

Abstract
Hematopoiesis is a process capable of generating millions of cells every second, as distributed in many cell types. The process is regulated by a number of transcription factors that regulate the differentiation along the distinct lineages and dictate the genetic program that defines each mature phenotype. Myc was first discovered as the oncogene of avian leukemogenic retroviruses; it was later found translocated in human lymphoma. From then on, evidence accumulated showing that c-Myc is one of the transcription factors playing a major role in hematopoiesis. The study of genetically modified mice with overexpression or deletion of Myc has shown that c-Myc is required for the correct balance between self-renewal and differentiation of hematopoietic stem cells (HSCs). Enforced Myc expression in mice leads to reduced HSC pools owing to loss of self-renewal activity at the expense of increased proliferation of progenitor cells and differentiation. c-Myc deficiency consistently results in the accumulation of HSCs. Other models with conditional Myc deletion have demonstrated that different lineages of hematopoietic cells differ in their requirement for c-Myc to regulate their proliferation and differentiation. When transgenic mice overexpress c-Myc or N-Myc in mature cells from the lymphoid or myeloid lineages, the result is lymphoma or leukemia. In agreement, enforced expression of c-Myc blocks the differentiation in several leukemia-derived cell lines capable of differentiating in culture. Not surprising, MYC deregulation is recurrently found in many types of human lymphoma and leukemia. Whereas MYC is deregulated by translocation in Burkitt lymphoma and, less frequently, other types of lymphoma, MYC is frequently overexpressed in acute lymphoblastic and myeloid leukemia, through mechanisms unrelated to chromosomal translocation, and is often associated with disease progression.
Keywords: Myc, hematopoietic stem cells, differentiation, leukemia
Overview of Hematopoiesis
Hematopoiesis is a process capable of generating 300 millions of cells per minute in the bone marrow of an adult human. These cells belong to many cell types, which widely differ in biological functions and number in the blood. All these morphologically, phenotypically, and functionally distinct cell types arise from pluripotent hematopoietic stem cells (HSCs) that are capable of self-renewal and differentiation through life.1,2 The self-renewal and quiescence of hematopoietic stem cells are controlled by a highly orchestrated integration of environmental signals, most of which originate from the stem cell niche.3–5 The so-called long-term HSCs (LT-HSCs) have the ability to self-renew and sustain hematopoiesis.4 This fraction of HSCs remains largely quiescent or even dormant throughout the lifetime of an organism. LT-HSCs differentiate into multipotent progenitors (MPPs), which are cells with diminished self-renewal that can give rise to common lymphoid progenitors (CLPs) and common myeloid progenitors, which in turn differentiate to produce T- or B-lymphocyte precursors and granulocyte/monocyte precursors (GMPs) or megakaryocyte/ erythroid precursors (MEPs), respectively.6 These multipotent progenitors undergo several steps of differentiation that give rise to committed progenitors and, ultimately, the diverse mature blood cells (Figure 1). The myeloid lineage includes erythrocytes, megakaryocytes producing platelets, different subclasses of granulocytes (neutrophils, eosinophils, basophils), monocytes–macrophages, and mast cells. The lymphoid lineage consists of T cells, B cells, and natural killer cells. Dendritic cells can be derived from either the myeloid or lymphoid pathway.
Figure 1.
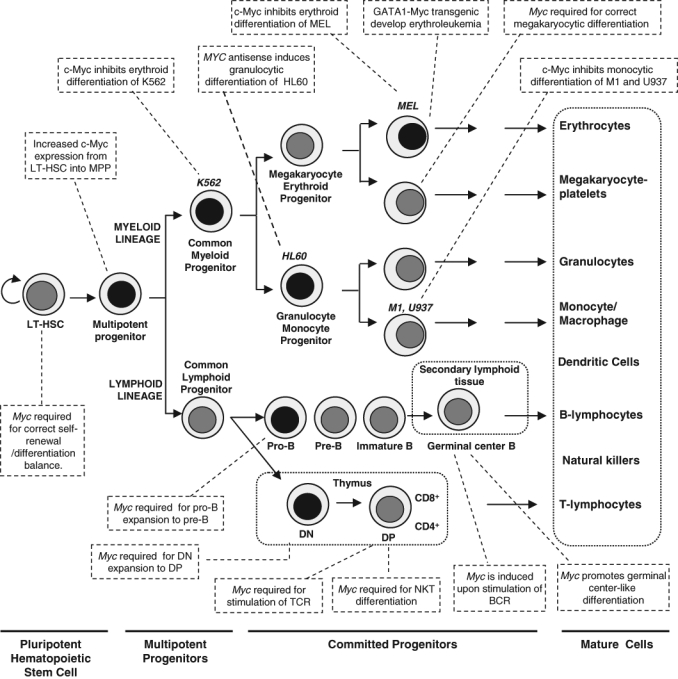
Classical model of hematopoiesis and the involvement of c-Myc in the process. Hematopoiesis begins at the level of pluripotent hematopoietic stem cells, which diverge into myeloid and lymphoid multipotent progenitors. These cells differentiate into committed progenitors and, finally, mature blood cells. T cells and B cells undergo further maturation in secondary lymphoid organs, such as spleen, lymph nodes, and mucosa-associated lymphoid tissues. Several leukemia-derived cell lines (MEL, K562, HL60, U937, M1), used as models to study c-Myc functions, are shown at their approximate stage of differentiation. Some reported effects of c-Myc on the differentiation of hematopoietic lineages are indicated in the boxes. Black nuclei indicate higher Myc expression. See the text for details and references. LT-HSC = long-term hematopoietic stem cells; MPP = multipotent progenitor; DN = double-negative T cell (CD4-, CD8-); DP = double-positive T cell; BCR = B-cell receptor; NKT = natural killer T cells.
Hematopoietic cell differentiation is controlled by intercellular and intracellular signaling mechanisms that target transcriptional regulators that in turn establish complex transcriptional networks. Therefore, lineage commitment relies on the timely activation of appropriate transcription factors and the silencing of inappropriate ones.7 Comprehensive reviews have dealt with the transcriptional control in the development of erythroid,8,9 myeloid,10 B-cell,11 and T-cell12 lineages. Moreover, the hematopoietic system must respond to important loss of tissue (i.e., by bleeding after injuries) to replenish the lost cells in the appropriate relative numbers. This means that hematopoiesis must be exquisitely regulated by a large group of cytokines by cell–cell interactions in the bone marrow niche, and by the concerted activity of transcription factors. The data accumulated over recent years in cell and animal models indicate that c-Myc is one of the pivotal transcription factors regulating hematopoiesis. Figure 1 shows a schematic overview of hematopoiesis and the involvement of c-Myc in the process.
MYC Roles in Hematopoietic Cell Differentiation in Culture
Gene expression deregulation owing to chromosome aberrations or epigenetic alterations in hematopoietic stem cells or multipotent progenitors gives rise to different hematopoietic malignancies. Immortalized cell lines derived from leukemias or lymphomas have been extensively used as suitable model systems to study hematopoietic cell differentiation. Myeloid leukemia cell lines are arrested at different stages of maturation and can be induced to differentiate into several pathways that resemble normal counterparts, depending on their differentiation potential (e.g., multipotent K562 cells, bipotent HL60 cells, or unipotent U937 cells; see Figure 1). These hematopoietic models, among others, have helped to highlight the importance of c-Myc on cellular processes such as proliferation, differentiation, and apoptosis.
Inhibition of cell differentiation was one of the first biological effects described for c-Myc (reviewed in Reference 13). In 1986, three reports showed that c-Myc blocked the chemically induced erythroid differentiation of Friend murine erythroleukemia cells.14–16 In the same year, it was reported that B-lymphocyte development was impaired in young Eµ-Myc transgenic mice before the onset of lymphoma.17 Since then, c-Myc ectopic expression has been found to inhibit differentiation in a number of cell lines and primary cells, and about half of them are hematopoietic cell lines (Table 1). However, inhibition of differentiation is not a universal c-Myc activity. For instance, in the tripotent cell line K562, we found that whereas c-Myc impairs AraC-induced erythroid differentiation,18 it does not impair TPA-induced myelomonocytic differentiation or staurosporine-induced megakaryocytic differentiation.19 Consistent with those results, c-Myc-dominant inhibitory mutants enhance erythroid but not megakaryocytic differentiation.20
Table 1.
Leukemia-Derived Cell Lines Showing c-Myc-Mediated Inhibition of Differentiation
| Hematopoietic Cell | Cell Type (Species) | Differentiation Type (Inducer) | c-Myc Effect | References |
|---|---|---|---|---|
| MEL | Erythrocytic cells (mouse) | Erythroid (DMSO) | Inhibition of differentiation | 14–16 |
| U937 | Monoblastic cells (human) | Macrophage (TPA) | Inhibition of differentiation | 135, 136 |
| HL60 | Promyelocytic cells (human) | Granulocytic | MYC antisense induces differentiation | 137, 138 |
| HL60 | Promyelocytic cells (human) | Monocytic | MYC inhibitor induces differentiation | 139 |
| K562 | Multipotential myeloid cells (human) | Erythroid (AraC) (p27KIP1) | Inhibition of differentiation | 18, 140 |
| K562 | Multipotential myeloid cells (human) | Erythroid | Max and dominant negative MYC mutants induce differentiation | 20 |
| J774 | Myelomonocytic cells (mouse) | Monocytic (LPS, TPA) | Inhibition of differentiation | 141 |
| M1 | Myeloblastic cells (mouse) | Macrophage (IL6) | Inhibition of differentiation; MYC antisense induces differentiation | 142, 143 |
| 1137 | Myeloblasts (mouse) | Granulocytic | Repression of MYC induces differentiation | 144 |
In genetically defined models where erythroid differentiation is induced by p27KIP1, c-Myc blocks differentiation.21 In this model, c-Myc impairs the upregulation of many erythroid-specific genes, as well as that of transcription factors that determine erythroid lineage differentiation (including GATA1 and NFE2); but, strikingly, it does not reverse the proliferation arrest and the repression of CDK activity mediated by p27KIP1. This suggests that c-Myc operates through the regulation of several or many genes, which is to be expected in view of the broad changes in the gene expression profile induced by c-Myc. In a complementary approach using the leukemia K562 and U937 cells, it was shown that c-Myc is not downregulated when cells are growth arrested but not differentiated.22,23 All together, the available data indicate that c-Myc can block differentiation of hematopoietic cell models in culture through mechanisms distinct from those involved in cell cycle progression.
MYC Expression in Hematopoietic Progenitors and HSCs
The involvement of c-Myc in hematopoiesis regulation was soon suggested by 3 avenues: First, all three oncogenic Myc retroviruses originally isolated induced chicken hematopoietic tumors, which were a type of myeloid leukemia (myelocytomatosis)24; second, MYC deregulation was first observed in Burkitt lymphoma as well as in chemically induced murine plasmacytomas25; and, third, inhibition of hematopoietic cell differentiation in culture was one of the first biological effects of c-Myc. Later on, the development of several mice models with targeted mutation of c-Myc confirmed its important role in hematopoiesis. We briefly review the conclusions drawn from several of these models.
There is a differential expression of Myc family members in hematopoietic lineages: c-Myc and N-Myc transcripts are coexpressed at similar levels in LT-HSCs26 and are detected in most progenitor subsets. In contrast, L-Myc mRNA is not expressed in any stem/progenitor cells, and it is only modestly expressed in CLPs, megakaryocytes, and macrophages (3%–5% of total Myc). Note that no hematopoietic cell type expresses only N-Myc or L-Myc without c-Myc.26 The pervasive c-Myc expression in the hematopoietic system seems to be in correspondence with the absolute prevalence of c-Myc deregulation in human leukemia and lymphoma, as compared with the other family members (see below).
The expression of c-Myc at the protein level in hematopoietic progenitors and mature lineages was studied in a mouse line where the endogenous Myc locus was replaced with an allele encoding a GFP-c-Myc fusion protein. These MycG/G mice were viable and showed an apparently normal hematopoiesis.27 The highest expression of c-Myc (i.e., c-Myc–GFP in this model) was detected in the myeloerythroid progenitor fraction of the adult bone marrow, a cell population that originates directly from Lin−Sca1+c-Kit+ (LSK) cells and is actively proliferating.28 Adult LSK cells include LT-HSCs and MPPs.4,5 Interestingly, those LSK cells with lower c-Myc expression (i.e., GFP expression) corresponded to LT-HSCs (the more primitive and undifferentiated population), whereas the LSK expressing more c-Myc corresponded to MPPs.28 Note that the abundance of c-Myc protein was substantially greater in fetal liver LSK cells than in adult LSK cells,28 which seems in concordance with the fact that fetal liver HSCs actively proliferate during embryonic days 12.5 to 14.5.29 Interestingly, Myc mRNA is already expressed at significant levels in LT-HSCs, but the increase in c-Myc protein expression (i.e., c-Myc-GFP) from LT-HSCs into MPPs is not observed at the mRNA level.26 This suggests that the protein changes are due to posttranscriptional mechanisms, which complicates the interpretation of previous data obtained measuring mRNA levels.
MYC Roles in Hematopoietic Stem Cells: Lessons from Targeted Mutation in HSCs in Mice
Mice embryos in which both Myc alleles have been mutated by gene targeting in ES cells fail to thrive and die before midgestation (embryonic day ~10.5). Myc-deficient embryos are much smaller and exhibit several defects, with the impaired hematopoiesis and angiogenesis being the most prominent and reproduced in different models.30–32 Whether deletion of Myc impairs vasculogenesis is controversial.30,33 There are data that strongly suggest that defective hematopoiesis is the main reason for Myc-dependent embryonic lethality of Myc-deficient embryos. First, Myc-deficient embryos survive up to embryonic day 10.5, coincident with the onset of fetal liver hematopoiesis. Second, when Myc deletion was generated in the epiblast (i.e., with wild-type placenta), it was shown that Myc is specifically required for yolk sac primitive and intraembryonic definitive hematopoietic development, although nonhematopoietic defects—such as abnormalities of the heart, pericardium, and neural tube; delay in embryo turning; and small embryo size—were virtually absent in these embryos.33,34 Finally, embryos lacking Myc in hematopoietic lineages (deleted in cells expressing the hematopoietic gene Vav1) phenocopied those lacking Myc in the entire embryo, although they survived until embryonic day 11.5 to 12.5 (i.e., 1 to 2 days longer than the complete null embryo).33
Although the above complete Myc null mouse demonstrates a role of c-Myc in hematopoiesis at an early stage, there is more controversy to the precise role of c-Myc in the process. This question has been addressed by conditional knockouts targeting Myc in hematopoietic precursors. The conditional deletion of Myc in the bone marrow (and most other tissues) achieved by the postnatal induction of the Cre gene in MxCre; Mycflox/flox mice results in severe cytopenia and accumulation of LT-HSCs.35 It has been suggested that this accumulation is caused not by alterations in HSC self-renewal or survival but rather by a failure to initiate normal stem cell differentiation, likely caused by increased HSC–niche adhesion.35
Consistent with the phenotype of Myc knockouts, enforced Myc expression in HSCs results in lower HSC numbers,35 accompanied with reduced expression of N-cadherin and integrins. In this scenario, high c-Myc levels would lead to detachment of the HSCs from their bone marrow niche, reduced self-renewal activity, and increased proliferation and differentiation of the Myc-expressing HSCs, thus exhausting the pool of LT-HSCs.35 Murine HSCs (Lin−Sca1+ cells) retrovirally transduced with a Myc-ER gene consistently showed extended proliferation in culture when treated with the c-Myc activator (hydroxytamoxifen).36 In agreement with this c-Myc effect, the bone marrow of mice deficient for p27KIP1 and Mxd1 (formerly Mad1) genes has an expanded pool of quiescent HSCs, which is consistent with the phenotype observed in Myc−/− mice owing to the well-known activity of both proteins as c-Myc antagonists.37
Other investigators, using the same mice model, observed that c-Myc loss in adult bone marrow results in the transient accumulation of HSCs, followed by the progressive loss of bone marrow cells, which is maximal at 8 weeks after Myc deletion.38 Perinatal deletion of Myc (again using the Mx-Cre system) results in the accumulation of a population of HSCs with the phenotype Lin-negative, Sca1+-c-Kit− cells (versus the c-Kit-positive found in wild-type HSCs). These cells are deficient in differentiation, and it is suggested that they reflect a potential primitive progenitor.38
Myc-deficient HSCs produced by either perinatal or adult excision by the Cre-Mycflox/flox model fail to engraft in recipient mice in a competitive or noncompetitive setting. This demonstrates that the accumulated Myc-deficient HSCs are functionally defective, and the impaired hematopoiesis can be attributed to an intrinsic defect of the Myc-deficient cell rather than to an aberrant microenvironment in the bone marrow.35,38
The results commented above refer to c-Myc effects in HSC biology. As already mentioned, both c-Myc and N-Myc mRNAs are expressed at similar levels in HSCs, so it is interesting to compare the effects of both Myc genes and their concomitant deletion in HSCs. The deletion of N-Myc alone does not seem to affect HSCs or hematopoiesis, but the simultaneous deletion of Myc and N-Myc results in HSCs that proliferate less than c-Myc−/− HSCs and undergo cell death, meaning that N-Myc plays a role in HSC survival.26 Interestingly, replacement of Myc by N-Myc in mice results in normal development, hematopoietic, and lymphoid differentiation.39
The c-Myc role in HSCs has been confirmed in mice with targeted mutation of the ubiquitin ligase Fbw7, which is the major (and, probably, the only) ubiquitin ligase for Myc in HSCs.28 Furthermore, Fbw7 deletion in bone marrow and fetal liver HSCs results in elevated expression of c-Myc, accompanied with a reduced fraction of LT-HSCs.28,40 This is fully consistent with the accumulation in LT-HSCs observed in Myc-deficient mice described above.26,35,38 Moreover, concomitant Myc deletion rescues the effect of Fbw7 deficiency, expanding the number of HSCs.28 Collectively, these investigations indicate that enforced c-Myc leads to loss of self-renewal activity at the expense of differentiation, whereas c-Myc deficiency results in increased HSC self-renewal and accumulation of HSCs. In summary, in the HSC population, c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation.
MYC in the Differentiation of Lymphoid Cells In Vivo
MYC involvement in the lymphoid lineage development has received much attention because of its heavy involvement in human lymphoma. Both N-Myc and c-Myc mRNA are expressed during the maturation and expansion of the earliest B-cell precursors (pro–B cells) into pre–B cells. Thereafter, only c-Myc is expressed in mature B cells after B-cell activation.41–43 Myc expression consistently increases in response to B-cell receptor (BCR) stimulation in vitro.44,45 In mice, pro–B cells and pre–B cells proliferate in response to cytokines such as IL-7 produced by bone marrow stromal cells, and this response is enhanced in pre–B cells from Eµ-Myc mice.46
This is confirmed by measuring GFP-Myc protein in the lymphoid compartment of the GFP-Myc cell line described above. Flow cytometry studies showed that c-Myc expression is dynamically regulated in developing and mature B and T lymphocytes in vivo. Despite the broad cellular effects of c-Myc, one study showed a close correlation between GFP-c-Myc levels and proliferation, both in lymphocyte differentiation and upon activation of mature lymphocytes, suggesting that c-Myc is required to promote proliferation of lymphocytes.27
As commented below, transgenic mice overexpressing MYC (the human ortholog) in B cells via the Eµ enhancer develop lymphoma with relatively long latencies. During the prelymphomatous state, the constitutive c-Myc expression results in a higher number of pre–B cells and a lower number of mature B cells, but the B cells are able to mature relatively normally despite the presence of deregulated c-Myc.17
In contrast, mice deficient in c-Myc in early B-lymphocyte precursors (pro–B stage) have impaired B-cell differentiation (Moreno de Alboran, personal communication). Conditional deletion of Myc in mature B cells results in the impaired proliferation of Myc null B lymphocytes in response to CD40 and IL-4.47 N-Myc expression from the Myc locus is capable of driving B- and T- lymphocyte differentiation in knock-in mice.39 One step further, the conditional deletion of both c-Myc and N-Myc, using CD19-cre mice, inhibits B-cell development at the transition from pro–B cell to pre–B cell (i.e., the stage at which the combined signaling from the pre-BCR and IL-7 normally induce c-Myc and N-Myc expression).46 The data indicate that Myc stimulates B-cell differentiation and expansion at least from the pre-BCR stage.
As in the case of B cells, c-Myc and N-Myc are expressed in immature T lymphocytes, but c-Myc is the only Myc gene expressed after the pro–T cell stage and in mature T cells.27,41,43 In parallel with the B-cell scenario, c-Myc is upregulated upon T-cell receptor stimulation.42,48
Based on 2 different models, it has been shown that Myc−/− T cells (in LckCre Mycflox/flox mice) cannot populate the adult thymus and that subsequent thymocyte maturation is ineffective: The cells fail to proliferate normally at the late double-negative CD4−CD8− stage, but this is not required for the progression from double-negative into double-positive (CD4+CD8+) cells.42,49 However, conditional ablation of Myc in double-positive thymocytes (in CD4Cre Mycflox/flox mice) impairs natural killer T-cell (NKT) development arresting intrathymic NKT proliferation upon agonist selection, whereas conventional T-cell development is not affected.50,51
In response to infections, human B lymphocytes transiently pass through the germinal center, where antigen-specific B cells suffer somatic hypermutation, class switch recombination, and clonal expansion.52 In Burkitt lymphoma, which is the hallmark of Myc-dependent neoplasia in humans, B-cell differentiation into plasma cells is impaired. c-Myc overexpression in Burkitt lymphoma cells induces a constitutive centroblast-like phenotype in the ganglia germinal center, thus blocking the differentiation into mature B cells. This effect may be in part mediated by the MYC-dependent upregulation of the germinal center–specific transcription factor Bcl6.53 Thus, c-Myc seems to impair the differentiation program as the primary cause of this lymphoma.
MYC in the Differentiation of Myeloid Cells In Vivo
The expression of Myc genes in the myeloid progenitors has merited less attention than that of lymphoid progenitors, but the effects of Myc deletion in the myeloid compartment have been recently reported in the c-Myc conditional knockout mice.54 Besides the reduced lymphocyte number discussed above, adult c-Myc−/− mice show significant thrombocytosis, severe anemia, and grossly decreased neutrophil/monocyte number.54 Thus, c-Myc induces opposite effects in the differentiation of megakaryocytic versus monocytic and erythroid lineages. Moreover, these effects of c-Myc deletion in vivo on hematopoietic lineages correlates well with c-Myc expression in mouse hematopoietic cells: Cells expressing higher levels of c-Myc, such as GMPs, CLPs, and erythrocytic blasts, are significantly reduced, whereas cells expressing lower Myc levels (HSCs, MEPs, and megakaryocytes) are less affected or are increased in number.54 The data are roughly consistent with the observations in cell culture models where c-Myc impairs monocytic, granulocytic, and erythroid differentiation (see above and Table 1).
Megakaryocytes from c-Myc−/− mice are significantly smaller in size and lower in ploidy than those of control mice; however, because of the increase in megakaryocytic number and although fewer platelets are produced by each megakaryocyte, a greater-than-3-fold increase in platelet number was observed in Myc−/− mice. It is noteworthy that mice deficient in the RNA-binding motif protein 15 (Rbm15) gene (involved in human acute megakaryoblastic leukemia) develop a phenotype similar to that of Myc−/− mice: a lower number of LT-HSCs and a higher number of abnormal low-ploidy megakaryocytes.55 Interestingly, the megakaryocyte number increase in Rbm15 knockout mice could be partially reversed by ectopic c-Myc.55 The involvement of c-Myc in megakaryocytic differentiation is confirmed in transgenic mice with c-Myc overexpressed in the megakaryocytic lineage, attained with a Myc transgene under the control of the platelet factor 4 promoter. These mice show an increase in low-ploidy megakaryocytes owing to enhanced proliferation and survival, along with the blocking of differentiation.56
Effects of Enforced Expression of c-Myc in Hematopoietic Cells In Vivo
Transgenic Mice Developing Lymphoid Neoplasia
The oncogenic function of c-Myc in hematopoietic malignancies was first demonstrated in the Eµ-Myc transgenic mouse, in which c-Myc expression is targeted to the lymphoid compartment by the immunoglobulin heavy chain gene promoter and enhancer.57,58 Analysis of the immunoglobulin gene rearrangements of these tumor cells indicates that tumor development can commence at several points during B-lymphocyte differentiation,17,57 suggesting that the deregulated Myc may be active at several developmental stages. The majority of the Eµ-Myc tumors appear after a latency period of 2 to 5 months and harbor mutations in the Arf-Mdm2-p53 pathway,59,60 indicating the insufficiency of c-Myc to transform lymphoid cells by itself. Eµ-N-Myc mice also develop B-cell lymphoma.61 An interesting model is that of mice that conditionally express a MYC transgene (repressed by tetracycline) in lymphoid cells because the tTA gene is under the Eµ control.62 Most of the tumors induced in these mice are immature T-cell lymphoma. Interestingly, the tumors undergo a sustained regression upon inactivation of the transgene (i.e., adding tetracycline in the drinking water).62
Although the Eµ-Myc-induced lymphoma demonstrates the ability of c-Myc to transform B cells in mice, the resulting tumors do not faithfully reproduce the Burkitt lymphoma, the hallmark of MYC-induced human B-cell lymphoma. Additional transgenic mice lines have been constructed to have better models for human Burkitt lymphoma. Yeast artificial chromosome (YAC) technology was used to obtain mice carrying a single copy of the 240-kb IgH/c-Myc translocation region.63 B-cell tumorigenesis occurs in these translocus mice, even when the entire Eµ intron enhancer region is deleted. The phenotype of tumors from IgH/c-Myc YAC transgenic mice is reminiscent of B-cell acute lymphoblastic leukemia (B-ALL),63 which is equivalent to Burkitt lymphoma in leukemic phase and represents about 5% of acute lymphoblastic leukemia (ALL).64 Finally, a mouse transgenic was generated carrying a murine Myc cDNA inserted in the mouse IgH locus in a site that corresponds to the t(8;14) translocation break in Burkitt lymphoma or the t(12;15) in murine plasmacytoma.65 These mice developed lymphoblastic B-cell lymphomas with a Burkitt-like morphology, diffuse large B-cell lymphomas and plasmacytomas (i.e., tumors of mature B cells). Thus, these mice model Burkitt lymphoma more precisely than do the Eµ-Myc and YAC-Myc mice, which do not develop plasmacytomas.65
Signaling of the BCR cooperates with c-Myc in the genesis of B lymphomas.66 Interestingly, tumors that grow in mice expressing an activated BCR differ from those found in Eµ-Myc mice and resemble Burkitt lymphoma. In contrast, BCR itself (in the absence of antigen stimulation) cooperates with c-Myc in tumorigenesis. The resulting tumors differ from those in the Eµ-Myc mice and those in Eµ-Myc/activated BCR and resemble a subset of chronic B-cell lymphocytic leukemia.66 A parallel model to study c-Myc involvement in B-cell differentiation targets Myc to the IgH Cα locus in mice. In contrast to Eµ-Myc mice, Cα-Myc mice do not develop early B-cell lymphoma but show impaired primary and secondary humoral immune responses, failing to generate mature antibody-secreting plasma cells owing to increased apoptosis.67 Only when deregulated c-Myc is combined with enforced expression of the antiapoptotic Bcl-xL gene do the mice develop plasmacytomas that reflect many features of human multiple myeloma. This transgenic model is the counterpart of mice carrying the t(12;15) translocation (IgH-Myc), which also develop plasmacitomas.68
Transgenic Mice Developing Myeloid Neoplasia
Although there is less information on the impact of c-Myc on myeloid transformation, transgenic mice with Myc overexpression in the myeloid cells demonstrate the carcinogenic potential of c-Myc in the myeloid compartment. Mice carrying the human MYC proto-oncogene under the control of the promoter of murine GATA-1 promoter (an erythroid-specific gene) developed an early-onset, rapidly progressive erythroleukemia that resulted in death 30 to 50 days after birth.69 More recently, Smith and coworkers generated several transgenic mice lines carrying the Myc gene under the control of the Vav promoter, which is active throughout all hematopoietic cell lineages (precursors and mature cells). Interestingly, the neoplasia lineage varied with the Myc expression level achieved in the transgenic mice. Aggressive T-cell lymphomas, as well as other hematopoietic abnormalities, predominated in the highest Myc-expressing transgenic mice, as expected from the broad expression of the transgene.70 In contrast, in the lines expressing lower c-Myc levels, most tumors were late-onset monocytic tumors.71 It is noteworthy that a 2-fold increase in c-Myc levels switched the phenotype from monocytic tumors to exclusively T-cell tumors. Thus, relatively low c-Myc levels can transform monocyte–macrophages but are insufficient to transform T lymphocytes.71
Retroviral-Mediated Expression of MYC in Murine Hematopoietic Precursors
Several reports illustrated the leukemogenic effects of c-Myc in hematopoietic cells after the enforced expression in murine hematopoietic precursors via retroviral infection, although the neoplasia induction varies, depending on the virus, infected cells, and Myc gene. Bone marrow retrovirally transduced with Myc resulted in rapid development of acute myeloid leukemia (AML),72 and fetal liver cells infected with a different retroviral vector resulted in long-latency lymphoma.73 The infection of bone marrow progenitors in a p53−/− background also resulted in lymphoma.74 Interestingly, upon culturing in vitro, a number of lymphoma-derived cells underwent myeloid differentiation. These cells switched from myeloid to lymphoid lineage and induced B-cell lymphomas when returning to in vivo conditions.75 In another study, mice bone marrow cells transduced with N-Myc developed monoclonal and transplantable AML, but c-Myc retrovirus was not leukemogenic in the same system.76 This model shows that N-Myc overexpression is highly oncogenic in mouse myeloid cells. In a recent report, bone marrow for lethally irradiated cells were repopulated with bone marrow cells expressing Myc, and the mice developed an AML-like disease.77 The coexpression of several antiapoptotic genes of the BCL2 family accelerated leukemogenesis but did not change the myeloid phenotype of the leukemia.77 Thus, in experimental models, c-Myc is able to induce lymphoid and myeloid neoplasia. The data agree with the observation that Myc is upregulated in radiation-induced AML in mice78 and with the deregulation of MYC found in human leukemia, as discussed below.
Deregulation of MYC in Hematopoietic Cell Neoplasia
The original finding of translocations involving MYC in Burkitt lymphoma fueled intense research in other lymphoma and leukemia. MYC translocations have also been found at low frequencies in other lymphomas but not in lymphoblastic cell leukemia, except in the Burkitt leukemia variant (former acute lymphocytic leukemia L3). In addition, a significant fraction of MYC in Burkitt lymphoma carry mutations in the coding sequence, with several of them resulting in more stable mutant c-Myc proteins, thus increasing the c-Myc level.79–81 However, no recurrent MYC translocations have been reported in other tumors, including other lymphoblastic or myeloblastic leukemia.82 Table 2 shows a summary of MYC alteration in human leukemia and lymphoma.
Table 2.
Summary of Myc Alteration in Human Hematological Malignanciesa
| Lymphoma/Leukemia | Myc Involvement | References |
|---|---|---|
| Lymphoid neoplasms | ||
| Diffuse large B-cell lymphoma (DLBCL) | MYC translocation (6–16%) | 145 |
| Burkitt lymphoma (including Burkitt leukemia variant) | MYC translocation and overexpresion (> 90%) | 146, 147 |
| DLBCL | c-Myc protein expression by immunochemistry (25%) | 148 |
| DLBCL | MYC amplification correlated with mRNA overexpression (38%) | 112 |
| Acute lymphoblastic leukemia (ALL) | MYC translocations t(8;14), t(8;22) t(2;8) (5%) | 84 |
| B-ALL (pediatric) | Coordinately elevated MYC and/or MYCN and TEL2 levels (30%) | 86 |
| B-ALL | MYC rearrangement/amplification (47–52%) | 145 |
| Plasma cell myeloma / multiple myeloma | MYC translocation (15-50%) | 98, 99 |
| Primary plasma cell leukemia | MYC translocation (13%) | 98, 145 |
| Chronic lymphocytic leukemia | Low c-Myc expression | 95 |
| Myeloid neoplasms | ||
| Acute myeloid leukemia (AML) | MYC mRNA overexpression by microarray analysis | 113 |
| AML (without translocations) | MYC mRNA overexpression by microarray (20%) | 149 |
| AML (pediatric) | MYCN overexpression (24–40%) | 76, 115 |
| AML (therapy related) | MYC mRNA overexpression | 150 |
| Essential thrombocythemia | MYC mRNA overexpression | 120 |
| Chronic myeloid leukemia | MYC mRNA overexpression | 130, 131 |
Myc in Human Lymphoid Leukemia
Lymphoid neoplasms can be broadly divided into those originating from immature lymphoid cells (T- and B-lymphoblastic leukemia and lymphoma) and the mature B- and T-cell neoplasms. The former include common leukemia, such as chronic lymphocytic leukemia (CLL), follicular lymphoma, diffuse large B-cell lymphoma, plasma cell myeloma (multiple myeloma), and Burkitt lymphoma, the first human tumor where MYC deregulation was identified.83
ALL is a heterogeneous disease comprising different entities that show clonal expansion of leukemic lymphoblasts.83,84 In human ALL, upregulation of c-Myc through different mechanisms has been reported. Translocations t(8;14), t(8;22), and t(2;8) involving MYC deregulation are present in 5% of adult ALL and 2% to 5% of children ALL.84 Aberrant c-Myc stability has been reported in lymphoblastic leukemia cell lines and bone marrow samples from pediatric ALL patients.85 Prolonged c-Myc protein half-life was not correlated with the MYC-stabilizing mutation, as found in Burkitt lymphomas.85 The ETS family factor TEL2 has been shown to accelerate lymphoma development in Eµ-Myc transgenic mice, and interestingly, TEL2 and c-Myc expression levels were coordinately elevated in pediatric B-ALL patients, suggesting that both oncogenes cooperate in B lymphomagenesis.86 Finally, more than 50% of human T-cell acute lymphoblastic leukemia (T-ALL) have activating mutations of NOTCH1,87 and MYC is a direct transcriptional target of oncogenic Notch1.88,89 Thus, it has been suggested that Notch1 mediates T-cell transformation at least in part by sustaining c-Myc levels (reviewed in References 90 and 91). Increased expression of MYC and c-Myc targets were recently found in T-ALL concomitant with LEF1 inactivation.92
CLL is the most frequent leukemia in adults (almost 25% of all leukemia in the United States and Europe).93,94 CLL is a slow-progression disease, with a median survival of 10 years, but it presents a marked variability: About one third of patients show a more aggressive form of the disease, with shorter survival periods. Interestingly, MYC mRNA expression is low in CLL,95 a result confirmed at the protein level (JM Caraballo and J León, unpublished results). MYC expression is similar in the bad and good prognosis groups of CLL. In contrast, in the malignant lymphoma form of the disease, Richter syndrome, MYC expression increases.96 Moreover, MYC translocation in CLL is associated with poor prognosis.97
Rearrangements of the MYC oncogene are present in 15% to 50% of primary human multiple myeloma tumors, in many cases involved in complex rearrangements.98,99 Frequent upregulation of MYC is also observed in plasma cell leukemia, a monoclonal gammopathy that can evolve from multiple myeloma.100
c-Myc in Human Myeloid Leukemia
Myeloid neoplasms belong to 3 major groups: myeloproliferative neoplasms (including chronic myeloid leukemia [CML]), myelodysplastic syndromes, and AML.101 Moreover, human AML is actually a heterogeneous group of neoplasias affecting the myeloid lineage. The former FAB (French-American-British) classification system of AML (M0 to M8 subtypes, attending to the differentiation type and stage) has been superseded by the World Health Organization classification, which identifies 15 diseases characterized by clinical presentation and recurrent chromosomal aberrations.101 Understandably, most molecular studies to date are carried out with a relatively small number of cases of each myeloid disease or do not make distinctions among the different entities. Therefore, the information on MYC expression in myeloid leukemia (or expression of any gene, except those involved in recurrent translocations) refers to a rather heterogeneous group of diseases, and its possible involvement in a particular myeloid neoplasm has not been properly addressed.
The overexpression of MYC in bone marrow and peripheral blood in sporadic AML cases was observed early on.102,103 Microarray-based studies and RT-qPCR studies showed that MYCN is over expressed in AML patients (as compared with normal bone marrow).76 Recurrent translocations in AML generate fusion proteins that are leukemogenic transcription factors.101 At least 3 of these—RUNX1-ETO, PML-RARα, and PLZF-RARα—induce c-Myc expression, suggesting that c-Myc is a downstream target of these oncogenes.104,105
Amplifications of MYC in AML have been reported.106–108 Double minute chromosomes and homogeneously staining regions containing amplified segments from chromosome band 8q24, including the MYC gene, have also been described.106,109 However, it has been reported that a high MYC copy number does not result in higher c-Myc expression,110,111 a situation that seems to conflict with the correlation between MYC amplification and expression observed in lymphoma.112
MYC expression appeared elevated in a microarray-based study in 5 AML samples, as validated by RT-PCR.113 In contrast, MYC was not detected as a major overexpressed gene in other microarray-based studies on AML samples.114–116 Note, however, that microarray-based studies are done at the mRNA level; thus, changes in c-Myc protein level are not evaluated. Also note that MYC expression changes less than 2-fold are usually filtered out by the statistical analysis of microarray data but that a 2-fold expression change of c-Myc may be relevant. For instance, a mere 2-fold change means a major difference for c-Myc transformation ability of rat fibroblasts and mouse embryonic stem cells,30,117 as well in transgenic animals where c-Myc dosage can be modulated.118 It is noteworthy that in Burkitt lymphoma, the paradigm of MYC activation in human cancer, c-Myc increase in expression can be only 2-fold with respect to normal lymphocytes.119 Finally, the great diversity of AML commented above makes the analysis of homogeneous sample cohorts difficult.
MYC mRNA is overexpressed in bone marrow cells from essential thrombocythemia, a myeloproliferative syndrome.120 This is in contrast with the lack of MYC overexpression in another common myeloproliferative syndrome, the polycythemia vera (PV).120 This is striking because more than 95% of PV carry activating mutation in the tyrosine kinase JAK2, which has been reported to induce MYC expression in cell lines,121,122 suggesting that the pathway is not operative in PV primary cells.
CML is a common myeloproliferative disorder that progresses in 2 phases: Most patients are diagnosed in a relatively benign chronic phase, which is followed by an accelerated and, finally, blastic crisis phase. The molecular hallmark of all CML phases is the expression of the Bcr-Abl kinase,123 which upregulates MYC expression122,124 and cooperates with c-Myc in transformation.125–127 Studies performed with a small number of cases showed that MYC mRNA levels were elevated in blastic crisis128,129 and in chronic phase versus healthy bone marrow.130,131 We recently observed the upregulation of c-Myc mRNA with CML progression (M Albajar et al., submitted). CML progression into blastic crisis is associated with cell survival, genomic instability, and differentiation arrest.123,132 We have also shown that enforced MYC expression in CML-derived cells as K562 results in aberrant DNA synthesis under imatinib stress and block imatinib-mediated differentiation (M Albajar et al., submitted), suggesting that c-Myc may contribute to CML transformation. Note that trisomy 8 and gain at 8q24 (where MYC maps) are among the most frequent cytogenetic alterations in CML,133,134 although their correlation with expression is unknown.
In conclusion, c-Myc function is pivotal for the correct hematopoiesis, helping to regulate the exquisite balance among self-renewal, differentiation, and proliferation required for blood formation. A reflection of c-Myc importance is the frequent finding of MYC deregulation in human leukemia and lymphoma, which would destroy this balance and transform hematopoietic cells by stimulating proliferation and blocking terminal differentiation.
Acknowledgments
We are grateful to Marta Albajar, Ana Batlle, and Ignacio Moreno de Alborán for a critical reading of parts of the article. We apologize to colleagues whose work was not cited in the form of their original articles but in reviews and whose work was not discussed because of space limitations or unintentional omission.
Footnotes
The work in our laboratories was supported by grants from the Ministerio de Ciencia e Innovacion of Spain (SAF08-01581) and the Red Temática de Investigación Cooperativa en Cancer (RD06/0020/0017) to J.L. and by a grant from Instituto Carlos III (FIS08/0829) to M.D.D.
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008;132:631-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 2008;453:306-13 [DOI] [PubMed] [Google Scholar]
- 3. Shizuru JA, Negrin RS, Weissman IL. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu Rev Med 2005;56:509-38 [DOI] [PubMed] [Google Scholar]
- 4. Kondo M, Wagers AJ, Manz MG, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol 2003;21:759-806 [DOI] [PubMed] [Google Scholar]
- 5. Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol 2006;7:333-7 [DOI] [PubMed] [Google Scholar]
- 6. Iwasaki H, Akashi K. Hematopoietic developmental pathways: on cellular basis. Oncogene 2007;26:6687-96 [DOI] [PubMed] [Google Scholar]
- 7. Miranda-Saavedra D, Gottgens B. Transcriptional regulatory networks in haematopoiesis. Curr Opin Genet Dev 2008;18:530-5 [DOI] [PubMed] [Google Scholar]
- 8. Kim SI, Bresnick EH. Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene 2007;26:6777-94 [DOI] [PubMed] [Google Scholar]
- 9. Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 2009;61:800-30 [DOI] [PubMed] [Google Scholar]
- 10. Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity 2007;26:726-40 [DOI] [PubMed] [Google Scholar]
- 11. Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity 2007;26:715-25 [DOI] [PubMed] [Google Scholar]
- 12. Rothenberg EV, Taghon T. Molecular genetics of T cell development. Annu Rev Immunol 2005;23:601-49 [DOI] [PubMed] [Google Scholar]
- 13. Leon J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009;8:1148-57 [DOI] [PubMed] [Google Scholar]
- 14. Coppola JA, Cole MD. Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nature 1986;320:760-3 [DOI] [PubMed] [Google Scholar]
- 15. Prochownik EV, Kukowska J. Deregulated expression of c-myc by murine erythroleukaemia cells prevents differentiation. Nature 1986;322:848-50 [DOI] [PubMed] [Google Scholar]
- 16. Dmitrovsky E, Kuehl WM, Hollis GF, Kirsch IR, Bender TP, Segal S. Expression of a transfected human c-myc oncogene inhibits differentiation of a mouse erythroleukaemia cell line. Nature 1986;322:748-50 [DOI] [PubMed] [Google Scholar]
- 17. Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell 1986;47:11-8 [DOI] [PubMed] [Google Scholar]
- 18. Delgado MD, Lerga A, Canelles M, Gomez-Casares MT, Leon J. Differential regulation of Max and role of c-Myc during erythroid and myelomonocytic differentiation of K562 cells. Oncogene 1995;10:1659-65 [PubMed] [Google Scholar]
- 19. Lerga A, Crespo P, Berciano M, et al. Regulation of c-Myc and Max in megakaryocytic and monocytic-macrophagic differentiation of K562 cells induced by protein kinase C modifiers: c-Myc is down-regulated but does not inhibit differentiation. Cell Growth Differ 1999;10:639-54 [PubMed] [Google Scholar]
- 20. Canelles M, Delgado MD, Hyland KM, et al. Max and inhibitory c-Myc mutants induce erythroid differentiation and resistance to apoptosis in human myeloid leukemia cells. Oncogene 1997;14:1315-27 [DOI] [PubMed] [Google Scholar]
- 21. Acosta JC, Ferrandiz N, Bretones G, et al. Myc inhibits p27-induced erythroid differentiation of leukemia cells by repressing erythroid master genes without reversing p27-mediated cell cycle arrest. Mol Cell Biol 2008;28:7286-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryan KM, Birnie GD. Cell-cycle progression is not essential for c-Myc to block differentiation. Oncogene 1997;14:2835-43 [DOI] [PubMed] [Google Scholar]
- 23. Gomez-Casares MT, Delgado MD, Lerga A, et al. Down-regulation of c-myc gene is not obligatory for growth inhibition and differentiation of human myeloid leukemia cells. Leukemia 1993;7:1824-33 [PubMed] [Google Scholar]
- 24. Sheiness D, Bishop JM. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J Virol 1979;31:514-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Varmus HE. The molecular genetics of cellular oncogenes. Annu Rev Genet 1984;18:553-612 [DOI] [PubMed] [Google Scholar]
- 26. Laurenti E, Varnum-Finney B, Wilson A, et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008;3:611-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang CY, Bredemeyer AL, Walker LM, Bassing CH, Sleckman BP. Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur J Immunol 2008;38:342-9 [DOI] [PubMed] [Google Scholar]
- 28. Reavie L, Gatta GD, Crusio K, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol 2010;11:207-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci U S A 1995;92:10302-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baudino TA, McKay C, Pendeville-Samain H, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 2002;16:2530-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 1993;7:671-82 [DOI] [PubMed] [Google Scholar]
- 32. Trumpp A, Refaeli Y, Oskarsson T, et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001;414:768-73 [DOI] [PubMed] [Google Scholar]
- 33. He C, Hu H, Braren R, et al. c-myc in the hematopoietic lineage is crucial for its angiogenic function in the mouse embryo. Development 2008;135:2467-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dubois NC, Adolphe C, Ehninger A, Wang RA, Robertson EJ, Trumpp A. Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 2008;135:2455-65 [DOI] [PubMed] [Google Scholar]
- 35. Wilson A, Murphy MJ, Oskarsson T, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 2004;18:2747-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Satoh Y, Matsumura I, Tanaka H, et al. Roles for c-Myc in self-renewal of hematopoietic stem cells. J Biol Chem 2004;279:24986-93 [DOI] [PubMed] [Google Scholar]
- 37. Walkley CR, Fero ML, Chien WM, Purton LE, McArthur GA. Negative cell-cycle regulators cooperatively control self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol 2005;7:172-8 [DOI] [PubMed] [Google Scholar]
- 38. Baena E, Ortiz M, Martinez AC, de Alboran IM. c-Myc is essential for hematopoietic stem cell differentiation and regulates Lin(-)Sca-1(+)c-Kit(-) cell generation through p21. Exp Hematol 2007;35:1333-43 [DOI] [PubMed] [Google Scholar]
- 39. Malynn BA, de Alboran IM, O’Hagan RC, et al. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev 2000;14:1390-9 [PMC free article] [PubMed] [Google Scholar]
- 40. Matsuoka S, Oike Y, Onoyama I, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev 2008;22:986-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zimmerman K, Alt FW. Expression and function of myc family genes. Crit Rev Oncog 1990;2:75-95 [PubMed] [Google Scholar]
- 42. Dose M, Khan I, Guo Z, et al. c-Myc mediates pre-TCR-induced proliferation but not developmental progression. Blood 2006;108:2669-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith RK, Zimmerman K, Yancopoulos GD, Ma A, Alt FW. Transcriptional down-regulation of N-myc expression during B-cell development. Mol Cell Biol 1992;12:1578-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klemsz MJ, Justement LB, Palmer E, Cambier JC. Induction of c-fos and c-myc expression during B cell activation by IL-4 and immunoglobulin binding ligands. J Immunol 1989;143:1032-9 [PubMed] [Google Scholar]
- 45. Larsson LG, Schena M, Carlsson M, Sallstrom J, Nilsson K. Expression of the c-myc protein is down-regulated at the terminal stages during in vitro differentiation of B-type chronic lymphocytic leukemia cells. Blood 1991;77:1025-32 [PubMed] [Google Scholar]
- 46. Habib T, Park H, Tsang M, et al. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J Cell Biol 2007;179:717-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Alboran IM, O’Hagan RC, Gartner F, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001;14:45-55 [DOI] [PubMed] [Google Scholar]
- 48. Lindsten T, June CH, Thompson CB. Multiple mechanisms regulate c-myc gene expression during normal T cell activation. EMBO J 1988;7:2787-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Douglas NC, Jacobs H, Bothwell AL, Hayday AC. Defining the specific physiological requirements for c-Myc in T cell development. Nat Immunol 2001;2:307-15 [DOI] [PubMed] [Google Scholar]
- 50. Dose M, Sleckman BP, Han J, Bredemeyer AL, Bendelac A, Gounari F. Intrathymic proliferation wave essential for Valpha14+ natural killer T cell development depends on c-Myc. Proc Natl Acad Sci U S A 2009;106:8641-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mycko MP, Ferrero I, Wilson A, et al. Selective requirement for c-Myc at an early stage of V(alpha)14i NKT cell development. J Immunol 2009;182:4641-8 [DOI] [PubMed] [Google Scholar]
- 52. Rajewsky K. Clonal selection and learning in the antibody system. Nature 1996;381:751-8 [DOI] [PubMed] [Google Scholar]
- 53. Scheller H, Tobollik S, Kutzera A, et al. c-Myc overexpression promotes a germinal center-like program in Burkitt’s lymphoma. Oncogene 2010;29:888-97 [DOI] [PubMed] [Google Scholar]
- 54. Guo Y, Niu C, Breslin P, et al. c-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood 2009;114:2097-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Niu C, Zhang J, Breslin P, Onciu M, Ma Z, Morris SW. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 2009;114:2087-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thompson A, Zhang Y, Kamen D, Jackson CW, Cardiff RD, Ravid K. Deregulated expression of c-myc in megakaryocytes of transgenic mice increases megakaryopoiesis and decreases polyploidization. J Biol Chem 1996;271:22976-82 [DOI] [PubMed] [Google Scholar]
- 57. Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985;318:533-8 [DOI] [PubMed] [Google Scholar]
- 58. Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: a pre-B-cell lymphoma model. Proc Natl Acad Sci U S A 1988;85:6047-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999;13:2658-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002;1:289-98 [DOI] [PubMed] [Google Scholar]
- 61. Sheppard RD, Samant SA, Rosenberg M, Silver LM, Cole MD. Transgenic N-myc mouse model for indolent B cell lymphoma: tumor characterization and analysis of genetic alterations in spontaneous and retrovirally accelerated tumors. Oncogene 1998;17:2073-85 [DOI] [PubMed] [Google Scholar]
- 62. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999;4:199-207 [DOI] [PubMed] [Google Scholar]
- 63. Palomo C, Zou X, Nicholson IC, Butzler C, Bruggemann M. B-cell tumorigenesis in mice carrying a yeast artificial chromosome-based immunoglobulin heavy/c-myc translocus is independent of the heavy chain intron enhancer (Emu). Cancer Res 1999;59:5625-8 [PubMed] [Google Scholar]
- 64. Magrath I. The pathogenesis of Burkitt’s lymphoma. Adv Cancer Res 1990;55:133-270 [DOI] [PubMed] [Google Scholar]
- 65. Park SS, Kim JS, Tessarollo L, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res 2005;65:1306-15 [DOI] [PubMed] [Google Scholar]
- 66. Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B cell antigen receptor and overexpression of MYC can cooperate in the genesis of B cell lymphomas. PLoS Biol 2008;6:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Khuda SE, Loo WM, Janz S, Van Ness B, Erickson LD. Deregulation of c-Myc Confers distinct survival requirements for memory B cells, plasma cells, and their progenitors. J Immunol 2008;181:7537-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst) 2006;5:1213-24 [DOI] [PubMed] [Google Scholar]
- 69. Skoda RC, Tsai SF, Orkin SH, Leder P. Expression of c-MYC under the control of GATA-1 regulatory sequences causes erythroleukemia in transgenic mice. J Exp Med 1995;181:1603-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith DP, Bath ML, Harris AW, Cory S. T-cell lymphomas mask slower developing B-lymphoid and myeloid tumours in transgenic mice with broad haemopoietic expression of MYC. Oncogene 2005;24:3544-53 [DOI] [PubMed] [Google Scholar]
- 71. Smith DP, Bath ML, Metcalf D, Harris AW, Cory S. MYC levels govern hematopoietic tumor type and latency in transgenic mice. Blood 2006;108:653-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 2005;106:2452-61 [DOI] [PubMed] [Google Scholar]
- 73. Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005;436:807-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu D, Thomas-Tikhonenko A. A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene 2002;21:1922-7 [DOI] [PubMed] [Google Scholar]
- 75. Yu D, Allman D, Goldschmidt MH, Atchison ML, Monroe JG, Thomas-Tikhonenko A. Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood 2003;101:1950-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kawagoe H, Kandilci A, Kranenburg TA, Grosveld GC. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res 2007;67:10677-85 [DOI] [PubMed] [Google Scholar]
- 77. Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 2009;28:1274-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hirouchi T, Takabatake T, Yoshida K, et al. Upregulation of c-myc gene accompanied by PU.1 deficiency in radiation-induced acute myeloid leukemia in mice. Exp Hematol 2008;36:871-85 [DOI] [PubMed] [Google Scholar]
- 79. Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J 1999;18:717-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000;95:2104-10 [PubMed] [Google Scholar]
- 81. Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 2000;20:2423-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee JW, Soung YH, Kim SY, et al. Mutational analysis of MYC in common epithelial cancers and acute leukemias. APMIS 2006;114:436-9 [DOI] [PubMed] [Google Scholar]
- 83. Jaffe ES, Harris NL, Stein H, Campo E, Pileri SA, Swerdlow SH. Introduction and overview of the calssification of the lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues (8th ed.). Lyon, France: International Agency for Research on Cancer; 2008. p. 158-66 [Google Scholar]
- 84. Faderl S, O’Brien S, Pui CH, et al. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer 2010;116:1165-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Malempati S, Tibbitts D, Cunningham M, et al. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 2006;20:1572-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cardone M, Kandilci A, Carella C, et al. The novel ETS factor TEL2 cooperates with Myc in B lymphomagenesis. Mol Cell Biol 2005;25:2395-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer 2006;6:347-59 [DOI] [PubMed] [Google Scholar]
- 88. Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006;20:2096-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 2006;103:18261-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sharma VM, Draheim KM, Kelliher MA. The Notch1/c-Myc pathway in T cell leukemia. Cell Cycle 2007;6:927-30 [DOI] [PubMed] [Google Scholar]
- 91. Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res 2008;14:5314-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gutierrez A, Sanda T, Ma W, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood 2010;115:2845-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yee KW, O’Brien SM. Chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc 2006;81:1105-29 [DOI] [PubMed] [Google Scholar]
- 94. Chiorazzi N, Allen SL, Ferrarini M. Clinical and laboratory parameters that define clinically relevant B-CLL subgroups. Curr Top Microbiol Immunol 2005;294:109-33 [DOI] [PubMed] [Google Scholar]
- 95. Greil R, Fasching B, Loidl P, Huber H. Expression of the c-myc proto-oncogene in multiple myeloma and chronic lymphocytic leukemia: an in situ analysis. Blood 1991;78:180-91 [PubMed] [Google Scholar]
- 96. Scandurra M, Rossi D, Deambrogi C, et al. Genomic profiling of Richter’s syndrome: recurrent lesions and differences with de novo diffuse large B-cell lymphomas. Hematol Oncol 2010;28:62-67 [DOI] [PubMed] [Google Scholar]
- 97. Huh YO, Lin KI, Vega F, et al. MYC translocation in chronic lymphocytic leukaemia is associated with increased prolymphocytes and a poor prognosis. Br J Haematol 2008;142:36-44 [DOI] [PubMed] [Google Scholar]
- 98. Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001;98:3082-6 [DOI] [PubMed] [Google Scholar]
- 99. Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A 2000;97:228-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chiecchio L, Dagrada GP, White HE, et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer 2009;48:624-36 [DOI] [PubMed] [Google Scholar]
- 101. Vardiman JW, Brunning RD, Arber DA, et al. , Introduction and overview of the classification of the myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues (4th ed.). Lyon, France: International Agency for Research on Cancer; 2008. p. 18-37 [Google Scholar]
- 102. Ferrari S, Narni F, Mars W, et al. Expression of growth-regulated genes in human acute leukemias. Cancer Res 1986;46:5162-6 [PubMed] [Google Scholar]
- 103. Calabretta B, Venturelli D, Kaczmarek L, et al. Altered expression of G1-specific genes in human malignant myeloid cells. Proc Natl Acad Sci U S A 1986;83:1495-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Muller-Tidow C, Steffen B, Cauvet T, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol 2004;24:2890-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rice KL, Hormaeche I, Doulatov S, et al. Comprehensive genomic screens identify a role for PLZF-RARalpha as a positive regulator of cell proliferation via direct regulation of c-MYC. Blood 2009;114:5499-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bruyere H, Sutherland H, Chipperfield K, Hudoba M. Concomitant and successive amplifications of MYC in APL-like leukemia. Cancer Genet Cytogenet 2010;197:75-80 [DOI] [PubMed] [Google Scholar]
- 107. Lee S, Kim M, Lim J, et al. Acute myeloid leukemia with MYC amplification in the homogeneous staining regions and double minutes. Cancer Genet Cytogenet 2009;192:96-8 [DOI] [PubMed] [Google Scholar]
- 108. Mathew S, Lorsbach RB, Shearer P, Sandlund JT, Raimondi SC. Double minute chromosomes and c-MYC amplification in a child with secondary myelodysplastic syndrome after treatment for acute lymphoblastic leukemia. Leukemia 2000;14:1314-5 [DOI] [PubMed] [Google Scholar]
- 109. Thomas L, Stamberg J, Gojo I, Ning Y, Rapoport AP. Double minute chromosomes in monoblastic (M5) and myeloblastic (M2) acute myeloid leukemia: two case reports and a review of literature. Am J Hematol 2004;77:55-61 [DOI] [PubMed] [Google Scholar]
- 110. Paulsson K, Lassen C, Kuric N, et al. MYC is not overexpressed in a case of chronic myelomonocytic leukemia with MYC-containing double minutes. Leukemia 2003;17:813-5 [DOI] [PubMed] [Google Scholar]
- 111. Storlazzi CT, Fioretos T, Surace C, et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 2006;15:933-42 [DOI] [PubMed] [Google Scholar]
- 112. Stasik CJ, Nitta H, Zhang W, et al. Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica 2010;95:597-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Court EL, Smith MA, Avent ND, et al. DNA microarray screening of differential gene expression in bone marrow samples from AML, non-AML patients and AML cell lines. Leuk Res 2004;28:743-53 [DOI] [PubMed] [Google Scholar]
- 114. Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 2004;350:1617-28 [DOI] [PubMed] [Google Scholar]
- 115. Ross ME, Mahfouz R, Onciu M, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 2004;104:3679-87 [DOI] [PubMed] [Google Scholar]
- 116. Stirewalt DL, Meshinchi S, Kopecky KJ, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer 2008;47:8-20 [DOI] [PubMed] [Google Scholar]
- 117. Bazarov AV, Adachi S, Li SF, Mateyak MK, Wei S, Sedivy JM. A modest reduction in c-myc expression has minimal effects on cell growth and apoptosis but dramatically reduces susceptibility to Ras and Raf transformation. Cancer Res 2001;61:1178-86 [PubMed] [Google Scholar]
- 118. Murphy DJ, Junttila MR, Pouyet L, et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008;14:447-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wilda M, Busch K, Klose I, et al. Level of MYC overexpression in pediatric Burkitt’s lymphoma is strongly dependent on genomic breakpoint location within the MYC locus. Genes Chromosomes Cancer 2004;41:178-82 [DOI] [PubMed] [Google Scholar]
- 120. Theophile K, Buesche G, Kreipe H, Bock O. The expression levels of telomerase catalytic subunit hTERT and oncogenic MYC in essential thrombocythemia are affected by the molecular subtype. Ann Hematol 2008;87:263-8 [DOI] [PubMed] [Google Scholar]
- 121. Watanabe S, Itoh T, Arai K. JAK2 is essential for activation of c-fos and c-myc promoters and cell proliferation through the human granulocyte-macrophage colony-stimulating factor receptor in BA/F3 cells. J Biol Chem 1996;271:12681-6 [DOI] [PubMed] [Google Scholar]
- 122. Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 2002;21:7137-46 [DOI] [PubMed] [Google Scholar]
- 123. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007;7:441-53 [DOI] [PubMed] [Google Scholar]
- 124. Gomez-Casares MT, Vaque JP, Lemes A, Molero T, Delgado MD, Leon J. C-myc expression in cell lines derived from chronic myeloid leukemia. Haematologica 2004;89:241-3 [PubMed] [Google Scholar]
- 125. Lugo TG, Witte ON. The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol 1989;9:1263-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992;70:901-10 [DOI] [PubMed] [Google Scholar]
- 127. Afar DE, Goga A, McLaughlin J, Witte ON, Sawyers CL. Differential complementation of Bcr-Abl point mutants with c-Myc. Science 1994;264:424-6 [DOI] [PubMed] [Google Scholar]
- 128. Handa H, Hegde UP, Kotelnikov VM, et al. Bcl-2 and c-myc expression, cell cycle kinetics and apoptosis during the progression of chronic myelogenous leukemia from diagnosis to blastic phase. Leuk Res 1997;21:479-89 [DOI] [PubMed] [Google Scholar]
- 129. Beck Z, Bacsi A, Kovacs E, et al. Changes in oncogene expression implicated in evolution of chronic granulocytic leukemia from its chronic phase to acceleration. Leuk Lymphoma 1998;30:293-306 [DOI] [PubMed] [Google Scholar]
- 130. Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene 2003;22:3952-63 [DOI] [PubMed] [Google Scholar]
- 131. Diaz-Blanco E, Bruns I, Neumann F, et al. Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia 2007;21:494-504 [DOI] [PubMed] [Google Scholar]
- 132. Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009;113:1619-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol 2002;107:76-94 [DOI] [PubMed] [Google Scholar]
- 134. Brazma D, Grace C, Howard J, et al. Genomic profile of chronic myelogenous leukemia: imbalances associated with disease progression. Genes Chromosomes Cancer 2007;46:1039-50 [DOI] [PubMed] [Google Scholar]
- 135. Larsson LG, Ivhed I, Gidlund M, Pettersson U, Vennstrom B, Nilsson K. Phorbol ester-induced terminal differentiation is inhibited in human U-937 monoblastic cells expressing a v-myc oncogene. Proc Natl Acad Sci U S A 1988;85:2638-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wu S, Cetinkaya C, Munoz-Alonso MJ, et al. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003;22:351-60 [DOI] [PubMed] [Google Scholar]
- 137. Bacon TA, Wickstrom E. Daily addition of an anti-c-myc DNA oligomer induces granulocytic differentiation of human promyelocytic leukemia HL-60 cells in both serum-containing and serum-free media. Oncogene Res 1991;6:21-32 [PubMed] [Google Scholar]
- 138. Holt JT, Redner RL, Nienhuis AW. An oligomer complementary to c-myc mRNA inhibits proliferation of HL-60 promyelocytic cells and induces differentiation. Mol Cell Biol 1988;8:963-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol 2006;34:1480-9 [DOI] [PubMed] [Google Scholar]
- 140. Acosta JC, Richard C, Delgado MD, et al. Amifostine impairs p53-mediated apoptosis of human myeloid leukemia cells. Mol Cancer Ther 2003;2:893-900 [PubMed] [Google Scholar]
- 141. Chisholm O, Stapleton P, Symonds G. Constitutive expression of exogenous myc in myelomonocytic cells: acquisition of a more transformed phenotype and inhibition of differentiation induction. Oncogene 1992;7:1827-36 [PubMed] [Google Scholar]
- 142. Nguyen HQ, Selvakumaran M, Liebermann DA, Hoffman B. Blocking c-Myc and Max expression inhibits proliferation and induces differentiation of normal and leukemic myeloid cells. Oncogene 1995;11:2439-44 [PubMed] [Google Scholar]
- 143. Shafarenko M, Liebermann DA, Hoffman B. Egr-1 abrogates the block imparted by c-Myc on terminal M1 myeloid differentiation. Blood 2005;106:871-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Johansen LM, Iwama A, Lodie TA, et al. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol 2001;21:3789-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006;16:318-30 [DOI] [PubMed] [Google Scholar]
- 146. Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene 2001;20:5595-610 [DOI] [PubMed] [Google Scholar]
- 147. Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood 2003;101:1220-35 [DOI] [PubMed] [Google Scholar]
- 148. Frost M, Newell J, Lones MA, Tripp SR, Cairo MS, Perkins SL. Comparative immunohistochemical analysis of pediatric Burkitt lymphoma and diffuse large B-cell lymphoma. Am J Clin Pathol 2004;121:384-92 [DOI] [PubMed] [Google Scholar]
- 149. Larramendy ML, Niini T, Elonen E, et al. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia: a microarray analysis. Haematologica 2002;87:569-77 [PubMed] [Google Scholar]
- 150. Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ hematopoietic stem/ progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci U S A 2002;99:14925-30 [DOI] [PMC free article] [PubMed] [Google Scholar]