

Abstract
The PI3K-Akt pathway is a major survival pathway activated in cancer. Efforts to develop targeted therapies have not been fully successful, mainly because of extensive internal intrapathway or external interpathway negative feedback loops or because of networking between pathway suppressors. The PTEN tumor suppressor is the major brake of the pathway and a common target for inactivation in somatic cancers. This review will highlight the networking of PTEN with other inhibitors of the pathway, relevant to cancer progression. PTEN constitutes the main node of the inhibitory network, and a series of convergences at different levels in the PI3K-Akt pathway, starting from those with growth factor receptors, will be described. As PTEN exerts enzymatic activity as a phosphatidylinositol-3,4,5-trisphosphate (PIP3) phosphatase, thus opposing the activity of PI3K, the concerted actions to increase the availability of PIP3 in cancer cells, relying either on other phosphoinositide enzymes or on the intrinsic regulation of PTEN activity by other molecules, will be discussed. In particular, the synergy between PTEN and the circle of its direct interacting proteins will be brought forth in an attempt to understand both the activation of the PI3K-Akt pathway and the connections with other parallel oncogenic pathways. The understanding of the interplay between the modulators of the PI3K-Akt pathway in cancer should eventually lead to the design of therapeutic approaches with increased efficacy in the clinic.
Keywords: PTEN, PI3K, Akt, synergy, protein interactions
The PI3K-Akt Pathway
Phosphoinositides are negatively charged constituents of lipid membranes formed by phosphorylation of hydroxyl groups on positions 3′, 4′, or 5′ of the inositol ring of phosphatidyl inositol (PI) byspecific kinases. The PI 3′-OH kinase (PI3K) family comprises 3 classes of proteins that phosphorylate PI, PI-4-phosphate (PI-4-P), or PI-4,5-bisphosphate (PI-4,5-P2 or PIP2) on position 3′.1 Class I PI3Ks primarily generate PI-3,4,5-trisphosphate (PIP3) from PIP2. PIP3 acts as a second messenger by binding to and activating pleckstrin homology (PH) domain–containing proteins, including the Ser/Thr kinase Akt/PKB. Conversely, PIP3 is hydrolyzed to PIP2 by PTEN that opposes PI3K and specifically dephosphorylates phosphoinositides in position 3′.2,3
The signal for PIP3 production stems from the activation of class IA PI3Ks by growth factor receptor tyrosine kinases (RTKs) and of class IB PI3Ks by G protein–coupled receptors (GPCRs).1 Class IA PI3Ks are the only PI3Ks that have been implicated in cancer thus far.4 They are heterodimers of a regulatory subunit (p85α, p55α, p50α, p85β, p55γ) and a catalytic subunit (p110α, p110β, p110δ).
The activation of Akt by PIP3 production triggers signaling through a multitude of Akt phosphorylation targets that control cell survival, growth, proliferation, and other cellular processes.5 Thus, binding of the Akt PH domain to PIP3 unmasks the kinase domain6 and allows the phosphorylation of Akt on Thr308 in the activation loop by phosphoinositide-dependent kinase 1 (PDK1) and on Ser473 in the carboxyl (C)–terminal hydrophobic motif by the mammalian target of rapamycin complex-2 (mTORC2).7,8 Conversely, 2 classes of phosphatases, protein phosphatase 2A (PP2A) and PH domain leucine-rich repeat protein phosphatases (PHLPP) 1 and 2, have been described to dephosphorylate and inactivate Akt.9,10
Akt phosphorylates a plethora of targets6,11,12 on RxRxxS/T consensus motifs.13 Interestingly, many of its effects reside in phosphorylating and inactivating inhibitors of cell cycle progression, survival, glycolysis, angiogenesis, and protein synthesis, thus unlocking most if not all the processes involved in oncogenesis14 (Fig. 1). For example, Akt phosphorylates and inhibits 2 upstream inhibitors of the small GTPase Ras homolog enriched in the brain (Rheb), tuberous sclerosis complex protein 2 (TSC2)15-17 and proline-rich Akt substrate of 40 kDa (PRAS40).18-20 In its turn, Rheb activates mTORC1 by inhibiting FKBP38, a negative regulator of mTORC1.21 The net effect of Akt activation is activation of mTORC1, which is responsible for the up-regulation of protein synthesis in cells.22 Another negatively regulated target of Akt is glycogen synthase kinase 3 (GSK3),23 whose phosphorylation induces Myc and cyclin D1 increased activity with subsequent increased proliferation.24,25 Akt also phosphorylates and inhibits the activity of the FOXO transcription factors26 that induce cell cycle arrest by coordinating the expression of multiple cell cycle regulators27,28 and apoptosis through the up-regulation of proapoptotic Bcl-2 family members.29,30
Figure 1.
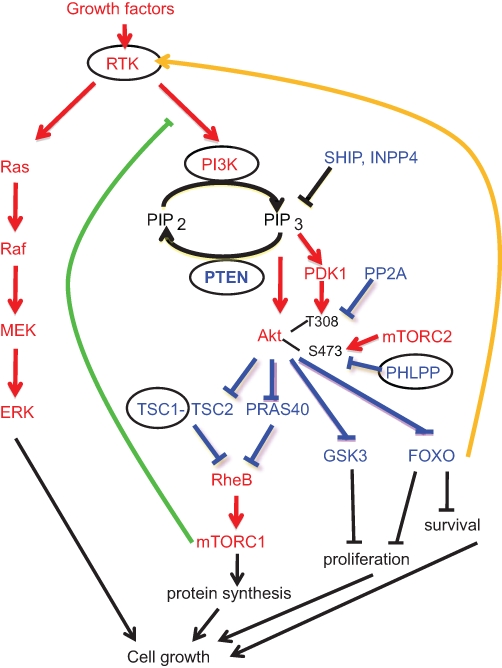
Nonlinear signaling through the PI3K-Akt pathway. Growth factors activate receptor tyrosine kinases (RTKs) that further activate parallel growth pathways. The MAPK pathway, which signals via Ras and downstream effectors, is an example of a linear growth-promoting pathway. The PI3K-Akt pathway is an example of a nonlinear pathway that signals through protein and lipid effectors and through layers of oncoproteins (red) and growth suppressors (blue). Negative feedback loops of the PI3K-Akt pathway are shown in green (repressing) or orange (activating). The effectors of the pathway whose alterations show combinatorial additive effects with PTEN inactivation in cancers are encircled.
Interestingly, 2 important negative feedback loops are triggered downstream of mTORC1 and FOXO transcription factors, blunting the effects of the pharmacological inhibition of the pathway (Fig. 1). mTORC1 activation results in transcriptional repression and inhibitory phosphorylation of the adaptor protein insulin receptor substrate 1 (IRS-1) with subsequent decreased PI3K-Akt signaling.31,32 The inhibition of mTORC1 by rapamycin would therefore trigger activation of PI3K-Akt through inhibition of this negative feedback loop. The FOXO-induced negative feedback loop works by increasing the expression of the insulin receptor with downstream activation of the PI3K-Akt pathway and has been initially identified in Drosophila.33,34 Importantly, this feedback is activated upon pharmacological inhibition of Akt and represents another mechanism to resistance to PI3K-Akt pathway inhibitors.35
From the integrated analysis of the PI3K-Akt pathway, one observes immediately that this is not a linear growth pathway, as is the case for the mitogen-activated protein kinase (MAPK) pathway (Fig. 1). Instead, the signals are transmitted through protein-protein or lipid-protein activation, and the pathway is sandwiched with pathway activators between pathway inhibitors. As described below, cancer cells take advantage of the complexity of this pathway to deregulate it at multiple levels in a combinatorial fashion.
PTEN Tumor Suppressor
PTEN/MMAC1 (phosphatase and tensin homolog, deleted on chromosome ten) has been identified by positional cloning as a candidate tumor suppressor gene located on chromosome 10q23.36,37 PTEN is frequently inactivated by mutation with loss of heterozygosity (LOH) in a number of cancers including brain, prostate, and uterine cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic). PTEN can also be inactivated by other mechanisms in somatic cancers, including promoter methylation,38 micro-RNA interference39 with or without pseudogene loss,40 phosphorylation,41 and delocalization from the plasma membrane.42 PTEN transcriptional regulation is also a possibility in tumors, and c-Jun, NF-κB, and HES-1 have been shown to repress PTEN transcription downstream of Ras, MKK4, and Notch activation, respectively,43-47 whereas EGR1, which activates PTEN transcription,48 has been shown to be excluded from the nucleus in tumors with reduced PTEN expression.49,50 Because of its high frequency of inactivation in somatic cancer, PTEN is ranked the second most mutated tumor suppressor gene after p53. Similarly to p53 and other bona fide tumor suppressors, germline mutations in PTEN gene cause Cowden and Bannayan-Riley-Ruvalcaba cancer predisposition syndromes.51,52 Cowden syndrome (incidence of 1 in 200,000 births) is characterized by intestinal hamartomas, mucocutaneous lesions, macrocephaly, fibrocystic disease, and increased risk for developing breast, thyroid, and endometrial cancer.
Initial studies have shown that the expression of PTEN induces a marked decrease of proliferation because of cell cycle arrest in G1 phase53,54 attributed to an increase of p27Kip154 and decreased level and nuclear localization of cyclin D1.55 PTEN also inhibits the migration of cells,56 likely by involvement of Rac and cdc42, but not of RhoA.57 All these effects are most likely mediated via the hydrolysis of PIP3 by PTEN2,58 and the repression of downstream pathways activated by protein interactions with PIP3. Recently, a structural role has been proposed for PTEN, in the maintenance of apical-basal polarity in polarized epithelial cells, by maintaining a segregated apical pool of PIP2 that would compartmentalize PIP2-binding proteins to the apical membrane.59-61 It is possible that loss of this function contributes to the epithelial-mesenchymal transition (EMT) observed in the progression of epithelial cancers.
PTEN Loss and Intrapathway Additive Activation
Fourteen years after its discovery as a tumor suppressor,36,37 PTEN reveals itself as a highly regulated tumor suppressor that behaves differently in different types of tumors. In some tumors, such as glioblastoma in which 10q chromosome deletion is present in 70% of cases,62,63 mutation with LOH of PTEN eliminates both alleles and therefore completely eliminates its expression. This situation conforms to Knudson’s 2-hit hypothesis for a tumor suppressor in which the complete gene elimination is required for tumor growth.64 In other types of tumors, PTEN shutdown is not complete. Mutations of one allele, transcriptional repression, epigenetic or posttranslational mechanisms, all of which would achieve partial inactivation of PTEN, and a combination of these mechanisms are also possible, leading to a continuum of lower than normal levels of functional PTEN in tumors. These mechanisms of reduced expression would exemplify a haploinsufficient tumor suppressor model for PTEN, and examples for such a behavior are found in both Cowden syndrome, in which not all tumors show LOH, and in most somatic cancers. In mice heterozygous for PTEN, both principles apply in that some tumors lose the PTEN second allele and some tumors do not.65,66 The fact that the levels of PTEN inversely correlate with tumorigenesis and Akt activation has been experimentally proven in mice with hypomorphic and hypermorphic PTEN alleles.67 It appears that even a small reduction of PTEN levels confers growth advantage to tumor cells, but the higher the reduction is, the more rapidly the tumor develops. This result explains why cancer cells target one or more of the plethora of mechanisms regulating PTEN levels and activity.
An interesting twist on the understanding of the PI3K-Akt pathway activation in tumors came from the discovery of apparently redundant coexisting PTEN inactivating mutations and PI3K activating mutations in endometrial cancer.68 In a linear pathway, such as MAPK or Wnt-APC-β-catenin, mutations in proteins activating the pathway in the same way are mutually exclusive in tumors because either a second mutation does not confer growth advantage to cells or perhaps this second mutation is even detrimental to cells in the context of a “just right” level of pathway activation.69 In the PI3K-Akt pathway, coexisting PI3K and PTEN mutations are present (Fig. 1), and at least, in endometrial, breast, and colorectal cancer, they seem to occur with a cumulative frequency equal to the product of the 2 frequencies, as if the 2 alterations are independently selected for.4 Although the cumulative frequency does not indicate synergism between mutations in PI3K and PTEN, it also excludes redundancy or mutual exclusivity, as in linear pathways. As these mutations are likely to occur sequentially, they might represent an alternative mechanism of eliminating both alleles of PTEN. In addition, the 2 proteins might have different functions other than PIP3 accumulation and might also respond to different genetic configurations of the cells at the time when the mutations occur.
PTEN is not the only phosphatase that contains the levels of PIP3. The ubiquitously expressed phosphoinositide 5′ phosphatase SHIP-2 that hydrolyzes PIP3 to PI-3,4-P2 has been previously shown to attenuate the activation of Akt in PTEN-negative glioblastoma cells.70 Recently, INPP4B, a 4′ phosphoinositide phosphatase that specifically dephosphorylates PI-3,4-P2, and by this further inhibits Akt, has been described as a tumor suppressor in breast and prostate cancers.71-73 It would be worth investigating whether SHIP-2 or INPP4B inactivation coexists with PTEN loss in a subset of these tumors, leading to higher Akt activation and thus to another convergent mechanism of PI3K-Akt activation.
Another example of additive activation of the pathway is the coexistence of RTK mutations or amplifications and PTEN mutations74 (Fig. 1). These events have been studied mainly in breast cancer, in which PTEN mutations confer resistance to HER2 inhibition,75-77 and also in glioblastoma, in which EGFR mutations or amplifications coexist with PTEN mutations.62,78 In this case, the signaling through the RTKs activates multiple parallel pathways involved in growth, including the PI3K-Akt pathway.
Theoretically, PTEN inactivation could cooperate with Akt oncogenic activation to boost the pathway in tumors. However, even if Akt amplifications and activating mutations have been reported in cancers,1,79 they have a low frequency, as compared to the activating mutations of PI3K.80 What appears though to be an important cooperation at the level of Akt activation is the coexistence of PTEN loss with PHLPP loss that synergistically activates Akt in glioblastoma (our unpublished observations). Other cooperative effects of PTEN with downstream modulators of the PI3K-Akt pathway are not yet well defined. Of note is that in bladder cancer, besides the coexistence of PTEN and PI3K double alterations, double PTEN and TSC1, or even triple PTEN, PI3K and TSC1 alterations have been reported, again suggesting nonredundancy by multiple hits in the activation of the PI3K-Akt pathway81 (Fig. 1).
Structure-Function Analysis of PTEN
PTEN encodes a 403–amino acid protein that comprises a phosphatase domain, a C2 domain that binds phospholipid membranes,82 and a C-terminal tail that contains 2 PEST sequences with Ser/Thr phosphorylation sites and a PDZ (PSD95/Dlg/ZO1) binding motif83,84 (Fig. 2). The importance of the phosphatase activity in tumor progression is underscored by a number of mutations occurring in the catalytic site that solely disrupt the enzymatic activity of PTEN.85,86 Other modifications shown to reversibly inhibit PTEN’s catalytic site are oxidation of the cysteine (Cys124)87-89 or acetylation of the lysines (Lys125 and Lys128)90 in the catalytic pocket. It is possible that these reversible modifications take place dynamically in tumor cells as dictated by the microenvironment conditions. A host of other mutations, especially in the C2 domain, but also in the phosphatase domain, cause misfolding of the protein and significantly reduce both its half-life and enzymatic function.83,91 Following these studies on PTEN protein stability, 2 mechanisms of PTEN degradation were identified, cleavage by caspase-392 and ubiquitination by NEDD4-1.93
Figure 2.

Diagram of PTEN domain structure.
Tumor-derived mutations that do not affect the enzymatic activity of PTEN in vitro were also identified. These were studied and found to affect the compartmentalization of PTEN in the cell, either by sequestering PTEN in the nucleus94 or importantly by interfering with PTEN’s recruitment to the plasma membrane. Besides the C2 domain, PTEN contains 2 other regions with lipid-binding properties, an N-terminal PIP2-binding motif and a positively charged region in the phosphatase domain.95,96 Mutations found in the C2 domain or the PIP2-binding motif disrupt the binding to phospholipid membranes and affect the tumor suppressor function of PTEN.82,91,96
The C-terminal tail of PTEN is also the target of mutations in tumors. As mentioned, this region contains the main phosphorylation sites mapped to residues Ser362, Thr366, Ser370, Ser380, Thr382, Thr383, and Ser385, and the kinases involved are casein kinase 2 (CK2), GSK3β, LKB1, and MAST.84,97-101 The phosphorylation of the tail has been shown to enhance PTEN stability but at the same time decrease its phosphatase activity.84 This apparent contradiction has been recently reconciled by the finding that the phosphorylated tail interacts with the rest of the molecule to confer a “closed” stable conformation that would be less interactive with phospholipid membranes and therefore less active.102 Of the kinases that inactivate PTEN according to this mechanism, CK2 has been shown to be overexpressed in T cell acute lymphoblastic leukemia and to participate in PTEN inactivation.41 The C-terminal PDZ-binding motif of PTEN is also targeted for deletions or mutations in tumors. Its function has been related to the involvement of PTEN in suppression of cell migration,103 inhibition of protein synthesis,104 and stabilization of PTEN at the plasma membrane.42,105
PTEN-Interacting Proteins as Regulators of Cancer
The activity of PTEN is modulated by posttranslational modifications that include oxidation, acetylation, phosphorylation, ubiquitination, and proteolytic cleavage and by protein-protein interactions. All these influences affect the enzymatic activity of PTEN directly or indirectly by modifying the protein conformation, stability, lipid membrane binding, and subcellular distribution. It is therefore straightforward to examine in tumors the deregulations of the enzymes that act on PTEN or of its interacting proteins that alter its activity, stability, and subcellular localization, in an attempt to delineate posttranslational PTEN-inactivating occurrences. Besides the effects on PTEN physiology, protein-protein interactions may also lead to the modification of the partner by PTEN. The molecules that interact with PTEN can be classified as 1) proteins that regulate PTEN, 2) proteins that are regulated by PTEN, and 3) potentially “2-way” proteins (Fig. 3). Examples for the 3 categories are provided below.
Figure 3.
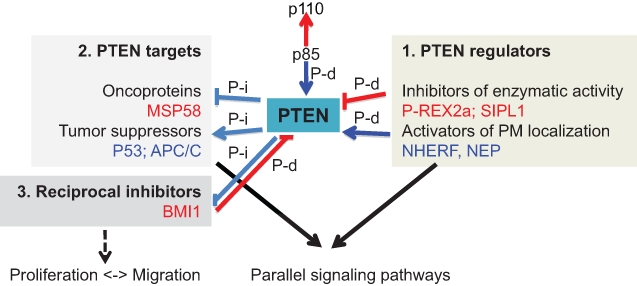
Three categories of PTEN-interacting proteins. Examples in each category are shown in red (oncoproteins) or blue (tumor suppressor proteins) and discussed in the text. PTEN phosphatase-dependent (P-d) or phosphatase-independent (P-i) connections resulting in oncogenic and tumor suppressor effects relevant for human cancers are shown with red and blue arrows, respectively.
1) In the first category, there are proteins that interact and inhibit PTEN enzymatic activity or, conversely, ligands that increase PTEN activity through stabilization at the plasma membrane. PIP3-RAC-exchanger-2a (P-REX2a) binds tightly to PTEN and inhibits its enzymatic activity.106 In breast cancers, P-REX2a is up-regulated preferentially in the PTEN wild-type tumors, and of these, especially in those exhibiting PI3K mutations, demonstrating a 2-tiered selectivity correlated with an incremental Akt activation. The P-REX2a-PTEN protein-protein inhibition thus constitutes an excellent example of both an alternate mechanism to genetic or epigenetic inhibition of PTEN and of the use of this mechanism for additive effects within the PI3K-Akt pathway. Another example of a direct inhibitor of PTEN phosphatase activity is Shank-interacting protein-like 1 (SIPL1), which has been shown to be elevated in PTEN wild-type cervical cancer.107 My group studies the Na+/H+-exchanger-3-regulatory-factor (NHERF) PTEN-interacting proteins NHERF1 and NHERF2, which bind to PTEN through PDZ domain interactions, and stabilizes it to the plasma membrane to contain the activation of PI3K.42,108,109 NHERF1 is either displaced from the membrane or lost in cancers of different origins, such as glioblastoma, breast, colorectal, and hepatocellular cancers.42,110-114 NHERF1 loss has been shown to contribute to the activation of Akt in tumors42,115,116 but also to the activation of other oncogenic pathways, such as the Wnt-β-catenin pathway.113,117,118 Therefore, NHERF1 alterations may simultaneously up-regulate parallel oncogenic pathways, including the PI3K-Akt pathway, conferring an enhanced growth advantage to tumor cells. Another example of a multitasking interactor and activator of PTEN is neutral endopeptidase 24.11 (NEP/CD10).119 NEP cleaves a host of physiologically active peptides through its extracellular domain, limiting their signaling, and binds to PTEN through its intracellular domain, recruits it to the plasma membrane, and increases PIP3 hydrolysis and Akt suppression.120 Interestingly, the phosphorylation of NEP by CK2 decreases the binding of PTEN and increases Akt activation.121 This finding implicates the kinase CK2 as a double-negative regulator of PTEN via diminished recruitment to the plasma membrane by NEP and via maintenance of a PTEN “closed” conformation by direct phosphorylation of PTEN’s tail.
2) From the second category of PTEN-interacting proteins, PTEN inhibits the transformation induced by the MSP58 oncogenic protein through interaction via PTEN’s tail.122 In this case, a hit on PTEN would deregulate both PTEN and its ligand, thus involving the PI3K-Akt pathway through PTEN and also a parallel oncogenic pathway through PTEN’s ligand. A couple of examples in this category define a role for PTEN in the nucleus by interacting and regulating the stability of p53 tumor suppressor123,124 or by increasing the activity of the anaphase-promoting complex/cyclosome (APC/C) E3 ubiquitin ligase through interaction with its components, leading to increased degradation of the cell cycle apparatus involved in the M-G1 transition.125 Importantly, the effect of PTEN on all the interacting proteins in this category is independent of its phosphatase activity and dependent only on an intact interaction region in PTEN, usually situated in the C-terminal half of the molecule.
3) The interactions in the third category are still to be characterized. BMI1, a polycomb protein that maintains the proliferation potential of hematopoietic and neural stem cells through the repression of cell cycle inhibitors, has been shown to interact with PTEN.126 Similarly to the interactions with p53 and APC/C, this interaction takes place in the nucleus and reduces BMI1’s function independently of PTEN’s phosphatase activity. BMI1 has also been shown to reduce PTEN’s inhibition of Akt, most likely by sequestration of PTEN in the nucleus. Finally, the colocalization of the 2 proteins appears to be significantly enhanced in prostatic intraepithelial neoplasia and carcinoma as compared to normal prostate epithelial cells.126 It is not clear what is the net effect on growth due to this type of interaction because PTEN would exert opposing influences either in the cytoplasm or in the nucleus. Similarly to the other nuclear effects of PTEN, the phosphatase-independent functions of PTEN might actually represent inhibitory cross-talks of the PI3K-Akt pathway, necessary to shut down cell proliferation during other cellular processes, such as cell migration that requires PI3K-Akt activation. In fact, we have recently shown that this proliferation-migration switch takes place in vivo in invading glioblastoma cells, even in the absence of PTEN.127 These migratory cells up-regulate Akt activation and stem cell behavior, which would be compatible with the activation of both Akt and BMI1 in the absence of PTEN. In this case, the suppression of MAPK via an inhibitory Akt-mediated phosphorylation most likely accounts for the reduction in proliferation.128
An intriguing protein-protein interaction is the one between PTEN and the p85 regulatory subunit of PI3K.129,130 p85 interacts with the dephosphorylated active form of PTEN in a high molecular weight complex that comprises also p110β but not p110α.130 The p85-PTEN interaction increases the enzymatic activity of PTEN and is enhanced after stimulation with epidermal growth factor.129 Hence, p85 regulates both the increase and the decrease of PIP3 levels following growth factor stimulation via association with p110 PI3K and PTEN, respectively. How this interaction regulates PTEN in cancer is still to be determined.
Conclusions
The discovery of PTEN as a tumor suppressor gene and the breakthrough finding that it functions as a PIP3 phosphatase have put the PI3K-Akt pathway on the map of important cancer pathways.2,36,37 In the years that followed, the PI3K-Akt signaling has been delineated as a nonlinear pathway that contains multiple levels of regulation and feedback loops. This elucidation process is still ongoing, and many regulatory mechanisms are being discovered, as a result of diagnostic studies or of specific therapies for different nodes in this pathway that are being developed and tested in clinical trials. PTEN remains the main negative regulator of the PI3K-Akt pathway and the target of a host of modulating proteins that are also affected in cancer. As discussed, the disruption of PTEN regulators may affect parallel oncogenic pathways besides the PI3K-Akt pathway, interconnecting these influences at the level of PTEN. In turn, PTEN is not only dedicated to inhibit the PI3K-Akt pathway but also, through protein-protein interactions, inhibits other growth pathways, thus becoming a major growth signaling inhibitor. Other emerging functions of PTEN, such as regulator of cell polarity or, as hypothesized here, regulator of a switch between cell proliferation and migration states, could also have key influences on the progression on various types of cancer. In conclusion, the integration of all these functions and connections makes of PTEN a central inhibitory node with a major impact in cancer.
Acknowledgments
I am very grateful to my postdoctoral mentor, Dr. Hidesaburo Hanafusa, for his honesty, open-mindedness, and unfailing example as a scientist, as well as for encouraging me in studying the regulation of the PTEN tumor suppressor. This review is dedicated to him. I also thank all the alumni of the Hanafusa laboratory for their friendship and scientific exchanges. Warm thanks for encouragement on this project go to Kathrin Kirsch, my bench mate during my postdoctoral years and good friend thereafter, and to Tomo Shishido and Tsuyoshi Akagi, who helped me with advice, reagents, and constant friendship. I also thank all the members of my laboratory who further developed the PTEN project and especially Yoko Takahashi, Fabiana Morales, Jennifer Molina, and Yuho Hayashi. Special thanks for insightful discussions go to my collaborators T.J. Liu, Gilbert Cote, Seth Corey, Xiaomin Chen, and Randy Legerski.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National Cancer Institute [grant number CA107201] and corresponding ARRA supplement and by bridge funds from the MD Anderson Cancer Center.
References
- 1. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606-19 [DOI] [PubMed] [Google Scholar]
- 2. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375-8 [DOI] [PubMed] [Google Scholar]
- 3. Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247-79 [DOI] [PubMed] [Google Scholar]
- 4. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257-62 [DOI] [PubMed] [Google Scholar]
- 6. Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473-88 [DOI] [PubMed] [Google Scholar]
- 7. Alessi DR, Kozlowski MT, Weng QP, Morrice N, Avruch J. 3-phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol. 1998;8:69-81 [DOI] [PubMed] [Google Scholar]
- 8. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101 [DOI] [PubMed] [Google Scholar]
- 9. Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93:5699-704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13-24 [DOI] [PubMed] [Google Scholar]
- 11. Brazil DP, Park J, Hemmings BA. PKB binding proteins: getting in on the Akt. Cell. 2002;111:293-303 [DOI] [PubMed] [Google Scholar]
- 12. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541-51 [PMC free article] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70 [DOI] [PubMed] [Google Scholar]
- 15. Dan HC, Sun M, Yang L, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277:35364-70 [DOI] [PubMed] [Google Scholar]
- 16. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648-57 [DOI] [PubMed] [Google Scholar]
- 17. Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658-65 [DOI] [PubMed] [Google Scholar]
- 18. Kovacina KS, Park GY, Bae SS, et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189-94 [DOI] [PubMed] [Google Scholar]
- 19. Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903-15 [DOI] [PubMed] [Google Scholar]
- 20. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316-23 [DOI] [PubMed] [Google Scholar]
- 21. Bai X, Ma D, Liu A, et al. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977-80 [DOI] [PubMed] [Google Scholar]
- 22. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9-22 [DOI] [PubMed] [Google Scholar]
- 23. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785-9 [DOI] [PubMed] [Google Scholar]
- 24. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-68 [DOI] [PubMed] [Google Scholar]
- 27. Ho KK, Myatt SS, Lam EW. Many forks in the path: cycling with FoxO. Oncogene. 2008;27:2300-11 [DOI] [PubMed] [Google Scholar]
- 28. Kops GJ, Medema RH, Glassford J, et al. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol. 2002;22:2025-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201-4 [DOI] [PubMed] [Google Scholar]
- 30. van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxid Redox Signal. 2011;14:579-92 [DOI] [PubMed] [Google Scholar]
- 31. Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009;21:827-35 [DOI] [PubMed] [Google Scholar]
- 32. Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35-42 [DOI] [PubMed] [Google Scholar]
- 33. Puig O, Marr MT, Ruhf ML, Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003;17:2006-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Puig O, Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev. 2005;19:2435-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943-7 [DOI] [PubMed] [Google Scholar]
- 37. Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356-62 [DOI] [PubMed] [Google Scholar]
- 38. Wiencke JK, Zheng S, Jelluma N, et al. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007;9:271-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H, Huang W, Jiang X, Pennicooke B, Park PJ, Johnson MD. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci U S A. 2010;107:2183-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118:3762-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Molina JR, Morales FC, Hayashi Y, Aldape KD, Georgescu MM. Loss of PTEN binding adapter protein NHERF1 from plasma membrane in glioblastoma contributes to PTEN inactivation. Cancer Res. 2010;70:6697-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beck SE, Carethers JM. BMP suppresses PTEN expression via RAS/ERK signaling. Cancer Biol Ther. 2007;6:1313-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hettinger K, Vikhanskaya F, Poh MK, et al. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ. 2007;14:218-29 [DOI] [PubMed] [Google Scholar]
- 45. Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vasudevan KM, Burikhanov R, Goswami A, Rangnekar VM. Suppression of PTEN expression is essential for antiapoptosis and cellular transformation by oncogenic Ras. Cancer Res. 2007;67:10343-50 [DOI] [PubMed] [Google Scholar]
- 47. Xia D, Srinivas H, Ahn YH, et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J Biol Chem. 2007;282:3507-19 [DOI] [PubMed] [Google Scholar]
- 48. Virolle T, Adamson ED, Baron V, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol. 2001;3:1124-8 [DOI] [PubMed] [Google Scholar]
- 49. Di Loreto C, Tell G, Pestrin M, Pandolfi M, Damante G, Puglisi F. PTEN and Egr-1 expression in thyroid proliferative lesions. Cancer Lett. 2005;224:105-9 [DOI] [PubMed] [Google Scholar]
- 50. Yamamoto C, Basaki Y, Kawahara A, et al. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:8715-25 [DOI] [PubMed] [Google Scholar]
- 51. Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4:492-502 [DOI] [PubMed] [Google Scholar]
- 53. Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002-8 [PubMed] [Google Scholar]
- 54. Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A. 1998;95:15406-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell-cycle arrest by decreasing the protein level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23:6139-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614-7 [DOI] [PubMed] [Google Scholar]
- 57. Liliental J, Moon SY, Lesche R, et al. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol. 2000;10:401-4 [DOI] [PubMed] [Google Scholar]
- 58. Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29-39 [DOI] [PubMed] [Google Scholar]
- 59. Martin-Belmonte F, Gassama A, Datta A, et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Martin-Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol. 2008;20:227-34 [DOI] [PubMed] [Google Scholar]
- 61. Pinal N, Goberdhan DCI, Collinson L, et al. Regulated and polarized PtdIns(3,4,5)P3 accumulation is essential for apical membrane morphogenesis in photoreceptor epithelial cells. Curr Biol. 2006;16:140-9 [DOI] [PubMed] [Google Scholar]
- 62. Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683-710 [DOI] [PubMed] [Google Scholar]
- 63. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Suzuki A, de la Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169-78 [DOI] [PubMed] [Google Scholar]
- 67. Alimonti A, Carracedo A, Clohessy JG, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669-73 [DOI] [PubMed] [Google Scholar]
- 69. Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787-90 [DOI] [PubMed] [Google Scholar]
- 70. Taylor V, Wong M, Brandts C, et al. 5′ phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol Cell Biol. 2000;20:6860-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010;107:22231-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hodgson MC, Shao LJ, Frolov A, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477-85 [DOI] [PubMed] [Google Scholar]
- 75. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395-402 [DOI] [PubMed] [Google Scholar]
- 76. Fujita T, Doihara H, Kawasaki K, et al. PTEN activity could be a predictive marker of trastuzumab efficacy in the treatment of ErbB2-overexpressing breast cancer. Br J Cancer. 2006;94:247-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117-27 [DOI] [PubMed] [Google Scholar]
- 78. Li L, Dutra A, Pak E, et al. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol. 2009;11:9-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439-44 [DOI] [PubMed] [Google Scholar]
- 80. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 81. Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008-17 [DOI] [PubMed] [Google Scholar]
- 82. Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323-34 [DOI] [PubMed] [Google Scholar]
- 83. Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A. 1999;96:10182-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A. 1997;94:12479-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Myers MP, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336-42 [DOI] [PubMed] [Google Scholar]
- 88. Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Meuillet EJ, Mahadevan D, Berggren M, Coon A, Powis G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch Biochem Biophys. 2004;429:123-33 [DOI] [PubMed] [Google Scholar]
- 90. Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281:26562-8 [DOI] [PubMed] [Google Scholar]
- 91. Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033-8 [PubMed] [Google Scholar]
- 92. Torres J, Rodriguez J, Myers MP, et al. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3: implications for the control of protein stability and PTEN-protein interactions. J Biol Chem. 2003;278:30652-60 [DOI] [PubMed] [Google Scholar]
- 93. Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007;26:3930-40 [DOI] [PubMed] [Google Scholar]
- 95. Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100:7491-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J. 2004;379:301-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280:35195-202 [DOI] [PubMed] [Google Scholar]
- 98. Mehenni H, Lin-Marq N, Buchet-Poyau K, et al. LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet. 2005;14:2209-19 [DOI] [PubMed] [Google Scholar]
- 99. Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG. Direct identification of PTEN phosphorylation sites. FEBS Lett. 2002;528:145-53 [DOI] [PubMed] [Google Scholar]
- 100. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus: implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993-8 [DOI] [PubMed] [Google Scholar]
- 101. Valiente M, Andres-Pons A, Gomar B, et al. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem. 2005;280:28936-43 [DOI] [PubMed] [Google Scholar]
- 102. Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci U S A. 2009;106:480-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mounir Z, Krishnamoorthy JL, Robertson GP, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal. 2009;2:ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wu X, Hepner K, Castelino-Prabhu S, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci U S A. 2000;97:4233-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fine B, Hodakoski C, Koujak S, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325:1261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. He L, Ingram A, Rybak AP, Tang D. Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J Clin Invest. 2010;120:2094-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Morales FC, Takahashi Y, Momin S, Adams H, Chen X, Georgescu MM. NHERF1/EBP50 head-to-tail intramolecular interaction masks association with PDZ domain ligands. Mol Cell Biol. 2007;27:2527-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Takahashi Y, Morales FC, Kreimann EL, Georgescu MM. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J. 2006;25:910-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cardone RA, Bellizzi A, Busco G, et al. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol Biol Cell. 2007;18:1768-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dai JL, Wang L, Sahin AA, Broemeling LD, Schutte M, Pan Y. NHERF (Na+/H+ exchanger regulatory factor) gene mutations in human breast cancer. Oncogene. 2004;23:8681-7 [DOI] [PubMed] [Google Scholar]
- 112. Hayashi Y, Molina JR, Hamilton SR, Georgescu MM. NHERF1/EBP50 is a new marker in colorectal cancer. Neoplasia. 2010;12:1013-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S. EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology. 2003;38:178-86 [DOI] [PubMed] [Google Scholar]
- 114. Song J, Bai J, Yang W, Gabrielson EW, Chan DW, Zhang Z. Expression and clinicopathological significance of oestrogen-responsive ezrin-radixin-moesin-binding phosphoprotein 50 in breast cancer. Histopathology. 2007;51:40-53 [DOI] [PubMed] [Google Scholar]
- 115. Georgescu MM. NHERF1: molecular brake on the PI3K pathway in breast cancer. Breast Cancer Res. 2008;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pan Y, Weinman EJ, Dai J. NHERF1 (Na+/H+ exchanger regulatory factor 1) inhibits platelet-derived growth factor signaling in breast cancer cells. Breast Cancer Res. 2008;10:R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Georgescu MM, Morales FC, Molina JR, Hayashi Y. Roles of NHERF1/EBP50 in cancer. Curr Mol Med. 2008;8:459-68 [DOI] [PubMed] [Google Scholar]
- 118. Kreimann EL, Morales FC, de Orbeta-Cruz J, et al. Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene. 2007;26:5290-9 [DOI] [PubMed] [Google Scholar]
- 119. Sumitomo M, Shen R, Nanus DM. Involvement of neutral endopeptidase in neoplastic progression. Biochim Biophys Acta. 2005;1751:52-9 [DOI] [PubMed] [Google Scholar]
- 120. Sumitomo M, Iwase A, Navarro D, et al. Synergy in tumor suppression by direct interaction of neutral endopeptidase with PTEN. Cancer Cell. 2004;5:67-78 [DOI] [PubMed] [Google Scholar]
- 121. Siepmann M, Kumar S, Mayer G, Walter J. Casein kinase 2 dependent phosphorylation of neprilysin regulates receptor tyrosine kinase signaling to Akt. PLoS One. 2010;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Okumura K, Zhao M, Depinho RA, Furnari FB, Cavenee WK. Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc Natl Acad Sci U S A. 2005;102:2703-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117-30 [DOI] [PubMed] [Google Scholar]
- 124. Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736-42 [DOI] [PubMed] [Google Scholar]
- 125. Song MS, Carracedo A, Salmena L, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144:187-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Fan C, He L, Kapoor A, et al. PTEN inhibits BMI1 function independently of its phosphatase activity. Mol Cancer. 2009;8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Molina JR, Hayashi Y, Stephens C, Georgescu MM. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12:453-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741-4 [DOI] [PubMed] [Google Scholar]
- 129. Chagpar RB, Links PH, Pastor MC, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010;107:5471-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rabinovsky R, Pochanard P, McNear C, et al. p85 associates with unphosphorylated PTEN and the PTEN-associated complex. Mol Cell Biol. 2009;29:5377-88 [DOI] [PMC free article] [PubMed] [Google Scholar]