Partial Clearance of Cells in Lumen Due to Decreased Apoptosis
MCF-10A is a spontaneously immortalized but untransformed mammary epithelial cell line. MCF-10A cells in three-dimensional culture form acini-like spheroids that exhibit many aspects of glandular architecture in vivo (17). Many oncogenes, such as ErbB2, can alter MCF10A acinar architecture in a manner that recapitulates their properties in vivo (20–22). Thus, whether mutant p53 can alter MCF-10A cell polarity in three-dimensional culture was used to measure mutant p53 gain of function. Here, we showed that parental MCF-10A cells formed normal acinar architectures along with hollow lumen (Fig. 1, A and C–E, To-Pro-3 panels; To-Pro-3 was used for nuclei staining), consistent with previous published reports (17, 19, 21). Fig. 1B is a schematic diagram of serial confocal cross-sections (x-y axis) through a hypothetical MCF-10A acinus. The diagrams overlaying each section illustrate the relative position of the optical section with respect to the z axis. We also showed that a polarized staining pattern primarily at lateral cell-cell junctions was detected for E-cadherin (Fig. 1C), consistent with the normal expression pattern of E-cadherin, a key component of adherens junctions. Similarly, a normal polarized staining pattern primarily at the lateral membranes was detected for β-catenin (Fig. 1D). β-catenin is a cytoskeletal marker and an adaptor protein of E-cadherin and cooperates with E-cadherin to mediate hemophilic adhesion with adjacent cells (23, 24). β-Catenin also is a co-activator of transcription factor T-cell-specific transcription factor/lymphoid enhancer binding factor (TCF/LEF) (25, 26). To further demonstrate that MCF-10A acini have a normal architecture, we examined the expression pattern of laminin V, one of the principal extracellular matrix proteins (25, 26). We found that laminin V was secreted from polarized epithelial cells surrounding a hollow lumen and deposited at the basal surface of the spheroid (Fig. 1E). Furthermore, we found that active caspase-3 was expressed in the lumen of early stage acini (Fig. 1F), which is known to be responsible for hollow lumen formation of late stage acini (23, 24). Thus, based on the cell morphology and expression patterns of E-cadherin, β-catenin, laminin V, and active caspase-3, we concluded that a normal acinus-like architecture for MCF-10A cells is well recapitulated in our three-dimensional culture condition.
FIGURE 1.

MCF-10A cells form acinar-like architectures with hollow lumen. A, selective images of MCF-10A cells in two-dimensional (a, 100×; b, 200×) and three-dimensional (c) cultures. B, schematic diagrams of serial confocal cross-sections (x-y axis) through a hypothetical MCF-10A acinus. The diagrams overlaying each section illustrate the relative position of the optical section with respect to the z axis. C, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against E-cadherin. MCF-10A cells were cultured on Matrigel for 20 days and immunostained with antibody against E-cadherin (green). E-cadherin is normally expressed at lateral cell-cell junctions. To-Pro-3 was used for nuclei staining. D, serial confocal cross sections of an acinus stained with To-Pro-3 and antibody against β-catenin. β-Catenin is expressed normally at cell-cell junctions. E, serial confocal cross sections of an acinus stained with To-Pro-3 and antibody against laminin V. Laminin V is normally deposited at the basal surface of the spheroid. F, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against active cleaved caspase-3. The experiment was performed as in C except that anti-cleaved caspase-3 was used, and MCF-10A cells were cultured in a three-dimensional culture for 10 day. Active caspase-3 is normally expressed in the lumen of early stage acini.
MCF-10A cells contain wild-type p53 (27). A report showed that 14-3-3ζ, which indirectly targets p53 protein stability, appears to alter MCF-10A cells to form normal acinus-like architectures (28). Another study showed that upon degradation of endogenous wild-type p53 by HPV16 E6 protein, mammary epithelial cells at early passages become resistant to apoptosis in three-dimensional culture (29). Due to other oncogenic functions inherent in 14–3-3ζ and HPV16 E6 in addition to targeting p53 protein stability, it is still uncertain whether endogenous wild-type p53 directly plays a role in formation of acinus-like architectures. To test this, we generated multiple MCF-10A cell lines in which endogenous wild-type p53 was knocked down by shRNA (clones #5 and #6). We found that upon stable knockdown of endogenous wild-type p53 (p53-KD) (Fig. 2A), near-normal acini were formed by p53-KD MCF-10A cells (Fig. 2B). However, the lumen of the acini was incompletely cleared (Fig. 2, C–E, To-Pro-3 panels). Consistent with partially filled lumen, the expression patterns of E-cadherin, β-catenin, and laminin V were altered by p53-KD (Fig. 2, C–E) compared with that in the parental MCF-10A cells (Fig. 1, C–E). This includes partially scattered and nonpolarized staining for E-cadherin, cytoplasmic staining for β-catenin, and scattered lumen staining for laminin V. In addition, there was no detectable active caspase-3 staining in the lumen of early stage acini (Fig. 2F). These data suggest that endogenous wild-type p53 is necessary for proper clearance of MCF-10A cells in the lumen.
FIGURE 2.

Knockdown of endogenous wild-type p53 in MCF-10A leads to incomplete clearance of cells in the lumen due to decreased apoptosis. A, generation of MCF-10A cell lines in which endogenous wild-type p53 was stably knocked down (clones #5 and #6). Western blot analysis was prepared using extracts from p53-KD MCF-10A cells that were mock-treated (−) or treated with doxorubicin (Dox; +). The blots were probed with antibodies against p53 and actin, respectively. B, selective images of MCF-10A cells in two-dimensional (a, 100×; b, 200×) and three-dimensional (c) cultures. C, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against E-cadherin. D, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against β-catenin. E, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against laminin V. F, serial confocal cross-sections of an acinus stained with To-Pro-3 and antibody against active cleaved caspase-3.
Mutant G245S Has a Dominant Negative Activity toward Wild-type p53, but Possesses a Weak Gain of Function in MCF-10A Three-dimensional Culture Model
To examine the effect of mutant G245S on MCF-10A cell polarity in three-dimensional culture, we generated multiple MCF-10A cell lines in which G245S was expressed ectopically along with or without p53-KD (Fig. 3, A and F). We found that upon ectopic expression of G245S, a near-normal acinus-like spheroid was formed in three-dimensional culture along with near-normal cell morphology in two-dimensional culture regardless of p53-KD (Fig. 3, B and G). However, the mutant G245S prevented the lumen of the acini from being completely cleared (Fig. 3, C–E, To-Pro-3 panels), which is similar to that by p53-KD (Fig. 2, C–E). Interestingly, a combination of ectopic expression of G245S with p53-KD led to a less cleared lumen (Fig. 3, H–J, To-Pro-3 panels and supplemental Fig. S1). In addition, we found that the staining pattern for E-cadherin was scattered and fragmented along with lack of polarized staining at lateral membranes (Fig. 3, C and H). The staining pattern for β-catenin was also altered along with cytoplasmic and nuclear accumulation of β-catenin (Fig. 3, D and I). Furthermore, laminin V was secreted into the lumen of the acini (Fig. 3, E and J). Considering that G245S along with p53-KD had only slightly more severe effects on cell polarity than the mutant alone, our data suggest that G245S has a potent dominant negative effect but possesses a weak gain of function.
FIGURE 3.

Mutant G245S has a dominant negative activity toward endogenous wild-type p53 but possesses a weak gain of function in the MCF-10A three-dimensional model. A and F, generation of MCF-10A cell lines in which siRNA-resistant mutant G245S was expressed (A), along with knockdown of endogenous wild-type p53 (F, clones #1 and #12). Western blot analysis was prepared using extracts from MCF-10A cells that were mock-treated (−) or treated with doxorubicin (Dox; +). The blots were probed with antibodies against p53 and GAPDH, respectively. B and G, selective images of MCF-10A cells in two-dimensional (a, 100×; b, 200×) and three-dimensional (c) cultures. C and H, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against E-cadherin. D and I, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against β-catenin. White arrows in D and I indicate the accumulation and translocation of β-catenin in acinar structure. E and J, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against laminin V.
Ectopic Expression of Mutant R248W, R175H, and R273H Disrupts Normal Acinar Architectures with Filled Lumen Regardless of p53-KD
To examine the effect of the mutants R248W, R273H, and R175H on MCF-10A cell polarity in three-dimensional culture, we generated multiple MCF10A cell lines in which these mutants were expressed ectopically individually along with or without p53-KD (see panels A and F in Figs. 4 and 5 and panel A in Fig. 6). We found that upon ectopic expression of an individual mutant, irregular and multiacinar structures along with some near-normal acini were formed by MCF-10A cells (see panel B in Figs. 4–6) as well as by MCF-10A cells with p53-KD (see panel G in Figs. 4 and 5). Interestingly, some cells also exhibited elongated cell morphology in two-dimensional culture (see panel B in Figs. 4–6; and panel G in Figs. 4 and 5). In addition, we found that the lumen of the acini was nearly completely filled with cells (see panels C–E in Figs. 4–6, panels H–J in Figs. 4 and 5, and supplemental Fig. S1). Consistent with the filled lumen and irregular multiacinar structures, the staining pattern for E-cadherin was markedly altered, including scattered and nonpolarized staining or little if any lateral staining (see panel C in Figs. 4–6; see panel H in Figs. 4 and 5). The staining pattern for β-catenin was also altered as the majority of β-catenin was expressed in the cytoplasm and nucleus (panel D in Figs. 4–6; see panel I in Figs. 4 and 5). Furthermore, laminin V was secreted into the lumen of the acini and cell-cell junctions (see panel E in Figs. 4–6 and panel J in Figs. 4 and 5). Thus, R248W, R273H, and R175H completely altered the normal MCF-10A cell polarity in two-dimensional and three-dimensional cultures regardless of p53-KD, suggesting that these mutants possess a strong gain of function.
FIGURE 4.

Ectopic expression of mutant R248W disrupts normal acinus architecture with filled lumen regardless of p53-KD. A and F, generation of MCF-10A cell lines in which siRNA-resistant mutant R248W was expressed (A), along with knockdown of endogenous wild-type p53 (F). Western blot analysis was prepared using extracts from MCF-10A cells that were mock-treated (−) or treated with doxorubicin (Dox; +). The blots were probed with antibodies against p53 and GAPDH, respectively. B and G, selective images of MCF-10A cells in two-dimensional (a, 100×; b, 200×) and three-dimensional (B, c–f and G, c–d) cultures. C and H, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against E-cadherin. D and I, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against β-catenin. E and J, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against laminin V.
FIGURE 5.

Ectopic expression of mutant R273H disrupts normal acinar architectures with filled lumen regardless of p53-KD. A and F, generation of MCF-10A cell lines in which siRNA-resistant mutant R273H was expressed (A), along with knockdown of endogenous wild-type p53 (F). Western blot analysis was prepared using extracts from MCF-10A cells that were mock-treated (−) or treated with doxorubicin (Dox; +). The blots were probed with antibodies against p53 and GAPDH, respectively. B and G, selective images of MCF-10A cells in two-dimensional (B, a, 100×; G, a, 100×; b, 200×) and three-dimensional (c and d) cultures. C and H, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against E-cadherin. D and I, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against β-catenin. E and J, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against laminin V.
FIGURE 6.

Ectopic expression of mutant R175H disrupts normal acinus architectures with filled lumen. A, generation of MCF-10A cell lines in which mutant R175H was expressed. Western blot analysis was prepared using extracts from MCF-10A cells that were mock treatment (−) or treatment with doxorubicin (Dox; +). The blots were probed with antibodies against p53 and GAPDH, respectively. B, selective images of MCF-10A cells in two-dimensional (a, 100×; b, 200×) and three-dimensional (c) cultures. C, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against E-cadherin. D, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against β-catenin. White arrows indicate the accumulation and translocation of β-catenin in acinar structure. E, representative confocal images of cross-sections through the middle of acini stained with To-Pro-3 and antibody against laminin V.
Mutant p53 Alters Normal MCF-10A Cell Polarity in Three-dimensional Culture via EMT
Because the normal expression patterns for E-cadherin, β-catenin, and laminin V in MCF-10A three-dimensional culture were altered by mutant p53, we examined whether the expression levels of these proteins are also altered by mutant p53. We found that the level of E-cadherin was decreased markedly upon p53-KD and/or ectopic expression of mutant G245S, R248W, R273H, and R175H (Fig. 7A, compare lane 1 with lanes 2–9, respectively). Similarly, the level of ZO-1, a tight junction protein, was decreased (Fig. 7A). In contrast, the levels of laminin V and β-catenin were increased significantly by p53-KD and/or ectopic expression of these mutants (Fig. 7C, compare lane 1 with lanes 2–9, respectively). In addition, these observations were also detected in multiple MCF10A cell lines in which p53 mutants (G245S and R273H) were expressed ectopically along with p53-KD (Fig. 7, B and D, compare lane 1 with lanes 2–8, respectively). Together, the expression levels of E-cadherin, laminin V, and β-catenin are consistent with their staining patterns and intensity in acinar structures (Figs. 1–6).
FIGURE 7.

Markers for epithelial-to-mesenchymal transitions are regulated upon ectopic expression of siRNA-resistant mutant p53 regardless of stable knockdown of endogenous wild-type p53 in MCF-10A cells. A, C, and E, Western blots were prepared with extracts from parental MCF-10A cells (lane 1), p53-KD MCF-10A cells (lane 2), MCF-10A cells in which a mutant p53 was ectopically expressed (lanes 3, 5, 7, and 9) and MCF-10A cells in which a mutant p53 was ectopically expressed along with knockdown of endogenous wild-type p53 (lanes 4, 6, and 8). The blots were probed with antibodies against E-cadherin (A), ZO-1 (A), p53 (A, C, and E), laminin V (C), β-catenin (C), Snail (E), Slug (E), Twist (E), and actin (A, C, and E). B, the experiments were performed as in A except that two MCF10A cell lines in which mutant G245S or R273H was ectopically expressed along with knockdown of endogenous wild-type p53 were used. D, the experiments were performed as in C except that two MCF10A cell lines in which mutant G245S or R273H was ectopically expressed along with knockdown of endogenous wild-type p53 were used. F, the experiment was performed as in A except that the blots were probed with antibodies against p53, p21, Bax, and Puma, respectively.
Down-regulation of E-cadherin along with up-regulation of β-catenin and laminin V is considered a hallmark of EMT (26). Because Snail-1, Slug, and Twist are known to induce EMT by down-regulating E-cadherin expression (26), these transcription factors were examined in MCF-10A cells. We found that Snail-1 and Slug were increased by ectopic expression of G245S, R175H, R248W, and R273H regardless of p53-KD (Fig. 7E, compare lane 1 with lanes 2–9, respectively). Slug expression was also increased by p53-KD (Fig. 7E, compare lanes 1 and 2). In addition, Twist was increased by mutant G245S and R248W in the absence of endogenous wild-type p53 (Fig. 7E, compare lane 1 with lanes 4 and 6, respectively).
To further confirm the gain of function of mutant p53 in EMT, several p53 target genes, including p21, Puma, and Bax, were examined in MCF10A cells in which a p53 mutant (G245S, R248W, or R273H) was expressed ectopically along with or without p53-KD. We found that compared with MCF10A cells with wild-type p53, the levels of p21, Puma, and Bax were decreased markedly in MCF10A cells with p53-KD, regardless of ectopic expression of a p53 mutant (Fig. 7F, compare lanes 1 and 2; compare lane 2 with 4, 6, and 8, respectively). This suggests that mutant p53 gain of function in EMT is not due to dominant negative effect to the residual p53 activity caused by incomplete knockdown. Nevertheless, the level of Bax and Puma in MCF10 cells with wild-type p53 was decreased upon ectopic expression of mutant G245S, R248W, and R273H (Fig. 7E, compare lane 1 with 3, 5, and 7, respectively), suggesting that these mutants have a dominant negative effect to endogenous wild-type p53.
To rule out the effect of knockdown-mediated adaptation and/or selection of stable clones on EMT, the gain of function for mutant p53 in EMT was examined in cells in which endogenous wild-type p53 was transiently knocked down along with transient ectopic expression of mutant p53. We found that the level of E-cadherin was decreased markedly upon transient expression of mutant G245S, R248W, or R273H regardless of transient knockdown of wild-type p53 (Fig. 8A, compare lane 1 with lanes 2–8, respectively). In addition, an increase in the levels of laminin V and β-catenin was observed upon transient expression of mutant G245S, R248W, or R273H regardless of transient p53-KD (Fig. 8B, compare lane 1 with lanes 2–8, respectively). Furthermore, we showed that Snail-1, Slug, and Twist were increased markedly upon transient expression of mutant G245S, R248W, and R273H, regardless of transient p53-KD (Fig. 8C, compare lane 1 with lanes 2–8, respectively). These observations are consistent with that in MCF10A cell lines in which p53 mutants were stably expressed individually along with or without stable knockdown of endogenous wild-type p53.
FIGURE 8.
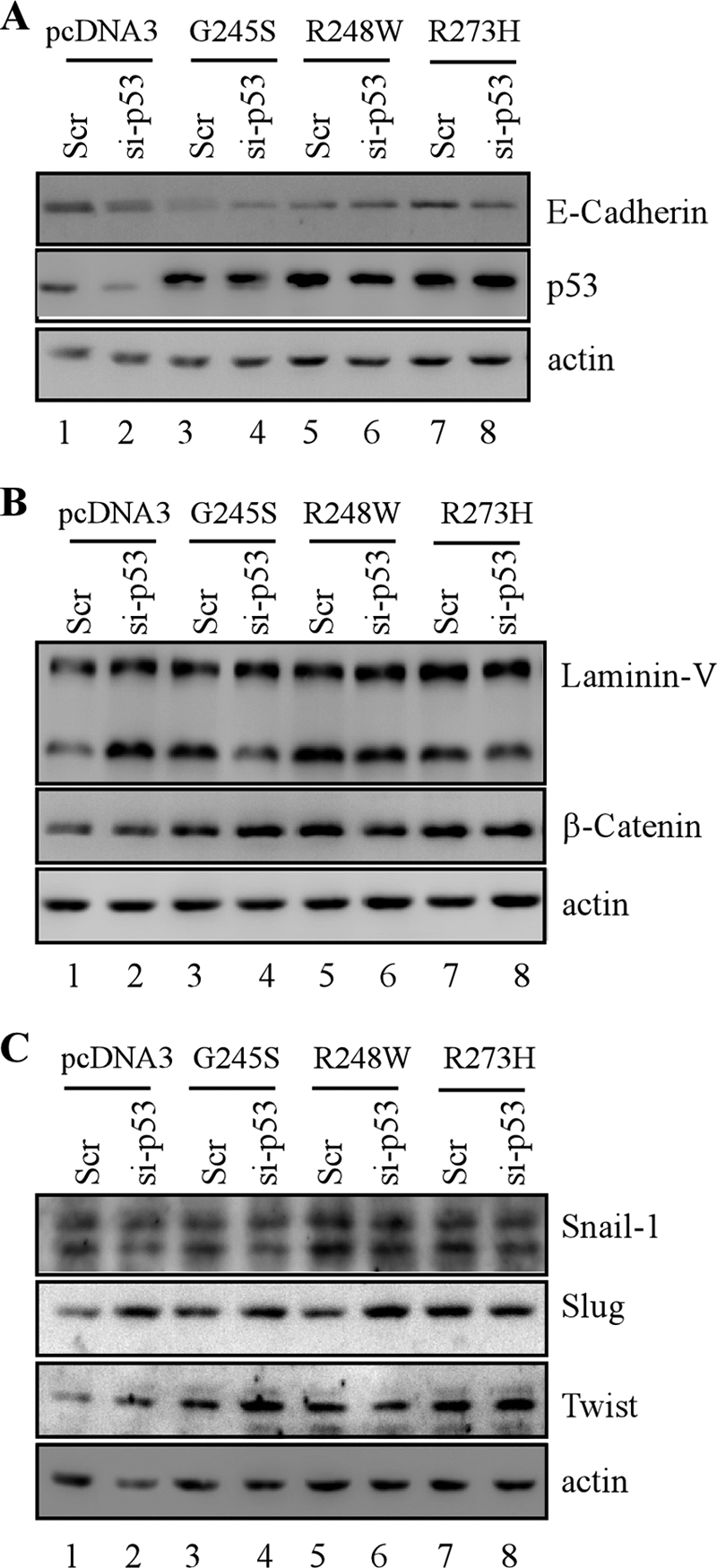
EMT markers are regulated upon transient overexpression of siRNA-resistant mutant p53 along with or without transient knockdown of wild-type p53 in MCF-10A cells. Western blots were prepared with extracts from MCF-10A cells in which scramble siRNA (Scr) was co-transfected with pcDNA3 empty vector (lane 1), G245S (lane 3), R248W (lane 5), or R273H (lane 7) and MCF-10A cells in which siRNA against p53 (si-p53) was co-transfected with pcDNA3 empty vector (lane 2), G245S (lane 4), R248W (lane 6), or R273H (lane 8). The blots were probed with antibodies against E-cadherin (A), p53 (A–C), laminin V (B), β-catenin (B), Snail (C), Slug (C), Twist (C), and actin (A–C), respectively.
DISCUSSION
Mutant p53 oncogenic properties have been investigated extensively, mostly via ectopic expression in p53-null transformed tumor cells (1). Because tumor cells, which carry no p53 or a mutant p53, may have already been transformed and thus, mutant p53 gain of function may not be required for the survival and invasive properties of transformed tumor cells. Thus, an ideal approach to investigate mutant p53 gain of function is to use immortalized near-normal cells, such as MCF-10A cells that form polarized and growth-arrested acinar structures with hollow lumen in a three-dimensional culture (17–19). Using the MCF-10A three-dimensional model of mammary morphogenesis, we made several novel observations. First, we found that endogenous wild-type p53 is not required for acinus formation, but knockdown of endogenous wild-type p53 leads to partial clearance of cells in the lumen due to decreased apoptosis. Second, we found that ectopic expression of mutant G245S leads to a phenotype remarkably similar to p53-KD, whereas a combination of ectopic expression of G245S with p53-KD only leads to a slightly less cleared lumen. This suggests that G245S has a potent dominant negative effect but possesses a weak gain of function. Third, we found that ectopic expression of R248W, R175H, and R273H disrupts normal acinar architectures with filled lumen along with formation of irregular and multiacinar spheroids regardless of p53-KD. Fourth, mutant p53 alters the normal expression pattern of E-cadherin, β-catenin, and laminin V along with increased expression of Snail, Slug, and Twist. Taken together, these data suggest that mutant R248W, R175H, R273H, and to a lesser extent, the mutant G245S and knockdown of endogenous wild-type p53 alter normal MCF-10 cell polarity via EMT.
Recent studies have demonstrated that loss of apoptotic response rather than hyperproliferative activity by an oncogene is likely to be responsible for filled lumen of MCF-10A acini (17). Previously, we showed that several hot-spot mutants, including G245S, R248W, R249S, and R273H, share a common property to substitute endogenous mutant p53 (R273H/P309S) in SW480 cells for cell survival and resistance to DNA damage (8). Here, we found that in the three-dimensional culture, the lumen of MCF-10A acini are partially cleared upon knockdown of endogenous wild-type p53 and/or ectopic expression of G245S (Figs. 2 and 3). This suggests that the decreased apoptotic response due to knockdown of endogenous wild-type p53 and dominant negative effect by G245S is likely responsible for incomplete clearance of cells in the lumen. In contrast, R175H, R248W, and R273H have a strong gain of function in altering MCF-10A cell polarity in three-dimensional culture, including irregular and multiacinar spheroids with filled lumen (Figs. 4–6). This implies that these p53 mutants not only abrogate p53-dependent but also p53-independent apoptotic responses. Together, these observations indicate that p53 mutants possess both common and distinct properties for their gain of function.
Here, we found that E-cadherin and ZO-1 were down-regulated, whereas β-catenin and laminin V up-regulated in MCF-10A cells upon ectopic expression of mutant p53. We also found that Snail, Slug, and Twist were up-regulated, consistent with the idea that E-cadherin is repressed by Snail, Slug, and Twist (25, 26). Collectively, the altered expression patterns of these EMT markers indicate that upon ectopic expression of mutant p53, MCF-10A cells undergo EMT, and thus, the cell polarity is altered by mutant p53 at least in part via EMT. Thus, we postulate that EMT represents a mutant p53 gain of function. It should be noted that EMT induction, including increased expression of Snail/Slug and decreased expression of E-cadherin, is associated with the acquisition of cancer cell invasive and metastatic properties and resistance to therapy (25). Overexpression of p53 mutants is correlated with increased the levels of EMT markers, consequentially enhanced EMT-associated stemness properties in MCF12A cells (13). These results suggested that mutant p53-mediated EMT pathway most likely accounts for cancer stem cell initiation, progression, or chemo-resistance. Consistent with this acquired function of EMT for mutant p53, knock-in mice, which carry a mutant p53 allele and a p53-null allele, develop aggressive tumors frequently metastasized to other organs, especially lung (5, 6).
In the process of EMT induction, Slug, Snail, and Twist transcriptional factors are invariably induced (25, 26). E-cadherin is known to be transcriptionally repressed by Snail, Slug, and Twist, leading to dissociation of β-catenin from adherens junctions and subsequent nuclear translocation of β-catenin. Nuclear β-catenin forms a transcriptional complex with TCF/LEF, which promotes robust expression of genes associated with cancer progression and metastasis, such as c-Myc (25, 26). Because Snail, Slug, and Twist are increased in MCF-10A cells by mutant p53, it is possible that mutant p53 may directly regulate Snail, Slug, and Twist expression, especially considering that mutant p53 gain of function is dependent on the ability of mutant p53 to transactivate or repress specific target genes (1). In an effort to identify target genes of mutant p53 in SW480 and MIA-PaCa-2 cells, Id2 and GRO1 were identified through multiple microarray analyses (7, 9). However, Snail, Slug, and Twist were not found to be regulated by mutant p53 in SW480 and MIA-PaCa-2 cells (data not shown). Thus, further study is warranted to address whether mutant p53 regulates a specific set of target genes in immortalized but untransformed cells, such as MCF-10A.
Supplementary Material
Acknowledgments
We thank Drs. Bo Liu, Julia Lee, and Roger Adamson for assistance in confocal microscopy.
This work was supported in part by National Institutes of Health Grant CA121137.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- EMT
- epithelial-to-mesenchymal transition
- KD
- knockdown.
REFERENCES
- 1. Brosh R., Rotter V. (2009) Nat. Rev. Cancer 9, 701–713 [DOI] [PubMed] [Google Scholar]
- 2. Ko L. J., Prives C. (1996) Genes Dev. 10, 1054–1072 [DOI] [PubMed] [Google Scholar]
- 3. Milner J., Medcalf E. A. (1991) Cell 65, 765–774 [DOI] [PubMed] [Google Scholar]
- 4. Willis A., Jung E. J., Wakefield T., Chen X. (2004) Oncogene 23, 2330–2338 [DOI] [PubMed] [Google Scholar]
- 5. Lang G. A., Iwakuma T., Suh Y. A., Liu G., Rao V. A., Parant J. M., Valentin-Vega Y. A., Terzian T., Caldwell L. C., Strong L. C., El-Naggar A. K., Lozano G. (2004) Cell 119, 861–872 [DOI] [PubMed] [Google Scholar]
- 6. Olive K. P., Tuveson D. A., Ruhe Z. C., Yin B., Willis N. A., Bronson R. T., Crowley D., Jacks T. (2004) Cell 119, 847–860 [DOI] [PubMed] [Google Scholar]
- 7. Yan W., Chen X. (2009) J. Biol. Chem. 284, 12178–12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan W., Chen X. (2010) J. Biol. Chem. 285, 14229–14238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yan W., Liu G., Scoumanne A., Chen X. (2008) Cancer Res. 68, 6789–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Irwin M. S., Kondo K., Marin M. C., Cheng L. S., Hahn W. C., Kaelin W. G., Jr. (2003) Cancer Cell 3, 403–410 [DOI] [PubMed] [Google Scholar]
- 11. Muller P. A., Caswell P. T., Doyle B., Iwanicki M. P., Tan E. H., Karim S., Lukashchuk N., Gillespie D. A., Ludwig R. L., Gosselin P., Cromer A., Brugge J. S., Sansom O. J., Norman J. C., Vousden K. H. (2009) Cell 139, 1327–1341 [DOI] [PubMed] [Google Scholar]
- 12. Sarig R., Rivlin N., Brosh R., Bornstein C., Kamer I., Ezra O., Molchadsky A., Goldfinger N., Brenner O., Rotter V. (2010) J. Exp. Med. 207, 2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang C. J., Chao C. H., Xia W., Yang J. Y., Xiong Y., Li C. W., Yu W. H., Rehman S. K., Hsu J. L., Lee H. H., Liu M., Chen C. T., Yu D., Hung M. C. (2011) Nat. Cell Biol. 13, 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hollstein M., Rice K., Greenblatt M. S., Soussi T., Fuchs R., Sørlie T., Hovig E., Smith-Sørensen B., Montesano R., Harris C. C. (1994) Nucleic Acids Res. 22, 3551–3555 [PMC free article] [PubMed] [Google Scholar]
- 15. Cho Y., Gorina S., Jeffrey P. D., Pavletich N. P. (1994) Science 265, 346–355 [DOI] [PubMed] [Google Scholar]
- 16. Rolley N., Butcher S., Milner J. (1995) Oncogene 11, 763–770 [PubMed] [Google Scholar]
- 17. Debnath J., Muthuswamy S. K., Brugge J. S. (2003) Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 18. Weaver V. M., Lelièvre S., Lakins J. N., Chrenek M. A., Jones J. C., Giancotti F., Werb Z., Bissell M. J. (2002) Cancer Cell 2, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muthuswamy S. K., Li D., Lelievre S., Bissell M. J., Brugge J. S. (2001) Nat. Cell Biol. 3, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carroll D. K., Carroll J. S., Leong C. O., Cheng F., Brown M., Mills A. A., Brugge J. S., Ellisen L. W. (2006) Nat. Cell Biol. 8, 551–561 [DOI] [PubMed] [Google Scholar]
- 21. Reginato M. J., Mills K. R., Paulus J. K., Lynch D. K., Sgroi D. C., Debnath J., Muthuswamy S. K., Brugge J. S. (2003) Nat. Cell Biol. 5, 733–740 [DOI] [PubMed] [Google Scholar]
- 22. Seton-Rogers S. E., Lu Y., Hines L. M., Koundinya M., LaBaer J., Muthuswamy S. K., Brugge J. S. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 1257–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feigin M. E., Muthuswamy S. K. (2009) Curr. Opin. Cell Biol. 21, 694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. St Johnston D., Ahringer J. (2010) Cell 141, 757–774 [DOI] [PubMed] [Google Scholar]
- 25. Singh A., Settleman J. (2010) Oncogene 29, 4741–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeisberg M., Neilson E. G. (2009) J. Clin. Invest. 119, 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Merlo G. R., Basolo F., Fiore L., Duboc L., Hynes N. E. (1995) J. Cell Biol. 128, 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danes C. G., Wyszomierski S. L., Lu J., Neal C. L., Yang W., Yu D. (2008) Cancer Res. 68, 1760–1767 [DOI] [PubMed] [Google Scholar]
- 29. Seewaldt V. L., Mrózek K., Sigle R., Dietze E. C., Heine K., Hockenbery D. M., Hobbs K. B., Caldwell L. E. (2001) J. Cell Biol. 155, 471–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.