

Abstract
Membrane metallo-endopeptidase (MME), also known as neutral endopeptidase 24.11 (EC 3.4.24.11), is involved in the metabolism of natriuretic peptides that play a key role in modulating cardiac structure and function. Common genetic variation in MME has not been addressed by resequencing the gene using DNA from different ethnic populations. We set out to identify and functionally characterize common genetic variation in MME in three ethnic groups. DNA samples from 96 European-American, 96 African-American, and 96 Han Chinese-American healthy subjects were used to resequence MME. Ninety polymorphisms, 65 novel, were identified, including 8 nonsynonymous single nucleotide polymorphisms (nsSNPs). Expression constructs for the nsSNPs were created and COS-1 cells were transfected with constructs for wild type (WT) and variant allozymes. Recombinant proteins were analyzed by quantitative Western blot analysis and by a one-step fluorometric assay. A significant reduction in enzyme activity (21% of WT) and immunoreactive protein (29% of WT) for the Val73 variant allozyme was observed. Proteasome-mediated degradation and autophagy participated in the degradation of this variant allozyme. The chaperone proteins, BiP and GRP94, were upregulated after transfection with Val73 MME, suggesting protein misfolding, compatible with conclusions based on the MME X-ray crystal structure. Multiple novel polymorphisms of MME were identified in three ethnic groups. The Val73 variant allozyme displayed a significant decrease in MME protein quantity and activity, with degradation mediated by both proteasome and autophagy pathways. This polymorphism could have a significant effect on the metabolism of natriuretic peptides.
Keywords: Natriuretic peptides, Membrane metallo-endopeptidase, Polymorphism, Pharmacogenetics
1. Introduction
The heart synthesizes two important peptides, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). ANP and BNP cause vasodilation and natriuresis by direct actions that are primarily cGMP-mediated. These two peptides play an important regulatory role in cardiovascular disease as well as a key role in modulating cardiac structure and function [1]. BNP is commercially available as nesiritide and is approved for intravenous use in the treatment of decompensated heart failure (HF). These peptides are degraded approximately equally by two mechanisms: enzymatic degradation catalyzed by membrane metalloendopeptidase (MME), or neutral endopeptidase (NEP 24.11; EC 3.4.24.11), and clearance by the natriuretic peptide receptor C (NPR-C) [2]. Sequence variation in the gene encoding the precursor of BNP (NPPB) has been associated with altered serum BNP levels [3], but the impact on BNP levels of MME gene sequence variation remains unknown. Measurement of serum BNP is useful for diagnostic and prognostic purposes, especially in heart failure [4]. Nesiritide (BNP) is used to treat acute decompensated heart failure, but response to its use is variable [5]. MME plays an important role in the metabolism of nesiritide [6], and the potential importance of MME genetic polymorphisms on nesiritide degradation is also unknown. Furthermore, compounds that inhibit MME by increasing endogenous peptides like natriuretic peptides and bradykinin have been developed, and have been used to treat hypertension and heart failure [7]. MME polymorphisms and their possible effect on the pharmacological actions of these drugs have not been studied. Important steps toward these translational studies would be the identification and characterization of common MME polymorphisms in “normal”, randomly selected populations. DNA samples from such subjects are studied because common genetic variation needs to be defined initially in “normal” individuals prior to studying drug effect and response [8].
MME is a 100 kDa, type II integral membrane protein containing a highly conserved zinc binding motif in its extracellular C-terminal domain [9]. The enzyme is present in polymorphonuclear leucocytes, brush border cells of the proximal tubule and podocytes of the kidney, and epithelial cells of the liver, breast, lung and brain. MME cleaves substrates on the amino side of hydrophobic amino acids by hydrolyzing peptide bonds, and, as a result, it inactivates several peptide hormones including glucagon, enkephalins, substance P, neurotensin, oxytocin, bradykinin, and natriuretic peptides. The MME gene encodes a 750 amino acid protein, is 45 kb in length, consisting of 23 exons and maps to chromosome 3q25.1–q25.2. The 5′-untranslated region of the gene is alternatively spliced, resulting in four separate 5′-UTR sequences [10]. However, the coding region is not affected by this alternative splicing. Because of the importance of MME as the final common pathway for natriuretic peptide metabolism and because of the importance of natriuretic peptides in cardiovascular disease, we resequenced MME in DNA from 288 subjects, identified novel coding and non-coding genetic polymorphisms in the MME gene, and subsequently characterized the functional implications of the coding polymorphisms.
2. Methods
2.1. DNA samples
DNA samples from 96 European-American (EA), 96 African-American (AA), and 96 Han Chinese-American (HCA) unrelated, healthy subjects (sample sets HD100CAU, HD100AA, HD100CHI) were obtained from the Coriell Cell Repository (Camden, NJ). All of these DNA samples were collected, anonymized and deposited by the National Institute of General Medical Sciences. All subjects had provided written consent for the use of their DNA for research purposes. The present study was reviewed and approved by the Mayo Clinic Institutional Review Board.
2.2. Gene resequencing
The MME gene was resequenced in the 288 DNA samples described in the preceding paragraph. Specifically, 14 PCR reactions were performed with primers that flanked exons for at least 200 bp on either side, as well as approximately 1 kb of the 5′-flanking region (5′-FR). Primer sequences are listed in Supplemental Table 1. Amplicons were then sequenced on both strands in the Mayo Molecular Biology Core Facility with an ABI 3700 DNA sequencer using BigDye™ dye terminator sequencing chemistry (Perkin-Elmer Life Science, Boston, MA). To exclude PCR-related artifacts, independent amplifications were performed for any SNP observed in only a single DNA sample or any sample with an ambiguous chromatogram. The sequencing chromatograms were analyzed using Mutation Surveyor version 2.2 (Softgenetics LLC, State College, PA). GenBank accession numbers for the MME reference sequences used in these experiments were NT_005612.15. All sequence data were deposited in PharmGKB with Accession Number PA30864.
2.3. MME expression in COS-1 cells
Human MME cDNA open reading frame (ORF) clone (NM_007289), transcript variant 2b, obtained from OriGene Technologies, Inc. (Rock-ville, MD), was used as wild type (WT) cDNA clone. This construct was cloned into the eukaryotic expression vector pCMV6-XL4. The insert was sequenced in both directions to verify construct sequence. Site-directed mutagenesis was then performed using the QuikChange II kit (Stratagene, La Jolla, CA) to create expression constructs for each of the variant allozymes. The sequences of primers used to perform site-directed mutagenesis are also listed in Supplemental Table 2. The sequences of all variant allozymes were also confirmed by sequencing in both directions. COS-1 cells were then transfected with expression constructs encoding WT and variant MME allozymes, as well as “empty” vector (EV) that lacked an insert as a control, using the Fugene 6 Transfection reagent (Roche Applied Science, Indianapolis, IN) at a charge ratio of 6:1. The cells were also cotransfected with pSV-ß-galactosidase (Promega, Madison, WI) to make it possible to correct for variation in transfection efficiency. After 48 h, the cells were harvested in a buffer containing 50 mM Tris–HCl (pH 7.4). The cells were then homogenized with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY), followed by centrifugation at 100,000×g for 1 h. After centrifugation, the supernatant was separated from the pellet and stored. The supernatant was used to assay β-galactosidase to make it possible to adjust for transfection efficiency. The pellet was homogenized after the addition of 50 mM Tris–HCl buffer (pH 7.4). Both cytosol and membrane preparations were stored at −80°C for use in the functional genomic studies described subsequently.
2.4. MME enzyme assay
MME activity was measured as previously described using a one-step fluorometric assay with Dansyl-d-Alanyl-Glycyl-p-nitrophenyla-lanylglycine (DAGNPG) as a substrate [11]. DAGNPG, 200 μM, was preincubated for 15 min at 37 °C in 50 μl of 50 mM Tris–HCl (pH 7.4). The reaction was initiated by adding 50 μM of the membrane preparation, and the mixture was incubated for 1 h at 37 °C. After incubation, the enzymatic reaction was stopped by boiling at 100 °C for 5 min, and the samples were diluted with 200 μl of Tris–HCl (pH 7.4). Optical density was determined using a 96 well plate reader (Tecan, Durham, NC, USA) using 342 nm as the excitation and 562 nm as the emission wavelength. A standard curve was prepared using known concentrations of d-Alanyl-Glycyl ranging from 4 to 64 μM. Substrate kinetic studies were performed using ten different DAGNPG concentrations ranging from 0.04 to 0.4 mmol/l.
2.5. Western blot analysis
Membrane preparations were subjected to electrophoresis on 10% Tris–HCl acrylamide gels loaded on the basis of co-transfected β-galactosidase activity measured with the Promega β-Galactosidase Enzyme Assay System (Promega). After electrophoresis, proteins were transferred to PVDF membranes (BioRad, Hercules, CA), and the membranes were incubated with rabbit polyclonal anti-MME antibody in (1:500) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), followed by a secondary antibody (1:5000). Immunoreactive proteins were detected using the ECL Western Blotting System (Amersham Pharmacia, Piscat-away, NJ). The IPLab Gel H (Biosystemetica, Plymouth, UK) system and the NIH image program (http://rsb.info.nih.gov/nih-image) were used to quantify immunoreactive proteins, and the data were expressed as a percentage of WT human MME protein intensity on the same gel. Three samples each for the WT, empty vector and variant allozymes were assayed for each allozyme studied.
2.6. Immunofluorescence microscopy and protein degradation studies
COS-1 cells were grown on coverslips for 24 h prior to transfection with expression constructs for the WT and Val73 variant allozymes. The cells were fixed and permeabilized as described previously, and were stained for 30 min with a rabbit polyclonal anti-MME antibody (1:100) (Santa Cruz Biotechnology, Inc.), followed by incubation for 30 min with an anti-rabbit antibody-fluorescein isothiocyanate (FITC) conjugate (1:200) (Santa Cruz Biotechnology, Inc.). DAPI nucleic acid stain (Invitrogen, Eugene, OR) and a mouse monoclonal antibody [RL90] to PDI (Abcam Inc., Cambridge, MA) were used to visualize the nuclei and endoplasmic reticulum (ER), respectively. Cells were then visualized with a Zeiss LSM 510 Confocal Laser Scanning Microscope. To quantify cells, we randomly selected three fields (each field containing approximately 200 cells), obtained photographs, and counted all cells in each field.
To test for proteasome-mediated degradation, after transfection with WT and Val73 MME constructs, COS-1 cells were treated with either DMSO or 20 μM of MG132, a proteasome inhibitor for 24 h. To determine whether autophagy might be responsible for clearance of the Val73 variant allozyme, transfected cells were treated with 10 μM of the autophagy inhibitor 3-methyladenosine (3MA) for 48 h. The possible interaction of the Val73 variant allozyme with chaperone proteins was determined by treating WT and Val73 transfected cells with purified mouse anti-BiP/GRP78 antibody (1:1000) (BD Transduction Laboratories, San Jose, CA) and rabbit polyclonal antibody to GRP94 (1:250) (Abcam Inc.). Binding protein (BiP) and GRP94 are major chaperone proteins in the endoplasmic reticulum. Immunoreactive MME protein was then quantified by Western blot analysis after normalization using plasma membrane calcium ATPase isoform 4 (PMCA4) as a membrane housekeeper protein standard. Mouse monoclonal antibody (JA9) to PMCA4ATPase was used in a 1:1000 dilution (Abcam Inc.).
2.7. Structural analysis
The 2.1 Å resolution crystal structure of human MME bound to the active site inhibitor phosphoramidon (PDB accession code 1DMT) [12] provided the starting point for computational modeling of the variant allozymes. Molecular visualization and modeling of the variant structures were performed using the computer program Coot [13]. Structural figures were prepared with Molscript and Raster3D [14,15]. Parallel molecular dynamics (MD) simulations of the WT and Val73 variant structures were performed with the GROMACS-4.0.3 software package and GROMOS-87 force field [16,17]. Using the SPC water model, the structures were solvated in a cubic periodic box extending at least 9 Å from the protein atoms and subjected to 30 ps of position-restrained MD calculations before an extended 6 ns of extended MD simulations. Energy was minimized in 500 steps using a steepest descents procedure. The temperature of the system was coupled to a reference bath of 300 K and a coupling time constant of 0.1 ps. Coordinates were saved every 1 ps for analysis.
2.8. Data analysis
Values for π, θ and Tajima's D were calculated as described by Tajima [18]. R2 values, a measure of linkage disequilibrium, were calculated as described by Hartl and Clark [19] and Hendrick [20]. Haplotype analysis was performed as described by Schaid et al. [21] using the E–M (expectation–maximization) algorithm [22]. Apparent Km values were determined using the GraphPad Prism 4.0 software program (GraphPad, San Diego, CA). Mean protein and Km values were compared using Student's t-test and ANOVA.
3. Results
3.1. Human MME resequencing
MME was resequenced using 288 anonymized DNA samples from subjects of three different ethnic groups. Ninety polymorphisms, including 86 SNPs and 4 indels, were observed; 62 in AA, 40 in EA, and 36 in HCA subjects (Table 1 and Fig. 1). Thirty-eight of these polymorphisms were “common”, with a minor allele frequency (MAF) of >1% in at least one ethnic group. There were 17 polymorphisms within exons, 8 of which were nonsynonymous SNPs (nsSNPs) resulting in the following changes in the encoded amino acids: Met8Val, Arg23Pro, Met73Val, Gly225Ala, Asn285Ser, Val345Ile, Met419Leu, Asn593Ser (Table 1). The alleles encoding Met8Val were observed in both EA and AA subjects. Met73Val, Gly225Ala, and Met419Leu encoding alleles were only observed in AA subjects. The alleles encoding Asn285Ser and Val345Ile were only observed in EA individuals, while those encoding Arg23Pro and Asn593Ser were observed only in HCA subjects. MAF for these nonsynonymous SNPs were less than 1% except for the alleles encoding Met8Val and Met419Leu. Sixty-five of the 90 MME polymorphisms were novel and were not present in public databases (www.ncbi.nlm.nih.gov/SNP) (Table 1). The MAFs of the known SNPs present in public databases (e.g. HapMap) were comparable to the MAFs that were observed. For example, the rs12696022 SNP had MAFs in HapMap of 0.192, 0.089 and 0.000 in CEU, HCB and YRI subjects, respectively, as compared to 0.156 in EA, 0.089 in HCA and 0.016 in AA subjects in our study.
Table 1.
MME polymorphism locations, alterations in nucleotide and amino acid sequences, as well as minor allele frequencies for all three ethnic groups studied are listed. SNPs in exons and 5′-FRs have been numbered on the basis of their locations in the cDNA, with the ‘A’ in the translation initiation codon assigned (+1). Negative numbers are located 5′, and positive numbers 3′ to this position. SNPs in introns have been numbered based on their distance to the nearest 5′ or 3′-exon–intron splice junctions, using positive and negative numbers, respectively. The final column and the gray shaded rows indicate whether the polymorphism was already present in dbSNP.
| Location | Nucleotide | Sequence change | Amino acid change | Frequency of variant allele | |||
|---|---|---|---|---|---|---|---|
| African-American | European-American | Han Chinese-American | RefSNPID | ||||
| 5′FR | −867 | G→A | 0.109 | 0.036 | 0.000 | rs1836912 | |
| 5′FR | −624 | deletion of G | 0.000 | 0.000 | 0.021 | ||
| 5′FR | −581 | G→A | 0.010 | 0.000 | 0.000 | ||
| 5′FR | −544 | G→A | 0.021 | 0.000 | 0.000 | ||
| 5′FR | −374 | T→C | 0.052 | 0.005 | 0.010 | ||
| 5′FR | −170 | T→G | 0.005 | 0.011 | |||
| Intron 1 | 148 | T→C | 0.042 | 0.000 | 0.000 | ||
| Intron 1 | 389 | A→T | 0.000 | 0.000 | 0.005 | ||
| Intron 1 | 402 | C→A | 0.000 | 0.005 | 0.000 | ||
| Intron 1 | 413 | C→T | 0.000 | 0.005 | 0.000 | ||
| Intron 1 | 592 | G→A | 0.005 | 0.031 | 0.000 | ||
| Intron 1 | 635 | G→C | 0.021 | 0.000 | 0.000 | ||
| Intron 1 | 691 | deletion of T | 0.068 | 0.000 | 0.000 | ||
| Intron 1 | 736 | G→T | 0.005 | 0.000 | 0.000 | ||
| Intron 1 | 799 | T→C | 0.120 | 0.047 | 0.010 | ||
| Intron 1 | −136 | T→A | 0.000 | 0.005 | 0.000 | ||
| Intron 1 | −123 | G→A | 0.000 | 0.010 | 0.000 | ||
| Intron 1 | −122 | C→G | 0.000 | 0.010 | 0.000 | ||
| Intron 1 | −115 | G→A | 0.073 | 0.219 | 0.031 | ||
| Intron 1 | −17 | A→G | 0.005 | 0.000 | 0.000 | ||
| Exon2 | 21 | G→A | 0.005 | 0.000 | 0.000 | ||
| Exon 2 | 22 | A→G | Met8Val | 0.005 | 0.026 | 0.000 | |
| Exon 2 | 66 | G→A | 0.000 | 0.005 | 0.000 | ||
| Exon 2 | 68 | G→C | Arg23Pro | 0.000 | 0.000 | 0.005 | |
| Intron 2 | 13 | C→T | 0.010 | 0.000 | 0.000 | ||
| Intron 2 | 33 | T→A | 0.005 | 0.000 | 0.000 | ||
| Intron 2 | 97 | G→C | 0.000 | 0.000 | 0.010 | rs12629968 | |
| Intron 2 | 135 | A→G | 0.010 | 0.000 | 0.000 | ||
| Intron 2 | 221 | G→A | 0.005 | 0.000 | 0.000 | ||
| Intron 2 | 338 | A→C | 0.010 | 0.000 | 0.000 | ||
| Intron 2 | 346 | G→C | 0.016 | 0.156 | 0.089 | rs12696022 | |
| Intron 2 | −106 | T→G | 0.099 | 0.151 | 0.073 | rs28617565 | |
| Intron 2 | −74 | A→T | 0.005 | 0.000 | 0.000 | ||
| Exon 3 | 162 | T→C | 0.000 | 0.010 | 0.000 | rs35926730 | |
| Intron 3 | 37 | T→C | 0.078 | 0.073 | 0.089 | rs4607069 | |
| Intron 3 | 61 | T→C | 0.000 | 0.000 | 0.005 | ||
| Intron 3 | 133 | T→G | 0.026 | 0.000 | 0.000 | ||
| Intron 3 | −15 | T→G | 0.078 | 0.000 | 0.000 | ||
| Exon 4 | 217 | A→G | Met73Val | 0.005 | 0.000 | 0.000 | |
| Intron 4 | 74 | G→A | 0.005 | 0.000 | 0.000 | ||
| Intron 4 | 84 | deletion of GGAATGT | 0.078 | 0.000 | 0.000 | ||
| Intron 4 | 97 | A→C | 0.000 | 0.000 | 0.005 | ||
| Exon 8 | 674 | G→C | Gly225Ala | 0.005 | 0.000 | 0.000 | |
| Intron 8 | 45 | G→T | 0.000 | 0.000 | 0.005 | ||
| Intron 8 | −133 | A→T | 0.000 | 0.000 | 0.005 | ||
| Exon 9 | 786 | G→A | 0.005 | 0.000 | 0.000 | rs34147103 | |
| Exon 9 | 854 | A→G | Asn285Ser | 0.000 | 0.005 | 0.000 | |
| Intron 9 | 6 | A→T | 0.000 | 0.005 | 0.000 | rs41267907 | |
| Intron 9 | −83 | C→T | 0.021 | 0.104 | 0.010 | rs10935999 | |
| Intron 10 | 71 | A→T | 0.005 | 0.000 | 0.000 | ||
| Intron 10 | 77 | C→T | 0.198 | 0.188 | 0.068 | rs28758831 | |
| Exon 11 | 1033 | G→A | Val345Ile | 0.000 | 0.005 | 0.000 | |
| Exon 11 | 1047 | A→G | 0.005 | 0.000 | 0.000 | rs35152996 | |
| Exon 12 | −5 | C→T | 0.000 | 0.000 | 0.005 | rs2304504 | |
| Exon 13 | 1191 | C→T | 0.0 10 | 0.005 | 0.000 | rs35227300 | |
| Exon 13 | 1255 | A→C | Met419Leu | 0.016 | 0.000 | 0.000 | rs34931605 |
| Intron 13 | 205 | G→T | 0.005 | 0.000 | 0.000 | ||
| Intron 13 | 132 | C→A | 0.005 | 0.000 | 0.000 | ||
| Intron 13 | 111 | C→T | 0.000 | 0.000 | 0.010 | ||
| Intron 13 | 110 | G→A | 0.016 | 0.005 | 0.005 | ||
| Intron 14 | 144 | T→A | 0.260 | 0.365 | 0.474 | rs6797911 | |
| Exon 15 | 1482 | T→C | 0.000 | 0.000 | 0.005 | ||
| Intron 15 | 52 | A→C | 0.000 | 0.005 | 0.000 | ||
| Intron 15 | 133 | G→A | 0.005 | 0.000 | 0.000 | ||
| Intron 15 | 160 | G→A | 0.000 | 0.000 | 0.005 | ||
| Intron 15 | 235 | C→T | 0.000 | 0.000 | 0.016 | ||
| Intron 15 | 320 | A→G | 0.005 | 0.021 | 0.078 | rs16824618 | |
| Intron 15 | 335 | C→T | 0.026 | 0.000 | 0.000 | ||
| Intron 16 | 11 | T→C | 0.464 | 0.349 | 0.198 | rs1436633 | |
| Intron 16 | 80 | C→A | 0.005 | 0.021 | 0.115 | rs3732886 | |
| Intron 16 | 103 | A→C | 0.464 | 0.349 | 0.198 | rs1436632 | |
| Intron 16 | −86 | G→A | 0.005 | 0.000 | 0.000 | ||
| Intron 16 | −20 | deletion of A | 0.323 | 0.271 | 0.073 | rs35450782 | |
| Intron 17 | 36 | T→C | 0.026 | 0.000 | 0.000 | ||
| Exon18 | 1778 | A→G | Asn593Ser | 0.000 | 0.000 | 0.005 | |
| Intron 18 | 64 | T→C | 0.005 | 0.000 | 0.000 | ||
| Intron 18 | 195 | G→A | 0.000 | 0.010 | 0.000 | ||
| Intron 18 | 263 | G→A | 0.010 | 0.000 | 0.000 | ||
| Intron 18 | 305 | C→T | 0.016 | 0.000 | 0.000 | ||
| Intron 18 | 309 | T→C | 0.036 | 0.193 | 0.000 | ||
| Intron 18 | 396 | TgA | 0.333 | 0.641 | 0.646 | rs9864287 | |
| Intron 18 | −3 | TgC | 0.182 | 0.068 | 0.146 | rs3736187 | |
| Intron 19 | 29 | T→C | 0.005 | 0.000 | 0.000 | ||
| Intron 19 | −12 | C→G | 0.010 | 0.000 | 0.000 | ||
| Intron 20 | 80 | A→G | 0.000 | 0.000 | 0.005 | ||
| Intron 21 | 36 | CgA | 0.010 | 0.021 | 0.135 | rs41267911 | |
| Intron 21 | −119 | GgA | 0.385 | 0.271 | 0.068 | rs3816803 | |
| Exon 22 | 2133 | G→A | 0.005 | 0.005 | 0.000 | ||
| Intron 22 | 21 | A→G | 0.000 | 0.005 | 0.000 | ||
| Intron 22 | −22 | AgG | 0.073 | 0.151 | 0.177 | rs16824656 | |
Fig. 1.
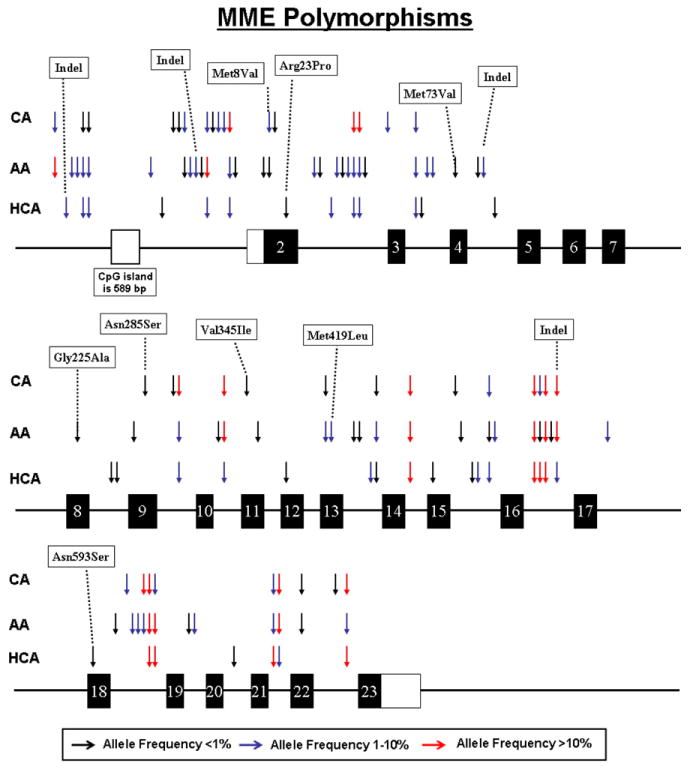
Human MME genetic polymorphisms. The figure shows a schematic representation of the structure of MME, with arrows indicating locations of polymorphisms. Exons encoding the open reading frame are represented as dark rectangles and portions of exons that encode UTR sequences are indicated by open rectangles. Colors of arrows indicate minor allele frequencies.
We also determined nucleotide diversity, a quantitative measure of genetic variation, adjusted for allele frequency, in all three ethnic groups by calculating θ, a population mutation measure that is theoretically equal to the neutral mutation variable, and π, average heterozygosity per site. Values for Tajima's D, a test of the neutral mutation hypothesis, were also estimated (Supplemental Table 3). Although negative values for Tajima's D indicate a departure from neutrality, none of the values listed in Supplemental Table 3 were statistically significant.
3.2. Linkage disequilibrium and haplotype analysis
Population-specific linkage disequilibrium analysis was performed for each ethnic group. All possible pairwise SNP comparisons were used to compute R2 values. Data for all pairwise SNP comparisons with R2 values≥0.7 for each ethnic group are listed in Supplemental Table 4. Haplotype analysis was also performed for MME, and a total of 27 haplotypes with frequencies of over 1% were observed (Supplelemental Table 5). Haplotype designations were based on the encoded amino acid sequence of the allozyme, with the most common amino sequence in AA subjects being designated as *1. Letter designations were then added based on decreasing frequencies, using AA samples as the ‘base’. Therefore, the *1 haplotype encodes the WT allozyme and the *2, *3, and *4 haplotypes contain SNPs that encode variant amino acids Met8Val, Arg23Pro, and Met73Val, respectively. None of the haplotypes listed encoded more than more than one variant amino acid. Haplotypes containing nsSNPs that had a frequency of less than 1% were included in Supplemental Table 5 because of the variant amino acid sequence. Haplotype frequencies, like MAFs, differed significantly among the three ethnic groups. Many haplotypes were observed in only one ethnic group. There were 6 haplotypes with an allele frequency of >1% in AA subjects, whereas 13 and 16 such haplotypes were observed in EA and HCA individuals, respectively (Supplemental Table 5). The *1C haplotype was observed with a relatively high frequency in all three populations, whereas *1D was observed in high frequency in the EA and HCA subjects, but not in AA subjects (Supplemental Table 5). Graphical representations of population-specific R2 values across the MME gene for the three populations studied are shown in Supplemental Fig. 1.
3.3. MME allozyme activity and protein levels
Expression constructs were created for the MME WT and eight variant allozymes to make it possible to determine the effect of the nsSNPs on levels of protein, enzyme activity, and substrate kinetics. These MME expression constructs were transfected into a mammalian expression system, COS-1 cells, to ensure that mammalian post translation modification and protein degradation systems would be present. Immunoreactive protein levels were then measured by quantitative Western blot analysis and levels of enzyme activity were measured in WT and all variant allozyme samples. The levels of MME immunoreactive protein, as well as the enzyme activity of the variant allozymes as a percentage of WT are shown graphically in Fig. 2A. The Val73 allozyme showed a decrease in protein levels to approximately 25% of WT. Similar results were observed when enzyme activity of the Val73 allozyme was assayed with DAGNPG as a substrate. Conversely, immunoreactive protein levels for the Ser285 allozyme were approximately 60% greater than that of WT, and a corresponding increase as compared to WT was seen in enzyme activity for this allozyme. No statistically significant differences were detected for the other six variants. There was a significant correlation between protein and enzyme activity levels for this group of MME allozymes (R2=0.7798, p=0.0016) (Fig. 2B). Because of the possibility that part of the difference in level of enzyme activity that we had observed for the Val73 and Ser285 variant allozymes might be due to alterations in substrate kinetics, substrate kinetic studies were also performed. The apparent Km values observed for DAGNPG for WT, Val73 and Ser285 allozymes were 236.5±5.5, 91.0±4.6 and 228.6±5.2, respectively. Although the Km values for WT and Ser285 allozyme did not differ significantly, the Val73 allozyme had a significantly decreased apparent Km value for DAGNPG as compared to WT (p≤0.005). However, this decrease in apparent Km could not explain the lower activity for this allozyme. Due to markedly reduced immunoreactive protein levels and enzymatic activity of the Val73 variant as compared to WT and the other variants we sought to delineate the processes that could lead to degradation of this variant protein.
Fig. 2.
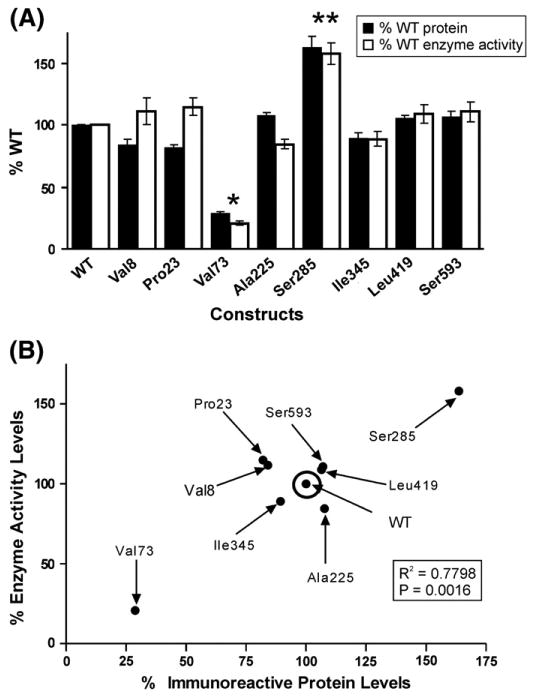
MME functional genomics. (A) Levels of MME immunoreactive protein and allozyme enzyme activity as a percentage of wild type (WT). Each bar represents the average of six independent transfections (mean±SE). All values are corrected for transfection efficiency. *p<0.001 and **p<0.05 when compared with the WT value. (B) Correlation between average levels of MME immunoreactive protein and enzyme activity.
3.4. Proteasome-mediated degradation of Val73
The decrease in levels of Val73 immunoreactive protein could be due to accelerated protein degradation, perhaps involving the ubiquitin-proteasome system and/or autophagy [23]. To delineate the possible role of proteasome-mediated degradation, the effect of the proteasome inhibitor MG132 was determined. Fig. 3A shows a graphic representation of significantly decreased Val73 immunoreactive protein values as compared to WT. After treatment with MG132, the average Val73 protein level increased almost 6-fold but no significant change was seen for the WT allozyme, indicating that proteasome-mediated degradation plays an important role in the degradation of the variant allozyme. A representative Western blot showing these results after normalizing for membrane protein PMCA4ATPase, is shown in Fig. 3B. In addition, the number of cells that stained for MME after MG132 treatment was far greater for Val73 (7±0.8% in control arm and 22±3% in treatment arm, p=0.02) than WT transfected COS-1 cells (15±2% in control arm and 19±3% in treatment arm, p=0.64) as shown graphically in Fig. 3C.
Fig. 3.
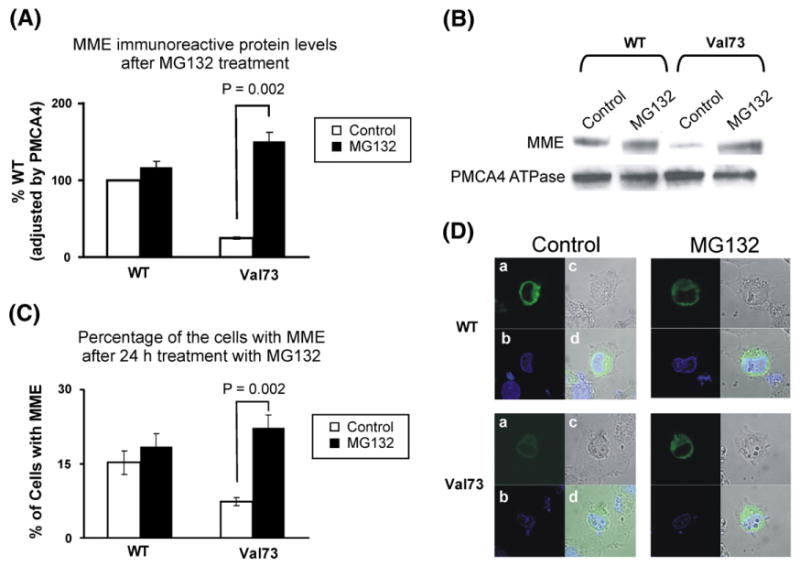
Proteasome mediated MME degradation. (A) Levels of WT and Val73 MME allozyme immunoreactive protein expressed as a percentage of WT after exposure to MG132, a proteasome inhibitor. The white bars represent “control” and the black bars represent samples exposed to the proteasome inhibitor MG132. Each value is the mean±SE of six independent studies. (B) Representative Western blot analysis of WT and Val73 MME immunoreactive protein after treatment of the COS-1 cells with MG132. (C) The bar graph shows the percentage of COS-1 cells transfected with WT and Val73 MME allozymes before and after treatment with MG132. The white bars represent “control” and the black bars represent samples exposed to the proteasome inhibitor MG132. Each bar represents the average of 3 independent readings (mean±SE). (D) Immunofluorescence microscopy for WT and Val73 MME variant allozymes transfected into COS-1 cells before and after treatment with MG132. The panels demonstrate (a) MME staining (green) in the nuclear membrane (NM) and endoplasmic reticulum (ER); (b) Nuclear staining (blue) using DAPI; (c) COS-1 cell structure without staining; (d) A merged field of (a), (b), and (c) showing localization of MME around the NM and ER.
3.5. Autophagy, aggresome formation and interaction with chaperone proteins
Autophagy is also an important mechanism for protein degradation. To assess the possible contribution of autophagy to MME degradation, WT and Val73 transfected COS-1 cells were treated with 3MA, an inhibitor of autophagy. Mean levels of MME immunoreactive protein increased significantly after treatment with 3MA for both the WT and Val73 variant allozymes as shown in the bar graph in Fig. 4A and Western blot in Fig. 4B, indicating a role for autophagy in MME degradation. Furthermore, fluorescence microscopy showed the formation of both micro- and macroaggregates of MME after 3MA treatment (Fig. 4D). These aggregates were seen after 3MA treatment of cells transfected with both the WT and Val73 allozymes (Fig. 4D). However, the number of cells transfected with microaggregates after 3MA treatment was far greater for Val73 (9±0.5% increasing to 23±0.2%, p=0.002) than WT transfected COS-1 cells (5±0.4% increasing to 7±0.4%, p=0.06), as shown graphically in Fig. 4C. The macro-aggregates observed in these studies may represent aggresomes, perinuclear bodies composed of misfolded protein, structures observed when proteins misfold and/or the capacity of proteasome-mediated degradation is exceeded [24].
Fig. 4.
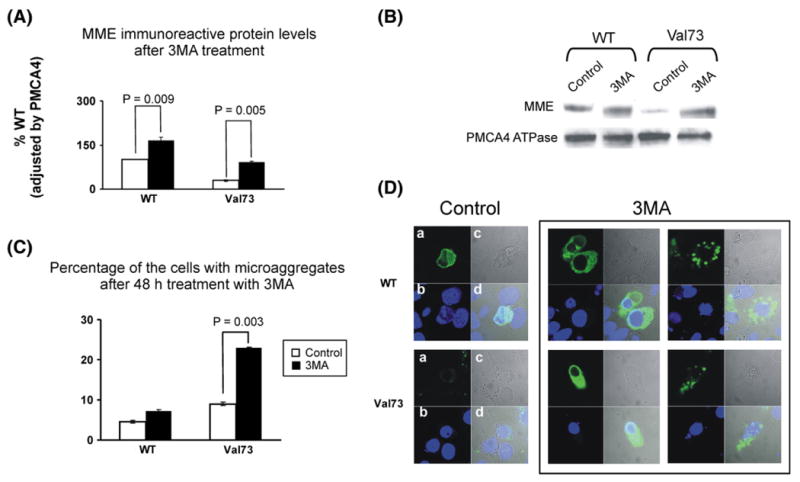
Autophagy, MME degradation and aggregate formation. (A) Levels of WT and Val73 MME allozyme immunoreactive protein expressed as a percentage of WT after exposure to 3MA, an autophagy inhibitor. Each bar represents the average of three independent transfections (mean±SE). (B) Representative Western blot analysis of WT and Val73 MME immunoreactive protein after treatment with 3MA, normalized for PMCA4 ATPase, a membrane protein. (C) The bar graph shows percentage of COS-1 cells transfected with WT and Val73 allozymes that stain for MME microaggregates before and after treatment with 3MA. Each bar represents the average of 3 independent determinations (mean±SE). (D) Immunofluorescence results for COS-1 cells transfected with WT and Val73 MME before and after treatment with 3MA using confocal epifluorescence microscopy. The panels to the extreme right show aggregate formation after treatment with 3MA in WT (top panel) and Val73 (bottom panel) transfected COS-1 cells.
Binding protein (BiP) is an ER lumen chaperone protein that binds misfolded secretory and/or membrane-bound proteins and ensures proper movement of these proteins from the ER to the Golgi apparatus. GRP94, an ER paralog of Hsp90, is also thought to regulate ER apoptotic signaling when there is an accumulation of misfolded protein in the ER lumen [25]. Both BiP and GRP94 are increased by conditions that produce misfolded proteins [25]. Fig. 5A shows increased immunoreactive protein levels of BiP and GRP94 for the Val73 variant as compared to the WT allozyme after transfection of COS-1 cells, implying that the former may misfold and that BiP and GRP94 may participate in Val73 protein degradation. The representative Western blot in Fig. 5B shows increased BiP and GRP94 after transfection with the Val73 variant allozyme as compared to WT, after normalizing for the membrane protein PMCA4ATPase. Because of these observations, we next used MME X-ray crystal structural information with regard to MME in an attempt to understand the structural implications of the Met73Val polymorphism.
Fig. 5.
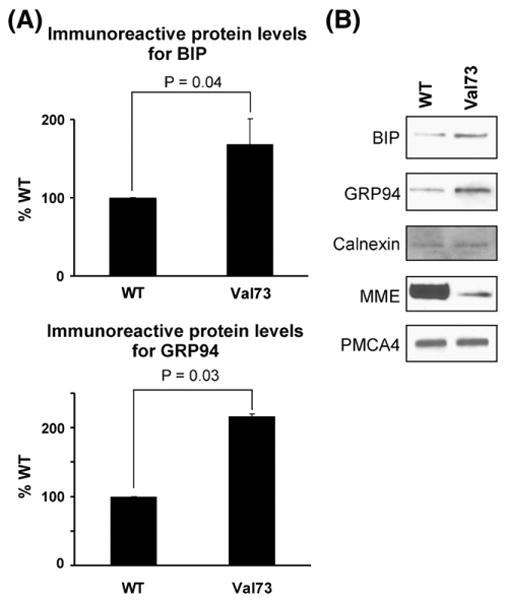
MME degradation and chaperone proteins. (A) The bar graph shows levels of BiP and GRP94, chaperone proteins in the endoplasmic reticulum for COS-1 cell lysates after transfection with WT and Val73 expression constructs. Protein levels are shown as a percentage of that seen after WT transfection. Each bar represents the average of 6 independent transfections (mean±SE). (B) Representative Western blot analysis for WT and Val73 COS-1 cell lysates showing BiP and GRP94, with the membrane proteins Calnexin and PMCA4 ATPase stained as “standards”.
3.6. Structural analysis of MME variant allozyme
The MME protein fold is bilobal, formed by two predominantly α-helical halves (Fig. 6A). These two lobes are primarily formed by the N-and C-terminal halves of the polypeptide, with several intertwining segments, including the N-terminus residues which pack against the C-terminal lobe. The inhibitor and the catalytic zinc ion shown in Fig. 6A are bound within an active site at the interface between the two lobes, with all residues directly interacting with the ligands located in the C-terminal half of the protein sequence. Residues altered in MME variant allozymes are located in both lobes. Most are distant from the active site, with the exception of Asn593, which is located 8.6 Å from the bound phosphoramidon. However, this is a surface exposed residue, and modeling of the Ser593 variant shows that substitution of the larger hydrophilic residue with a smaller one should be fully compatible with the WT protein structure.
Fig. 6.
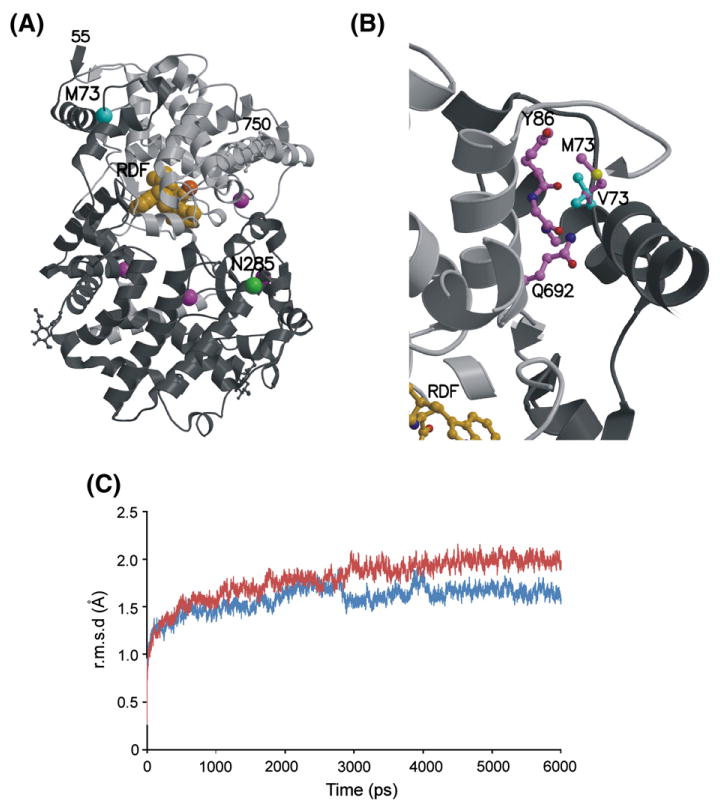
Structural modeling of MME variants. (A) X-ray crystal structure of human MME[12] showing the locations of residues altered by MME nsSNPs. The ordered protein fold contains residues 55–750, shown as a ribbon diagram with the N-terminal and C-terminal halves in dark and light gray, respectively. The active site contains a zinc ion (dark orange sphere) and phosphoramidon inhibitor (light orange spheres). Cα atoms of residues altered by the Met73Val (MT3) and Asn285Ser (N285) variants are highlighted as cyan and green spheres, respectively. The locations of other residues altered by variants with WT enzyme behavior (Gly225, Val345, Met419, and Asn593) are marked by purple spheres. Two variant residues, Met8 and Arg23 are located in the extended N-terminus which is not present in the MME crystal structure and, thus, cannot be modeled. (B) Closeup view of the modeled Met73Val (M73) variant. The side chains of the WT Met73 (crystal structure, magenta carbon atoms and bonds) and variant Val73 (V73) (theoretical model, cyan carbon atoms and bonds) interact with atoms in nearby residues such as the main chain and side chain atoms of Tyr86 (Y86), the main chain carbonyl of the adjacent Ala87, and the side chain of Gln692 (Q692) (all with magenta carbon atoms and bonds). Substitution of the unbranched Met73 with the branched Val73 introduces short contacts that are incompatible with the WT protein conformation. (C) MD simulation trajectories for WT and Val73 MME. Average main chain conformational differences between the initial MME crystal structure and energy minimized WT (blue) and Val73 (red) were calculated for structures sampled at 1 ps increments during the course of 6 ns simulations. Both trajectories plateaued, indicating stable equilibria, but the average r.m.s.d. for the Val73 simulation is significantly higher than for WT, revealing that the Val73 substitution is less compatible with WT conformation than the original Met73.
Two of the MME variant allozymes showed distinctive behavior during our functional experiments. Val73 had low expression and activity after transfection into COS-1 cells. This residue is located in the N-terminal region of the protein which interacts with the C-terminal lobe (Fig. 6A). The Met73 side chain is buried in a well-packed, predominantly hydrophobic environment. Computational substitution with the shorter but wider branched-chain Val73 introduces a number of sterically unfavorable short contacts that cannot be relieved by simple rotation of the side chain conformation (Fig. 6B). These short contacts involve residues in adjacent α-helices located in both the N-terminal region (Tyr86 and Ala87) and the C-terminal lobe (Gln692). Therefore, structural modeling indicated that Val73 was incompatible with the WT conformation and would likely result in a modified structure or compromise protein folding/stability. In contrast to Val73, the Ser285 variant resulted in elevated levels of enzyme expression and activity (Fig. 2A). This residue is located in the globular N-terminal lobe (Fig. 6A). Its side chain is exposed on the protein surface and interacts with three solvent molecules. Structural modeling of the Ser285 variant revealed that it was compatible with the WT protein fold and was unlikely to compromise protein folding or stability.
To further explore the possible structural consequences of the Val73 variant, parallel MD simulation calculations were carried out to explore energy-minimized conformations for both WT and Val73 variant MME protein. The simulations covered 6 ns, and molecular coordinates were saved every 1 ps for analysis. To compare the conformations between simulated structures and the initial WT crystal structure and model of Val73, the root mean square difference (r.m.s.d.) of superimposed backbone atoms was calculated. Plotting overall r.m.s.d. between simulated structures at different time points and the initial crystal structure for the two MD simulations reveals different trajectories for the WT and Val73 structures, with the variant deviating more significantly (r.m.s.d. ∼2.0 Å) from the starting crystal structure than the WT (r.m.s.d. ∼1.6 Å) (Fig. 6C). This confirms, as predicted by the initial modeling, that the Val73 is not compatible with the WT conformation. A plot of r.m.s.d. per residue, between the final simulated structures and the initial crystal structure, and visual comparison of the structures, shows that the most significant conformational deviation is in the variant region, involving residues Val73–Ala75 (Supplementary Fig. 2). This region shows minimal deviation in the WT simulation, indicating that the conformational shifts are due to the Met73Val substitution in the variant.
4. Discussion
MME plays an important role in the degradation of glucagon, enkephalins, substance P, neurotensin, oxytocin, bradykinin, and natriuretic peptides [26]. MME is expressed in cardiac and kidney tissue and is upregulated, with an increase in enzymatic activity, in patients with aortic stenosis and heart failure [27,28]. The upregulation in MME expression is thought to promote fibrosis in aortic stenosis [29]. MME also plays an important role in the local control of bradykinin in cardiac tissue by almost exclusively metabolizing bradykinin in human cardiac membranes [27]. Despite the importance of MME in cardiac disease and the role that it plays in metabolizing natriuretic peptides, a comprehensive attempt to identify common MME gene sequence variation in different ethnic groups by systematically resequencing the gene had not been undertaken previously. Genetic variation in MME has been reported to play a role in anxiety disorders, Alzheimer's disease and in the development of glomerulonephritis in the neonate [30–32]. For example, a dinucleotide polymorphism in the 5′-FR of MME results in altered MME activity with altered hydrolysis of enkephalin. This polymorphism has been reported to be significantly associated with anxiety and obsessive–compulsive disorder [30]. Amyloid beta peptide in the brain is degraded by MME and its accumulation is associated with Alzheimer's disease. A GT repeat polymorphism in the MME promoter has been linked to late onset Alzheimer's dementia [31]. In a kindred study, three families with cases of neonatal membranous glomerulonephritis were screened and found to have two different MME polymorphisms, 466delC and C1342T, resulting in a truncated protein leading to instability and absence of MME protein [32]. This MME sequence variation can cause alloimmunization during pregnancy, resulting in an anti-MME IgG antibody response in the mother when exposed to fetal MME antigens, causing glomerulonephritis in the neonate and early adulthood.
Genetic variation in the natriuretic peptide system has been associated with an increased risk of stroke, left ventricular hypertrophy, and essential hypertension [33,34]. A pharmacogenetic association of the T2238C variant of NPPA (atrial natriuretic precursor A), a gene encoding the precursor of atrial natriuretic peptide, has been demonstrated with the use of a diuretic, chlorthalidone as an anti-hypertensive agent [35]. Patients with this variant had a better response to blood pressure reduction and a lower risk for a coronary event, stroke and death when treated with a diuretic as compared to a calcium channel blocker. The natriuretic peptides play an important role in the structural and clinical features of heart failure and hypertension, and are metabolized, in part, by MME. However, the functional and clinical implications, if any, of common sequence variation in MME for these diseases remains unclear.
In the present study, we identified 90 MME SNPs, 8 of which were nsSNPs, by resequencing DNA samples from three different ethnic groups. Sixty-five of the polymorphisms found using this approach were not present in public databases, including seven of the eight SNPs that altered encoded amino acids. We then functionally characterized the non-synonymous SNPs in a mammalian cell-based system by expressing recombinant MME variant allozymes. These functional genomic studies revealed markedly reduced MME immunoreactive protein and enzyme activity for the Val73 variant. The finding that the other nsSNPs did not result in a change in MME protein level or activity is not surprising and has been demonstrated previously during functional genomic studies of resequencing data performed in our laboratory. These observations are likely related to the structural consequences of alterations in amino acid sequence [36–38]. The decreased expression of the MME Val73 variant allozyme was consistent with molecular modeling and MD simulation calculations performed with the X-ray crystal structure of human MME. Substitution of Val for Met73 introduced unfavorable interactions with surrounding residues that could potentially compromise protein folding and/or stability. More detailed MD simulations confirm that the Val73 substitution is not compatible with the WT protein conformation, especially in the local region around Met/Val73. Similar structural modeling of the Ser285 variant allozyme indicated that substitution of that residue would probably not be detrimental to protein folding. We also sought to delineate mechanisms that could potentially explain the low level of MME immunoreactive protein for the Val73 variant allozyme. We showed that this variant allozyme is degraded by both proteasome-mediated degradation and autophagy. It also resulted in aggregate formation and its presence was accompanied by increased levels of two ER chaperone proteins, BiP and GRP94, further suggesting that Val73 may be misfolded. Our results obviously do not include longitudinal clinical data on the multi-ethnic populations studied. These samples are part of the “Human Variation Panel” and were purposely collected from “healthy” subjects, anonymized and deposited by the National Institute of General Medical Sciences (NIGMS), one of the National Institutes of Health (NIH). The Coriell Cell Repository that provided these samples is funded, in part, by an award from the (NIGMS). The Pharmacogenomics Research Network mandates the use of these samples for pharmacogenomic studies and requires that the polymorphism data be deposited in a public repository (PharmGKB). The purpose of our study was not to identify SNPs in populations with specific diseases but rather to begin the process of defining common and rare variants found for MME, both for cardiovascular pathophysiology and response to drugs used in cardiovascular medicine, but also extending beyond cardiovascular medicine. These fundamental studies should be expanded in the future to include populations suffering from cardiovascular disease and patients treated with drugs that modulate the natriuretic peptide system. Another limitation of this study is the fact that the enzymatic activity of the variant MME allozymes with natriuretic peptides was not studied. However, we used an established and well described high throughput assay to detect enzymatic activity that is highly specific for MME, thus making these findings broadly applicable, not only to natriuretic peptides but also to other MME substrates.
In summary, we resequenced MME and identified multiple novel polymorphisms, including several SNPs that alter encoded amino acids. The genetic alteration of just one amino acid, Met73Val, resulted in a significant decrease in the quantity and activity of the enzyme protein. We then showed that accelerated protein degradation mediated by both proteasome and autophagy pathways was responsible for the decrease in Val73 MME protein. The Met73Val polymorphism, although rare, could have could have a significant effect on the metabolism of natriuretic peptides. Rare variants with MAFs that range from 0.1% to 3% like Val73 are often detected during comprehensive resequencing studies. There is an increasing recognition that rare variants are often population specific and can be functionally relevant with high penetrance to disease. The role of such rare variants in disease pathophysiology is the subject of vigorous pursuit at present [39]. A recent example is the existence of rare genetic variants for sialic acid acetylesterase, an enzyme that regulates B lymphocyte antigen receptor signaling, that had not been revealed by genome wide association studies but contributes significantly to the population attributable risk of autoimmune diseases like rheumatoid arthritis and type 1 diabetes mellitus [40]. Genetic variation in the natriuretic peptide system has already been shown to have important implications in cardiovascular disease, and nonsynonymous SNPs can play an important role in variation in drug response. The insight provided by the present study of genetic variation in MME in three different ethnic groups would help lay the foundation for understanding the role it plays in the pathophysiology of heart failure and clinical response to drugs that serve as substrates for MME like nesiritide or drugs which inhibit MME or NEP. Therefore, our resequencing MME and our studies of the functional implications of the nonsynonymous SNPs identified could provide a foundation for future studies of common sequence variation of other important candidate genes in the pharmacokinetic and pharmacodynamic pathways for drugs used to treat hypertension and heart failure that target the natriuretic peptide system.
Supplementary Material
Acknowledgments
This study was supported, in part by HL 84904 (Heart Failure Clinical Research Network), a Marie Ingalls Cardiovascular Career Development Award, a PhRMA Foundation “Center for Excellence in Clinical Pharmacology” Award (R.M. Weinshilboum), and NIH grants UL1RR24150 (N. L. Pereira), RO1 GM28157 (R.M. Weinshilboum), RO1 CA132780 (R.M. Weinshilboum) and UO1 GM61388 (The Pharmacogenetics Research Network). We thank Luanne Wussow for her assistance with the preparation of this manuscript.
Footnotes
Disclosures: None.
Appendix A. Supplementary data: Supplementary data to this article can be found online at doi:10.1016/j.yjmcc.2010.07.020.
References
- 1.Munagala VK, Burnett JC, Jr, Redfield MM. The natriuretic peptides in cardiovascular medicine. Curr Probl Cardiol. 2004;29(12):707–69. doi: 10.1016/j.cpcardiol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Rademaker MT, Charles CJ, Kosoglou T, Protter AA, Espiner EA, Nicholls MG, et al. Clearance receptors and endopeptidase: equal role in natriuretic peptide metabolism in heart failure. Am J Physiol. 1997 Nov;273(5 Pt 2):H2372–9. doi: 10.1152/ajpheart.1997.273.5.H2372. [DOI] [PubMed] [Google Scholar]
- 3.Lanfear DE, Stolker JM, Marsh S, Rich MW, McLeod HL. Genetic variation in the B-type natriuretic peptide pathway affects BNP levels. Cardiovasc Drugs Ther. 2007;21(1):55–62. doi: 10.1007/s10557-007-6007-5. [DOI] [PubMed] [Google Scholar]
- 4.Dao Q, Krishnaswamy P, Kazanegra R, Harrison A, Amirnovin R, Lenert L, et al. Utility of B-type natriuretic peptide in the diagnosis of congestive heart failure in an urgent-care setting. J Am Coll Cardiol. 2001;37(2):379–85. doi: 10.1016/s0735-1097(00)01156-6. [DOI] [PubMed] [Google Scholar]
- 5.Nesiritide Study Group. Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. N Engl J Med. 2000;343(4):246–53. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- 6.Florkowski CM, Richards AM, Espiner EA, Yandle TG, Sybertz E, Frampton CM. Low-dose brain natriuretic peptide infusion in normal men and the influence of endopeptidase inhibition. Clin Sci (Lond) 1997;92(3):255–60. doi: 10.1042/cs0920255. [DOI] [PubMed] [Google Scholar]
- 7.Corti R, Burnett JC, Jr, Rouleau JL, Ruschitzka F, Lüscher TF. Vasopeptidase inhibitors: a new therapeutic concept in cardiovascular disease? Circulation. 2001;104(15):1856–62. doi: 10.1161/hc4001.097191. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Weinshilboum RM. Pharmacogenomics: candidate gene identification, functional validation and mechanisms. Hum Mol Genet. 2008;17(R2):R174–9. doi: 10.1093/hmg/ddn270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23(3):261–9. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 10.D'Adamio L, Shipp MA, Masteller EL, Reinherz EL. Organization of the gene encoding common acute lymphoblastic leukemia antigen (neutral endopeptidase 24.11): multiple miniexons and separate 5′ untranslated regions. Proc Natl Acad Sci USA. 1989;86(18):7103–7. doi: 10.1073/pnas.86.18.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Florentin D, Sassi A, Roques BP. A highly sensitive fluorometric assay for “enkephalinase,” a neutral metalloendopeptidase that releases tyrosine–glycine–glycine from enkephalins. Anal Biochem. 1984;141(1):62–9. doi: 10.1016/0003-2697(84)90425-1. [DOI] [PubMed] [Google Scholar]
- 12.Oefner C, D'Arcy A, Hennig M, Winkler FK, Dale GE. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J Mol Biol. 2000;296(2):341–9. doi: 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- 13.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 14.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24(5):946–50. [Google Scholar]
- 15.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Meth Enzymol. 1997:505–24. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 16.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible and free. J Comp Chem. 2005;26:1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 17.van Gunsteren WF, Berendsen HJC. Biomos BV Nijenborgh. Vol. 4. AG Groningen, The Netherlands: 1987. Gromos-87 manual; p. 9747. [Google Scholar]
- 18.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics Nov. 1989;123(3):585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartl DL, Clark AG. Organization of genetic variation. 3rd. Sunderland, MA: Sinauer Associates; 2000. [Google Scholar]
- 20.Hendrick P. Genetics of populations. 3rd. Sudbury, Massachusetts: Jones and Bartlett Publishers; 2000. [Google Scholar]
- 21.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002 Feb;70(2):425–34. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long JC, Williams RC, Urbanek M. An E–M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet. 1995;56(3):799–810. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25(11):1629–38. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 24.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 25.Melnick J, Aviel S, Argon Y. The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J Biol Chem. 1992;267(30):21303–6. [PubMed] [Google Scholar]
- 26.Roques BP. Cell surface metallopeptidases involved in blood pressure regulation: structure, inhibition and clinical perspectives. Pathol Biol (Paris) 1998;46(3):191–200. [PubMed] [Google Scholar]
- 27.Fielitz J, Dendorfer A, Pregla R, Ehler E, Zurbrügg HR, Bartunek J, et al. Neutral endopeptidase is activated in cardiomyocytes in human aortic valve stenosis and heart failure. Circulation. 2002;105(3):286–9. doi: 10.1161/hc0302.103593. [DOI] [PubMed] [Google Scholar]
- 28.Knecht M, Pagel I, Langenickel T, Philipp S, Scheuermann-Freestone M, Willnow T, et al. Increased expression of renal neutral endopeptidase in severe heart failure. Life Sci. 2002;71(23):2701–12. doi: 10.1016/s0024-3205(02)01990-2. [DOI] [PubMed] [Google Scholar]
- 29.Helske S, Laine M, Kupari M, Lommi J, Turto H, Nurmi L, et al. Increased expression of profibrotic neutral endopeptidase and bradykinin type 1 receptors in stenotic aortic valves. Eur Heart J. 2007;28(15):1894–903. doi: 10.1093/eurheartj/ehm129. [DOI] [PubMed] [Google Scholar]
- 30.Comings DE, Dietz G, Gade-Andavolu R, Blake H, Muhleman D, Huss M, et al. Association of the neutral endopeptidase (MME) gene with anxiety. Psychiatr Genet. 2000;10(2):91–4. doi: 10.1097/00041444-200010020-00007. [DOI] [PubMed] [Google Scholar]
- 31.Yamada M, Sodeyama N, Itoh Y, Takahashi A, Otomo E, Matsushita M, et al. Association of neprilysin polymorphism with cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry. 2009;74(6):749–51. doi: 10.1136/jnnp.74.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet. 2004;364(9441):1252–9. doi: 10.1016/S0140-6736(04)17142-0. [DOI] [PubMed] [Google Scholar]
- 33.Rubattu S, Stanzione R, Di Angelantonio E, Zanda B, Evangelista A, Tarasi D, et al. Atrial natriuretic peptide gene polymorphisms and risk of ischemic stroke in humans. Stroke. 2004;35(4):814–8. doi: 10.1161/01.STR.0000119381.52589.AB. [DOI] [PubMed] [Google Scholar]
- 34.Sarzani R, Dessì-Fulgheri P, Salvi F, Serenelli M, Spagnolo D, Cola G, et al. A novel promoter variant of the natriuretic peptide clearance receptor gene is associated with lower atrial natriuretic peptide and higher blood pressure in obese hypertensives. J Hypertens. 1999;17(9):1301–5. doi: 10.1097/00004872-199917090-00010. [DOI] [PubMed] [Google Scholar]
- 35.Lynch AI, Boerwinkle E, Davis BR, Ford CE, Eckfeldt JH, Leiendecker-Foster C, et al. Pharmacogenetic association of the NPPA T2238C genetic variant with cardiovascular disease outcomes in patients with hypertension. JAMA. 2008;299(3):296–307. doi: 10.1001/jama.299.3.296. [DOI] [PubMed] [Google Scholar]
- 36.Aksoy P, Zhu MJ, Kalari KR, Moon I, Pelleymounter LL, Eckloff BW, et al. Cytosolic 5′-nucleotidase III (NT5C3): gene sequence variation and functional genomics. Pharmacogenet Genomics. 2009;19(8):567–76. doi: 10.1097/FPC.0b013e32832c14b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartman WR, Pelleymounter LL, Moon I, Kalari K, Liu M, Wu TY, et al. CD38 expression, function, and gene resequencing in a human lymphoblastoid cell line-based model system. Leuk Lymphoma. 2010;51(7):1315–25. doi: 10.3109/10428194.2010.483299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li F, Feng Q, Lee C, Wang S, Pelleymounter LL, Moon I, et al. Human betaine-homocysteine methyltransferase (BHMT) and BHMT2: common gene sequence variation and functional characterization. Mol Genet Metab. 2008;94(3):326–35. doi: 10.1016/j.ymgme.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40(6):695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Surolia I, Pirnie SP, Chellappa V, Taylor KN, Cariappa A, Moya J, et al. Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature. 2010;466(7303):243–7. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.