

Abstract
Chronic treatment with morphine results in a decrease in μ-opioid receptor sensitivity, an increase in acute desensitization, and a reduction in the recovery from acute desensitization in locus ceruleus neurons. With acute administration, morphine is unlike many other opioid agonists in that it does not mediate robust acute desensitization or induce receptor trafficking. This study compares μ-opioid receptor desensitization and trafficking in brain slices taken from rats treated for 6–7 d with a range of doses of morphine (60, 30, and 15 mg · kg−1 · d−1) and methadone (60, 30, and 5 mg · kg−1 · d−1) applied by subcutaneous implantation of osmotic minipumps. Mice were treated with 45 mg · kg−1 · d−1. In morphine-treated animals, recovery from acute [Met]5enkephalin-induced desensitization and receptor recycling was diminished. In contrast, recovery and recycling were unchanged in slices from methadone-treated animals. Remarkably the reduced recovery from desensitization and receptor recycling found in slices from morphine-treated animals were not observed in animals lacking β-arrestin-2. Furthermore, pharmacological inhibition of G-protein receptor kinase 2 (GRK2), although not affecting the ability of [Met]5enkephalin to induce desensitization, acutely reversed the delay in recovery from desensitization produced by chronic morphine treatment. These results characterize a previously unidentified function of the GRK/arrestin system in mediating opioid regulation in response to chronic morphine administration. They also suggest that the GRK/arrestin system, rather than serving as a primary mediator of acute desensitization, controls recovery from desensitization by regulating receptor reinsertion to the plasma membrane after chronic treatment with morphine. The sustained GRK/arrestin-dependent desensitization is another way in which morphine and methadone are distinguished.
Introduction
Activation of neuronal μ-opioid receptors (MORs) increases a potassium conductance in many areas of the nervous system (Williams et al., 2001). This receptor/effector pathway has been used in many studies to characterize the acute and chronic actions of opioids. After chronic treatment of animals with morphine, there is a modest shift (twofold to fourfold) in the concentration response to some opioid agonists and a decrease in the maximum response induced by others (Williams et al., 2001). Similar results were obtained when the inhibition of calcium current or the inhibition of transmitter release by opioid was measured. The aim of this study is to compare the adaptations in cellular signaling that result from the chronic treatment of animals with two distinctly different opioid agonists.
Receptor desensitization and internalization are two early events that are thought to mark the onset of tolerance at the single-cell level (Williams et al., 2001). The model for G-protein-coupled receptor (GPCR) desensitization is dependent on receptor phosphorylation by a G-protein receptor kinase (GRK) that facilitates binding of β-arrestin (βArr), AP-2, and clathrin, resulting in the targeting of this complex for internalization (Gainetdinov et al., 2004). This sequence of events is dependent on the agonist used (Blake et al., 1997; Yu et al., 1997; Borgland et al., 2003; Schulz et al., 2004; Bohn et al., 2000, 2004; Koch et al., 2005; Stafford et al., 2001; Walker and Young, 2001; Whistler et al., 1999; Finn and Whistler, 2001; Grecksch et al., 2006; Martini and Whistler, 2007).
Morphine and methadone are two opioids that have quantitatively different actions at the cellular level. Morphine is a partial agonist that is relatively poor at inducing both desensitization and internalization (Arttamangkul et al., 2008; Bailey et al., 2003). Methadone is a more efficacious and efficiently induces desensitization and internalization (Rodriguez-Martin et al., 2008). The present study examines receptor activation, desensitization, recovery from desensitization, and trafficking in locus ceruleus (LC) neurons from rats and mice after chronic treatment with morphine or methadone. When rats were treated with morphine or methadone, the concentration–response curve of [Met5]enkephalin (ME) measured in LC neurons was shifted to the right and acute desensitization was increased. The recovery from desensitization was reduced in slices from morphine-treated rats and mice, whereas the recovery from desensitization was not different from control in slices from methadone-treated animals. The recovery from desensitization was not changed in β-arrestin-2 knock-out animals that were treated with morphine. Likewise, the recovery from desensitization was the same as control in morphine-treated mice after the inhibition of GRK2. The results suggest that chronic morphine treatment reduced the recovery from desensitization by a GRK2/β-arrestin-2-dependent mechanism.
Materials and Methods
Tissue preparation.
For experiments using rats, adult (150–250 g) male Sprague Dawley rats (Charles River Laboratories) were used. Details of the method of slice preparation and recording have been published previously (Alvarez et al., 2002). Briefly, animals were anesthetized with isoflurane and killed. The brain was dissected, blocked, and mounted in a vibratome chamber to cut horizontal slices (200–260 μm thick) containing LC. Slices were stored at 35°C in an artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 d-glucose (while being continuously equilibrated with 95% O2/5% CO2). Slices were incubated for a minimum of 1 h to wash out drugs used in chronic treatment protocols that may have remained in brain tissue.
Recordings.
Slices (260 μm) were hemisected and transferred to the recording chamber (0.5 ml) where they were superfused with 35°C ACSF at a rate of 1.5 ml/min. Whole-cell recordings were made from rat LC neurons with an Axopatch 200B amplifier (Molecular Devices) in the voltage-clamp mode (cells held at −55 mV). Pipettes (1.7–2.1 MΩ) were filled with an internal solution containing the following (in mm): 115 methyl potassium sulfate, 20 NaCl, 1.5 MgCl2, 10 HEPES, 10 BAPTA, 2 Mg-ATP, 0.5 Na-GTP, and 10 phosphocreatine, pH 7.3. Intracellular recording in mouse LC were made with an Axoclamp 2A using sharp electrodes (50–60 MΩ) filled with KCl (2 m). Direct current (0–200 pA) was applied to inhibit spontaneous firing and hold the membrane potential at approximately −60 mV. Data were collected with PowerLab (Chart version 4.2.3). Analysis was performed with Prism and Kaleidagraph software. Values are presented as arithmetic mean ± SEM. Comparisons of ME-induced currents and hyperpolarizations and of acute desensitization between treatment groups was performed using one-way ANOVA, followed by Dunnett's post hoc test. The interaction between the time and extent of recovery between treatment groups was examined using two-way ANOVA, followed by Bonferroni's adjusted-α multiple comparison test to compare recovery at each time point. p values <0.05 were considered significant.
Drug treatment.
Rats were implanted with osmotic minipumps (Alzet 2ML1) to deliver morphine [National Institute on Drug Abuse (NIDA)–Neuroscience Center], methadone (NIDA–Neuroscience Center), or carrier (control). The minipumps have a 2 ml reservoir and deliver their contents for 7 d at the rate of 10 μl/h. Pumps were filled with the required concentration of drug and dissolved in water, based on the weight of the rat and the desired dosing parameter (morphine, 60, 30, and 15 mg · kg−1 · d−1; methadone, 60, 30, and 5 mg · kg−1 · d−1). Mice received 45 mg · kg−1 · d−1 of either morphine or methadone via osmotic minipumps that have a 200 μl reservoir and release at a rate of 1 μl/h for 7 d. Animals were anesthetized with isoflurane, and an incision was made in the mid-scapular region to insert the pump subcutaneously. Rats receiving 60 mg · kg−1 · d−1 of either morphine or methadone were first given intraperitoneal injections of 5 mg/kg at 9:00 A.M. and 7 mg/kg at 6:00 P.M. on day 1. On day 2, they received 7 mg/kg intraperitoneally at 9:00 A.M., and the osmotic minipump was implanted at 6:00 P.M. Animals were returned to their housing facility during recovery. Experiments were performed on day 6 or 7 after minipump implantation. Control animals consisted of naive animals and those implanted with vehicle-filled pumps.
Drug concentration analysis.
Brain and plasma samples were analyzed at the University of Utah, Center for Human Toxicology under the supervision of Dr. Roger Foltz in conjunction with NIDA. Plasma and whole brain samples were obtained for drug (morphine or methadone) concentration analysis at the time of brain slice preparation. After isoflurane anesthesia, 3 ml whole blood was obtained via cardiac puncture with a heparinized syringe. Blood was centrifuged, and plasma was collected. Brain tissue removed after blocking the LC was collected and homogenized in water. Samples were frozen at −20°C and shipped to University of Utah, Center for Human Toxicology for analysis. Samples were analyzed by liquid chromatography/tandem mass spectrometry using electrospray ionization and selected reaction monitoring. Samples from morphine-treated animals were analyzed for morphine and the metabolites morphine-3-glucuronide and morphine-6-glucuronide. The quantification range for these compounds was between 1.0 and 1000 ng/ml. Samples from methadone-treated animals were analyzed for R- and S-methadone and their respective metabolites R- and S-2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP). The quantification range for these compounds was between 2.5 and 500 ng/ml.
FlagMOR transgenic mice.
Mice were bred and genotyped as described previously (Arttamangkul et al., 2008). Data were collected from mice that were hemizygous for the FlagMOR transgene (Flag-TG/+) and backcrossed with C57BL/6 for at least five generations. Mice were also bred with MOR−/− or βArr2−/− mice to generate mice that were hemizygous for the transgene and homozygous knock-outs (Flag-TG/+MOR−/− and Flag-TG/+Arr−/−). Transgenic mice and transgenic mice that were crossed with the MOR knock-out animal were used. These mice expressed a somewhat greater number of receptors (Arttamankgul et al., 2008). The Bmax for MOR binding was twofold higher in the transgenic mouse, resulting in a fourfold shift in the concentration response curve to ME. The concentration–response curve approached that of the wild-type (WT) animals after the transgenic animal was crossed with the MOR knock-out animal. Given the small shift in the concentration–response curve to ME, it is unlikely that the small difference in the level of MOR expression has an impact on the interpretation of the results.
Receptor internalization and recycling.
Brain slices (200 μm) were incubated in ACSF containing M1 antibody (Sigma) that was conjugated to 10 μg/ml Alexa Fluor-594 (Invitrogen) for 25–45 min. Slices were visualized using an upright microscope with a custom-built two-photon apparatus. A 15 μm z-series was collected in 1 μm sections using ScanImage software. ME and calcium-free ACSF containing EGTA (0.5 mm, calcium-free) were applied by superfusion, and all experiments were performed at 35°C. A control image (C) was collected before drug application, and a second image was collected after perfusion of ME (30 μm, 10 min). This was followed by perfusion of the calcium-free solution (10 min) either immediately after washout of ME (I) or after a 30 min washout period (R). Images were analyzed offline using NIH ImageJ software. Images were z-projected using the sum-slices method. Five regions of interest were selected and averaged for background fluorescence. The average background fluorescence was subtracted from total fluorescence to yield the fluorescence intensity. Internalization was calculated as the fraction of fluorescence intensity remaining after perfusion of ME and the calcium-free solution compared with control (I/C). The total fluorescence remaining after a 30 min wash and calcium-free solution (R/C) was subtracted from the average internalization to determine the fraction of internalized receptors that recycled to the plasma membrane. One-way ANOVA was used to compare ME-induced internalization between treatment groups. Bidirectional Student's t test was used within each treatment group to compare differences in fluorescence intensity remaining after internalization and recycling.
GRK2as5 transgenic mouse.
To acutely regulate GRK2 activity in tissue, a “pseudo-knock-in” mouse that expresses a previously described analog-sensitive version of GRK2 (Kenski et al., 2005) in place of the endogenous kinase was generated. This mutant GRK2 is selectively inhibited by the nucleotide analog 1NaPP1 [1-(1,1-dimethylethyl)-3-(1-naphthalenyl)-1H-pyrazolo[3, 4-d]pyrimidin-4-amine (gift from Kevan Shokat, University of California, San Francisco, San Francisco, CA)]. The fosmid M1–1960 containing the entire murine Adrbk1 gene and conserved 5′ region was selected from the EpiFOS library (BACPAC; Children's Hospital Oakland Research Institute, Oakland, CA) for targeted mutagenesis. Mutations coding for C221V and L271G, which confer sensitivity to the small molecule inhibitor 1NaPP1 (Kenski et al., 2005), were introduced into Adrbk1 by homologous recombination using the Counter-Selection BAC Modification method (Genebridges). The modified fosmid was linearized and microinjected into C57BL/6 blastocysts (Gladstone Transgenic Core at University of California, San Francisco). Founders, identified by Southern blot and the presence of a silent mutation introducing a BstEII site in the transgene detectable by PCR amplification and restriction digest, were crossed to C57BL/6 WT animals to identify founders with germ-line transmission. Founders with endogenous levels of grk2as5 transgene expression were selected for crosses to Adrbk1+/− heterozygous knock-out mice in a C57BL/6 background (Jaber et al., 1996) (gift from Marc Caron, Duke University, Durham, NC), and additional crosses were continued until mice homozygous for the transgene in a homozygous Adrbk1−/− background were obtained. Verification that starting breeding pairs were double-homozygous mice was confirmed by mating each partner to WT mice, and all resulting offspring were double heterozygotes.
Drugs.
Drugs were applied by bath superfusion. ME, bestatin, and yohimbine were dissolved in water. 1NaPP1 and UK14304 [5-bromo-6-(2-imidazolin-2-ylamino)-quinoxaline] were dissolved in DMSO. Final concentrations of DMSO did not exceed 0.01%. For experiments with 1NaPP1, slices were incubated before recording and were also superfused with the inhibitor. Thiorphan was dissolved in ethanol (applied <0.00001%). Morphine sulfate and methadone hydrochloride used for treatment were obtained from NIDA–Neuroscience Center. All other drugs were obtained from Sigma-Aldrich.
Results
Chronic treatment with morphine and methadone in rats
Chronic treatment of rats with morphine produces tolerance that results in a twofold rightward shift of the dose–response curve to DAMGO (d-Ala2-N-Me-Phe4-glycol5-enkephalin) in locus ceruleus neurons (Christie et al., 1987). Concentration–response curves measuring the outward current were constructed for ME to determine whether treatment of rats with morphine or methadone resulted in comparable tolerance in LC neurons. Osmotic minipumps containing either morphine (60 mg · kg−1 · d−1) or methadone (60 mg · kg−1 · d−1) were implanted for 6 d, and the outward current induced by various concentrations of ME were measured (Fig. 1A,B). The ME-stimulated current was normalized to the peak current induced by the α-2 adrenoceptor agonist UK14304 (3 μm), which couples to the same population of G-protein-linked inwardly rectifying potassium channels. In rats that were opiate naive (control), the maximum ME-induced current was 145 ± 8% of the peak UK14304-induced current. The maximum current was not different after chronic morphine or chronic methadone treatment (153 ± 8 and 144 ± 7%, respectively). The concentration of ME that produced a half-maximal outward current (EC50) in slices from control was 281 ± 47 nm. Slices from chronic morphine-treated rats showed a twofold right shift in the ME concentration–response curve (578 ± 85 nm) compared with control, consistent with what has been reported previously after treatment of rats using morphine pellets (Christie et al., 1987). Chronic methadone treatment also resulted in a twofold right shift (500 ± 78 nm), suggesting that the doses of morphine and methadone used to treat rats produced a similar amount of cellular tolerance.
Figure 1.

Chronic morphine and methadone treatment resulted in reduced potency of ME in rat LC neurons. A, Representative trace showing outward currents produced by perfusion of varying concentrations of ME. At the end of the experiment, the outward current produced by UK14304 (3 μm) was measured and reversed by yohimbine (3 μm; Yoh). B, Concentration–response curves for ME in slices from untreated, chronic morphine (60 mg · kg−1 · d−1) and chronic methadone (60 mg · kg−1 · d−1) treated rats. Outward currents induced by ME (30 nm to 10 μm) were normalized to the peak current induced by UK14304. All points are mean ± SE. The EC50 values from control (281 ± 47 nm), chronic morphine (578 ± 85 nm), and chronic methadone (500 ± 78 nm).
After chronic treatment of rats with morphine, ME-induced receptor desensitization measured by the decline in the amplitude of the hyperpolarization was increased (Dang et al., 2004). To determine whether chronic methadone treatment produced a similar adaptation, ME-induced desensitization of outward currents was measured in slices from rats that were implanted with morphine or methadone pumps. To induce desensitization, a saturating concentration of ME (30 μm) was perfused, and the peak and decline in the outward current was measured during a 10 min application. In control slices, the outward current that remained after 10 min was 65 ± 2% of the initial peak amplitude (Fig. 2A). The outward current declined to 53 ± 2 and 54 ± 2% of the initial peak current after chronic morphine and chronic methadone treatment, respectively (Fig. 2B,C). Therefore, treatment with both morphine and methadone increased the amount of ME-induced acute desensitization (one-way ANOVA, F(2,22) = 8.067, p = 0.002) (Fig. 2D) and shifted the concentration–response curve.
Figure 2.

Enhanced desensitization observed after chronic treatment of rats with morphine or methadone. A–C, Representative traces showing the decline in the outward current during a 10 min application of a saturating concentration of ME (30 μm). The peak current (dashed line) declined during the 10 min application to a desensitized amplitude (double arrows). D, Bars represent the mean percentage of peak current ± SE in control (black), morphine treated (gray), and methadone treated (white). n is indicated in bars. One-way ANOVA followed by Dunnett's post hoc test showed a significant difference between morphine treated and methadone treated vs control (*p < 0.01).
Recovery from ME-induced desensitization was examined in slices from morphine- and methadone-treated rats by comparing the current amplitude resulting from a pulse of ME (300 nm) applied before and after desensitization. The amplitude of the ME (300 nm) current 5 min after desensitization was 22 ± 3% of the prepulse amplitude in control slices (Fig. 3A). The test pulse of ME (300 nm) was applied again at 15, 25, 35, and 45 min. In slices from untreated rats, the current recovered to 71 ± 5% of the prepulse amplitude after 45 min. After chronic morphine treatment, the ME-induced current measured 45 min after desensitization was 42 ± 4% of prepulse (Fig. 3B). In slices taken from animals that were treated chronically with methadone, the current recovered to 67 ± 2% of the prepulse amplitude after 45 min (Fig. 3C). Two-way ANOVA showed no interaction between time and treatment group (F(8,99) = 1.49, p = 0.17), suggesting that the rate of recovery was not significantly different between treatment groups. However, a significant reduction in the extent of recovery from desensitization at 45 min was observed after chronic morphine treatment (p < 0.001) but not after chronic methadone treatment (p > 0.05). Thus, chronic treatment with morphine resulted in a unique adaptive change that prevented recovery of MOR function that did not occur after chronic methadone treatment (Fig. 3D).
Figure 3.

Recovery from desensitization is impaired in slices from mice treated with chronic morphine but not methadone. A, Representative traces of outward currents in slices from untreated mice. ME (300 nm) was perfused before desensitizing with ME (30 μm, 10 min). ME (300 nm) was tested again 5 min after washout of the desensitizing concentration, and the amplitude of the current was compared with the prepulse amplitude (dashed line). ME (300 nm) was tested again at 15, 25, 35, and 45 min to measure recovery from desensitization (percentage of prepulse amplitude). At the end of the experiment, the current induced by UK14304 (3 μm; UK) was determined and reversed with yohimbine (3 μm; Yoh). B, C, Representative traces from morphine-treated and methadone-treated rats. D, Summary of recovery from desensitization (percentage of prepulse amplitude). □, Control; ○, morphine treated; ♦, methadone treated. Error bars represent SEM (n). Two-way ANOVA with Bonferroni's post hoc comparison (*p < 0.001).
To ensure that the observed differences between chronic morphine and methadone treatment was not dependent on the dose of drug, several doses of each agonist were used for treatment, and recovery from desensitization was examined. Chronic morphine treatment had no effect on the rate of recovery from desensitization (F(16,133) = 1.18, p = 0.29) but resulted in reduced recovery from desensitization after 45 min at all doses tested (p < 0.01) (Fig. 4A). The rate (F(12,144) = 0.40, p = 0.96) and the extent (p > 0.05) of recovery from desensitization after methadone treatment were not different from control at all doses tested (Fig. 4B). Drug levels in brain and plasma for the drugs were measured for the various doses and are presented in Table 1. Morphine brain concentrations as low as 9 ± 1 ng/ml were sufficient to cause changes in desensitization and recovery.
Figure 4.

The recovery from desensitization after morphine treatment of rats was independent of the dose but required more than 1 d of treatment. A, Plot of the recovery from desensitization, as a function of time, in slices taken from rats treated with morphine. When rats were treated for 6–7 d with different doses of morphine, the rate and extent of recovery from desensitization was reduced compared with untreated rats. In slices taken from rats given two injections of morphine on a single day, the recovery from desensitization was not different from untreated animals. B, In slices taken from rats treated with different doses of methadone, the rated and extent of recovery from desensitization was not different from untreated. Error bars represent SEM. Two-way ANOVA with Bonferroni's post hoc comparison (*p < 0.05).
Table 1.
The concentration (nanograms per milliliter) of morphine, methadone, and metabolites measured in plasma and brain samples taken from rats after a 6–7 d treatment period
| Dose | Morphine | M-3-G | M-6-G | |
|---|---|---|---|---|
| Morphine-treated | ||||
| Plasma | 60 | 317 ± 21 | >LOQ | 90 ± 20 |
| 15 | 65 ± 3 | 505 ± 85 | 28 ± 5 | |
| Brain | 60 | 36 ± 2 | 14 ± 2 | 3 ± 1 |
| 15 | 9 ± 1 | 3 | <LOQ |
| Dose | R-Methadone | S-Methadone | R-EDDP | S-EDDP | |
|---|---|---|---|---|---|
| Methadone-treated | |||||
| Plasma | 60 | 101 ± 6 | 140 ± 9 | 18 ± 1 | 2 ± 1 |
| 5 | 9 ± 1 | 8 ± 1 | <LOQ | <LOQ | |
| Brain | 60 | 150 ± 11 | 197 ± 11 | 4 | 4 |
| 5 | 9 | 12 ± 1 | <LOQ | <LOQ | |
Morphine doses of 60 and 15 mg · kg−1 · d−1 (n = 10 and 8, respectively); methadone doses of 60 and 5 mg · kg−1 · d−1 (n = 13 and 6, respectively) were delivered via osmotic minipump. Data are expressed as mean values ± SEM. Level of quantification (LOQ) for morphine, 1.0–1000 ng/ml; range of quantification for methadone, 2.5–500 ng/ml. M-3-G, Morphine-3-glucuronide; M-6-G, morphine-6-glucuronide.
To determine whether acute morphine was sufficient to induce the same changes in MOR desensitization and recovery as chronic treatment, rats were given two 10 mg/kg intraperitoneal injections of morphine, 9 h apart, and experiments were performed 24 h after the first injection. After ME-induced desensitization, the current induced by a test pulse of ME (300 nm) was 22 ± 2% of the prepulse at 5 min and recovered to 76 ± 4% at 45 min (Fig. 4A). Thus, recovery was not different from control (p > 0.05), indicating that 1 d of treatment was not sufficient to produce the changes observed by chronic treatment for 6 d.
Chronic treatment in mouse
Desensitization and recovery
Receptor trafficking is an important aspect of MOR regulation that may also be differentially regulated by chronic opioid treatment. To directly monitor trafficking of MOR, a transgenic mouse with the tyrosine hydroxylase promotor driving expression of a Flag-tagged MOR was used. Intracellular recording of membrane potential was used to measure the hyperpolarization induced by ME. In slices from these transgenic mice, ME (100 nm) produced a hyperpolarization of 16.8 ± 2.9 mV. The maximum hyperpolarization induced by ME (30 μm) was 32.3 ± 1.3 mV and declined to 77 ± 3% of the initial peak amplitude during the 10 min application (Fig. 5A). The test pulse of ME (100 nm) applied 5 min after desensitization was 27.9 ± 5% of the initial prepulse amplitude. Test pulses were applied again after 10, 20, and 30 min. After 30 min, the test pulse amplitude had recovered to 92.5 ± 3.3% of the prepulse amplitude.
Figure 5.

Recovery from desensitization impaired after chronic morphine treatment in Flag-TG/+ mice. A, Representative trace of membrane potential in slices from an untreated Flag-TG/+ mouse. ME (100 nm) was perfused before and 5, 10, 20, and 30 min after desensitizing with a saturating concentration of ME (30 μm). After 30 min, recovery from desensitization was nearly complete (dashed line). Representative trace of desensitization and recovery in slices from morphine-treated (B) and methadone-treated (C) mice. D, Summary of recovery from desensitization (percentage of prepulse amplitude). □, Control; ○, morphine treated; ◆, methadone treated. Error bars represent SEM (n). Two-way ANOVA with Bonferroni's post hoc comparison (*p < 0.05).
Desensitization and recovery from desensitization were measured in transgenic mice (Flag-TG/+) and in Flag-TG/+ mice lacking the endogenous receptor (Flag-TG/+MOR−/−). In slices from animals treated with morphine (45 mg · kg−1 · d−1, 6 or 7 d), the amplitude of the hyperpolarization produced by 100 nm ME was 16.5 ± 2.4 mV (t(11) = 0.08 p = 0.94) and 30 μm ME was 28.6 ± 1.9 mV (t(12) = 1.654, p = 0.124), not different from untreated mice. During the application of 30 μm ME, the hyperpolarization declined to 74.3 ± 3.7% (t(11) = 0.50, p = 0.62) of the maximum (Fig. 5C). Similar to what was observed in rat, the rate of recovery from desensitization was not altered by chronic morphine treatment (F(6,58) = 1.74, p = 0.13), but the extent of recovery was incomplete after 30 min (70.9 ± 5.5% of prepulse p < 0.01) (Fig. 5B).
Chronic treatment with methadone was performed only in mice lacking the endogenous receptor (Flag-TG/+MOR−/−, 45 mg · kg−1 · d−1, 6 or 7 d). This allowed the use of a high dose of methadone without special precautions to limit lethality. Results from methadone-treated mice were consistent with what was observed in the rat. The maximum hyperpolarization induced by ME was 27.8 ± 1.6 mV (t(10) = 2.27, p = 0.06) and declined to 77.1 ± 2% (t(9) = 0.08, p = 0.94) of the maximum during the 10 min application (Fig. 5D). Both the time course and the extent of recovery from desensitization in slices from chronic methadone-treated mice were not different from untreated (Fig. 5B) (p > 0.05). In summary, chronic treatment with morphine and methadone had no effect on the acute ME-induced desensitization as measured by the decline in the maximum hyperpolarization or by the more sensitive measure of comparing the amplitude of the hyperpolarization induced by a nonsaturating concentration of ME 5 min after desensitization. Chronic morphine treatment did, however, result in a reduced recovery from desensitization that was not observed after chronic methadone treatment, confirming that only morphine induced adaptive changes to MOR regulation.
Internalization and recycling
Receptor trafficking after chronic opioid treatment was examined using LC slices from Flag-TG/+ mice prepared as for electrophysiology experiments. Extracellular N-terminally tagged MORs were labeled with an M1 anti-Flag antibody conjugated to Alexa Fluor-594 (M1–A594) and imaged using a two-photon microscope. Initially, M1–A594 labeling was localized primarily to the plasma membrane of LC neurons. Application of ME (30 μm) for 10 min resulted in an increase in intracellular puncta that remained after stripping receptors from the plasma membrane with EGTA-buffered calcium-free solution (Fig. 6A). ME-induced internalization was quantified as the percentage of total fluorescence remaining after stripping the extracellular labeling compared with the initial control image. In untreated mice, ME induced internalization of 52.3 ± 2.6% of labeled receptors. In slices taken from animals that were treated chronically with morphine or methadone, ME-induced internalization was 58.2 ± 5 and 56.2 ± 3.5%, not different from untreated controls (one-way ANOVA, F(2,25) = 0.626, p = 0.54) (Fig. 6C).
Figure 6.

Internalization and recycling of FlagMOR in live brain slices. A, Representative images of ME-induced internalization. Control (C) is initial staining with an Alexa Fluor-594-conjugated anti-Flag M1 antibody before agonist treatment. Internalization was induced with a 10 min perfusion of ME (30 μm). Immediately after washout of ME, calcium-free EGTA solution was perfused to remove M1–A594 from receptors on plasma membrane (I). B, Control (C) image of initial staining. Internalization was induced by ME, and, after a 30 min wash in normal ACSF, some receptors recycled to plasma membrane. Ca-free EGTA solution was then applied to strip antibody from the plasma membrane (R). C, Summarized data of total fluorescence remaining after perfusion of Ca-free EGTA solution immediately after internalization was induced (I/C) or after 30 min (R/C) in slices from untreated (black), morphine-treated (gray,) and methadone-treated (white) mice. Dashed line indicates percentage fluorescence intensity remaining in drug-free experiment. Error bars represent SEM. I/C and R/C were compared within treatment groups using Student's t test (*p < 0.05).
Recycling of internalized receptors is thought to be important for recovery from desensitization. To examine recycling, normal ACSF was perfused for 30 min after washout of the ME (30 μm) solution. As described previously, this time point allowed near complete recovery from desensitization. After the 30 min wash, a redistribution of fluorescence labeling to the plasma membrane occurred, and this was removed using the calcium-free solution (Fig. 6B). In slices from untreated mice, the total intracellular fluorescence remaining after 30 min (R/C) was 34.1 ± 1.1%, significantly less than the intracellular fluorescence immediately after ME-induced internalization (t(26) = 7.55, p < 0.0001) (Fig. 6C). In slices from mice treated chronically with morphine, there was no significant difference in the intracellular fluorescence remaining immediately after internalization and 30 min later (58.2 ± 5 vs 49.7 ± 3.5%), suggesting that few receptors had recycled in the 30 min washout period (t(29) = 1.37, p = 0.18). In slices from mice treated chronically with methadone, 40 ± 2.5% of the total intracellular fluorescence remained after 30 min, which was significantly less than the fluorescence immediately after internalization (t(17) = 3.88, p < 0.001) (Fig. 6C). Therefore, chronic morphine treatment decreased the trafficking of receptors back to the plasma membrane, which was not observed after chronic treatment with methadone.
Role of arrestin in morphine-induced adaptations
β-Arrestin-2 has been identified as a molecule important for MOR regulation in cell lines and is implicated in contributing to morphine tolerance in vivo (Bohn et al., 2000). Desensitization and the recovery from desensitization were examined in FlagMOR mice that were crossed with β-arrestin-2 knock-out mice. The hyperpolarization produced by ME (100 nm) in untreated Flag-TG/+Arr−/− mice (13.7 ± 1.1 mV, t(15) = 1.16, p = 0.26) was not different from Flag-TG/+ mice, but the maximum hyperpolarization produced by a saturating concentration of ME was slightly less (28.4 ± 0.8 mV, t(16) = 2.76, p = 0.014) (Fig. 7A). As reported previously, the decline in the hyperpolarization during a 10 min application of ME (30 μm) was not affected by loss of β-arrestin-2 (79.1 ± 2% of peak, t(14) = 0.53, p = 0.61) (Arttamangkul et al., 2008). ME (100 nm) tested 5 min after desensitization produced a hyperpolarization that was 32.8 ± 2% of the initial prepulse amplitude and recovered to 94.4 ± 7% after 30 min. Therefore, desensitization and complete recovery were observed in the absence of β-arrestin-2.
Figure 7.

Reduced recovery from desensitization in chronic morphine-treated animals requires βArr2; experiments were performed in FlagMOR–βArr2−/− mice. A, Representative traces showing desensitization and recovery in an untreated (top) and morphine-treated (bottom) mouse. Dashed lines represent prepulse amplitude of the hyperpolarization produced by ME (100 nm). B, Summary of recovery (percentage of prepulse amplitude) for untreated (♦) and morphine-treated (•) mice. Two-way ANOVA with Bonferroni's post hoc comparison (*p < 0.05). KO, Knock-out.
Chronic morphine treatment in Flag-TG/+Arr−/− mice did not alter ME-induced hyperpolarization (12.6 ± 1.8 mV, t(15) = 0.55, p = 0.59 and 29.9 ± 1.3 mV, t(17) = 1.07, p = 0.3 for 100 nm and 30 μm, respectively) compared with untreated Flag-TG/+Arr−/−, nor did it affect the decline from the peak during a 10 min application (75.6 ± 4% of peak, t(13) = 0.75, p = 0.47). Chronic morphine treatment had no effect on the rate (F(3,42) = 0.2, p = 0.9) or the extent (p > 0.05) of recovery from ME-induced desensitization (Fig. 7B,C). Therefore, the reduced recovery from desensitization observed in rats and Flag-TG/+ mice after treatment with morphine was absent in slices from morphine-treated Flag-TG/+Arr−/− mice.
Internalization and recycling were compared in untreated and morphine-treated Flag-TG/+Arr−/− mice. Despite the role of arrestin in mediating receptor endocytosis, no change in internalization was observed in untreated Flag-TG/+Arr−/− mice relative to untreated Flag-TG/+ mice (59.3 ± 5.8%, t(15) = 1.22, p = 0.24). Likewise, internalization was not affected in slices from mice that were treated chronically with morphine (54.9 ± 4.5%, t(16) = 0.6 p = 0.56) (Fig. 8A,B). In slices from untreated and treated Flag-TG/+ Arr−/−, there was significantly less fluorescence remaining (39.8 ± 4.6 and 37.4 ± 3.3%, respectively) when Ca-free solution was perfused after 30 min compared with immediately after ME-induced internalization, suggesting that receptor recycling occurred in both groups (untreated, t(17) = 2.63, p = 0.02; morphine, t(27) = 3.17, p = 0.004) (Fig. 7C). Thus, in Flag-TG/+Arr−/− mice that were treated chronically with morphine, the recovery from desensitization and receptor recycling were the same as in untreated animals. These results suggest that adaptations in the processes that underlie the reduced recycling of receptors to the plasma membrane induced by chronic morphine treatment are β-arrestin-2 dependent.
Figure 8.

Internalization and recycling are not altered by chronic morphine treatment in FlagMOR-βArr2−/− mice. A, Internalization induced by ME. Control (C) is initial staining with an Alexa Fluor-594-conjugated anti-Flag M1 antibody before agonist treatment. Internalization was induced with a 10 min perfusion of ME (30 μm) and was followed immediately by Ca-free EGTA to remove M1–A594 from receptors on plasma membrane (I). B, Recycling of receptors after ME-induced internalization. Control (C) is initial staining, and internalization was induced with ME (30 μm). After a 30 min wash in normal ACSF, some receptors recycled to plasma membrane and Ca-free EGTA was applied to remove M1–A594 from receptors on plasma membrane. C, Summarized data of total fluorescence remaining after Ca-free EGTA solution was applied immediately after ME-induced internalization (I/C) and after 30 min wash (R/C) in untreated (white) and morphine-treated (black) Flag-TG/+Arr−/− slices. Error bars represent SEM, and Student's t test was used to compare I/C with R/C within groups (*p < 0.05).
GRK2 inhibition
Phosphorylation of GPCRs by GRK contributes to desensitization and facilitates recruitment of β-arrestin-2 (Gainetdinov et al., 2004). Desensitization and the recovery from desensitization were examined after inhibition of GRK2 to determine whether phosphorylation by GRK2 is required for MOR desensitization and the adaptations produced by chronic morphine treatment. A pseudo-knock-in mouse expressing a mutant GRK2 (GRK2as5) that is specifically inhibited by 1NaPP1 was used for these experiments. In the absence of 1NaPP1, the kinase activity of GRK2as5 is maintained (Kenski et al., 2005). Concentration–response curves for ME were constructed using slices from untreated GRK2as5 mice in the absence and presence of 1NaPP1. In the absence of 1NaPP1, ME produced a maximal membrane hyperpolarization of 28 ± 1.7 mV with an EC50 of 1.1 ± 0.51 μm (Fig. 9A). The maximum hyperpolarization in slices incubated with 1NaPP1 was not different (27.2 ± 1.1 mV, t(15) = 0.41, p = 0.69), and the EC50 was 917 ± 100 nm. This is similar to what has been reported in wild-type mice (Torrecilla et al., 2008).
Figure 9.
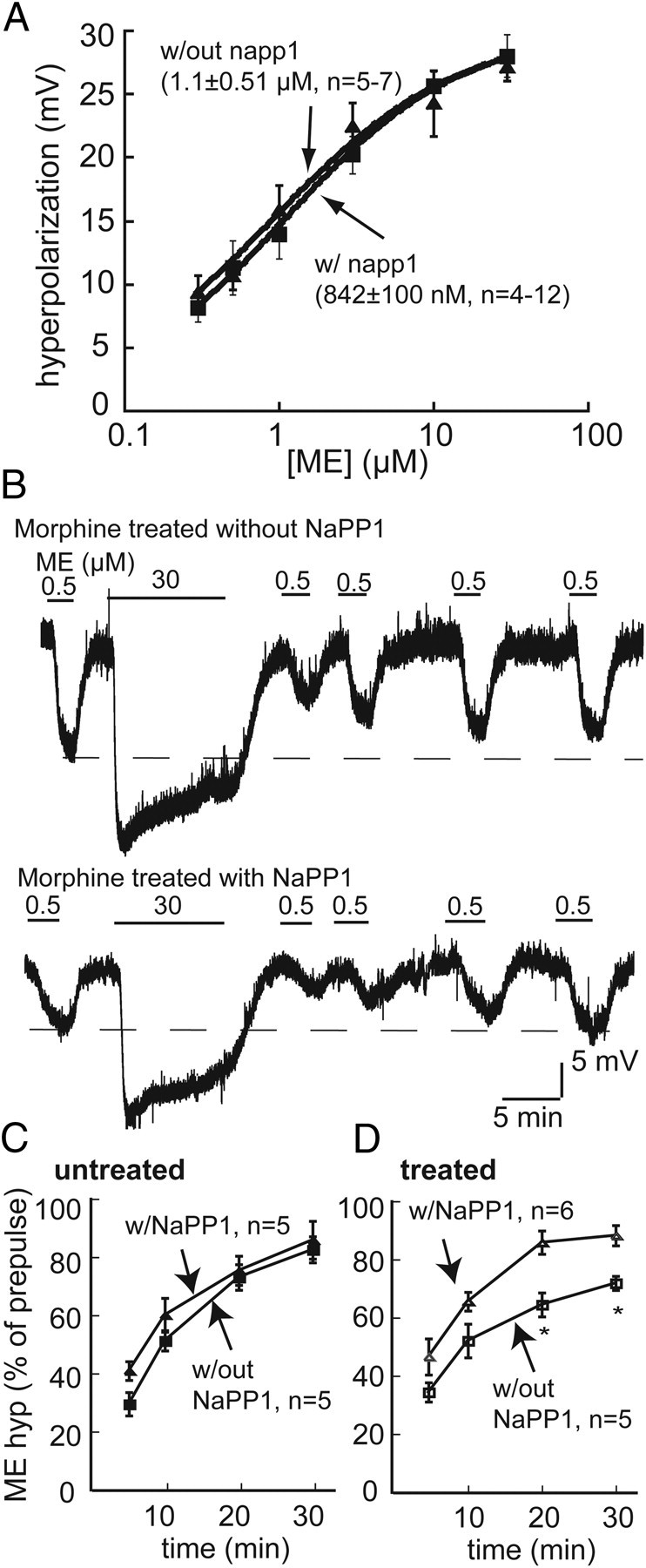
Inhibition of GRK2 with 1NaPP1 in the GRK2as5 mice reverses the effects of chronic morphine treatment on recovery from desensitization. A, Concentration–response curve for ME constructed from average membrane hyperpolarizations in slices from GRK2as5 mouse in the absence (■) or presence (▴) of 1NaPP1 (10 μm). B, Representative recordings in slices from morphine-treated GRK2aa5 mice without 1NaPP1 (top) and slices incubated with 1NaPP1 (bottom; 10 μm, 30 min) before desensitizing with ME. C, Summary of recovery (percentage of prepulse amplitude) in slices from untreated GRKas5 without (■) or with (▴) 1NaPP1. D, Summary of recovery in slices from morphine-treated GRKas5 without (□) and with (▵) 1NaPP1. Error bars represent SEM. Two-way ANOVA with Bonferroni's post hoc comparison (*p < 0.05).
To determine whether GRK2-dependent phosphorylation is required for desensitization and the recovery from desensitization, experiments were performed in either the absence or presence of the inhibitor 1NaPP1. In experiments without 1NaPP1, ME (500 nm) produced a membrane hyperpolarization of 11.3 ± 2.1mV. The maximum hyperpolarization produced by ME (30 μm, 28 ± 1.7 mV) declined to 74.3 ± 2.1% of the peak during the 10 min application. After a 5 min wash, ME (500 nm) produced a hyperpolarization that was 29.6% of the prepulse amplitude, and, after 30 min, the amplitude of the hyperpolarization was 82.6 ± 4.5% of the initial value. In slices that were incubated in 1NaPP1 (10 μm, 30 min) to inhibit GRK2, the hyperpolarization produced by ME (500 nm) was (10.7 ± 1.1 mV), and the hyperpolarization produced by ME (30 μm) was 26.9 ± 1.2 mV. In slices from untreated GRK2as5 animals, there was no effect of 1NaPP1 on ME-induced desensitization (76.4 ± 3.4% of peak amplitude, t(9) = 1.02, p = 0.34) or the time course (F(3,34) = 0.41, p = 0.75) and extent of recovery from desensitization (p > 0.05) (Fig. 9C). Thus, desensitization and the recovery from desensitization were independent of GRK2 activity in untreated mice.
After chronic morphine treatment, desensitization induced by ME was not affected by incubation with 1NaPP1 (74.3 ± 2.1% without and 67.8 ± 2.8% with 1NaPP1, t(8) = 1.64, p = 0.14). The test pulse amplitude 30 min after washing out the high concentration of ME was 72.3 ± 2.4% of the prepulse in the absence of 1NaPP1, and, in slices that were incubated in 1NaPP1, the recovery from desensitization had a similar rate (F(3,34) = 0.85, p = 0.48) but was more complete (93.1 ± 5.2% of prepulse, p < 0.01) than slices from morphine-treated animals in the absence of 1NaPP1 (Fig. 9B,D). Therefore, the activity of GRK2 was required to decrease the extent of recovery from desensitization after chronic morphine treatment.
Discussion
Acute morphine and methadone differentially affect receptor activation, desensitization, and internalization (Whistler et al., 1999; Alvarez et al., 2002; Arttamangkul et al., 2008). The results of this study confirm that chronic treatment with morphine decreased recovery from acute desensitization. In addition, an inhibition of receptor recycling was observed after chronic morphine treatment, an effect that was not present after chronic treatment with methadone, or in morphine-treated β-arrestin-2 knock-out mice. Likewise, inhibition of GRK2 in slices taken from morphine-treated mice resulted in complete recovery from desensitization. The results suggest that, after chronic treatment with morphine, β-arrestin-2 and the kinase activity of GRK2 prevent or slow the normal recovery from acute receptor desensitization, and this adaptive change is agonist dependent.
Agonist selectivity and chronic opioid adaptations
The reduced recovery from desensitization and recycling was unique to treatment with morphine, required more than a 24 h exposure, and was not induced by methadone treatment. Plasma and brain drug levels rule out failure of delivery from osmotic pumps as a source for these differences that were maintained over a wide range of doses (Table 1). There were some similarities in the adaptations produced by chronic morphine and methadone treatment. First, the concentration–response curves for ME-induced outward current in slices from rats were shifted twofold to the right after treatment with both agonists, suggesting a similar amount of cellular tolerance by treatment with morphine and methadone.
Adaptations induced by chronic morphine treatment have been studied extensively in rat locus ceruleus. The present study extends this work by describing agonist selectivity induced by chronic opioid regulation by morphine and methadone in mice, thus making it practical to apply transgenic and knock-out models to investigate mechanism. Although the overall observations found in rat and mouse experiments were similar, differences exist based on the method of recording. The studies performed in rat used whole-cell voltage-clamp recordings. In voltage clamp, the peak outward current was not dependent on the driving force for potassium ions, such that the peak current was a better reflection of the maximum activation of potassium conductance. Intracellular recording of membrane potential were used in the mouse because the amplitude of the outward current measured with voltage clamp was small whereas the amplitude of the hyperpolarization induced by ME was large; therefore, measurement of the recovery from desensitization was more accurate. The peak hyperpolarization induced by ME was, however, blunted because the membrane potential approached the potassium equilibrium potential. The same results have been found using the recording of membrane potential in the rat, indicating that the difference in desensitization measured with the two configurations was not dependent on species (Harris and Williams, 1991; Osborne and Williams 1995; Dang and Williams, 2004; Arttamangkul et al., 2008; Dang et al., 2009). There was also a difference in the extent of recovery between the two methods. The extent of recovery was always greater with the use of intracellular recording in recordings from both rats and mice (Harris and Williams, 1991; Osborne and Williams, 1995; Dang and Williams, 2004; Arttamangkul et al., 2008). This probably results from the dialysis of the intracellular milieu into the whole-cell pipette. Having validated the basic observation in the mouse, several genetic manipulations were applied to investigate the basis of the adaptations induced by chronic morphine treatment.
Endocytosis and recycling
The use of the FlagMOR transgenic animal allowed the analysis of receptor internalization and recycling. There was a correlation between the decreased recovery from desensitization, measured electrophysiologically, and receptor recycling assessed by fluorescence microscopy. This suggests that endocytic recycling of receptors contributes to the recovery from desensitization. The endocytic trafficking of opioid receptors has been proposed to be an important process in the development of tolerance among different opioid agonists (Koch et al., 2005; Martini and Whistler, 2007; Koch and Höllt, 2008). The present results are consistent with this general concept and reveal an unexpected role of the GRK/arrestin system specifically in response to chronic treatment with morphine.
Role of β-arrestin-2
Both desensitization and internalization were unaffected in the β-arrestin-2 knock-out animals. Although the expression of β-arrestin-1 in LC neurons may compensate for the loss of β-arrestin-2, there is increasing evidence that opioid receptor desensitization occurs independent of β-arrestin-2 (Walwyn et al., 2007; Arttamangkul et al., 2008; Dang et al., 2009). The results do, however, demonstrate that β-arrestin-2 affects components of the desensitization/internalization pathway in LC. After chronic morphine, the reduction in recovery and recycling was lost in mice lacking β-arrestin-2. Given that morphine is generally less effective than many agonists at the induction of arrestin translocation and receptor internalization, the role of β-arrestin-2 in morphine-induced adaptations is somewhat surprising. This could result from a compensatory mechanism induced by continued signaling, which affects the ability of receptors to recycle after ME-induced activation and endocytosis.
Role of GRK2
Inhibition of GRK2as5 with 1NaPP1 in slices from mice treated chronically with morphine resulted in complete recovery from desensitization. These results are consistent with the results obtained from the β-arrestin-2 knock-out animals, based on the present view that GRKs and arrestins function in series (Gainetdinov et al., 2004). It was remarkable that acute inhibition of GRK2 reversed the delayed recovery from desensitization induced by chronic morphine, because GRK2 was active throughout the morphine treatment period. It was also surprising that inhibition of GRK2 with 1NaPP1 had no effect on ME-induced receptor desensitization measured in slices from untreated or treated mice. This suggests that the major function of GRK2 activity, under the present experimental conditions, is to regulate recovery from, rather than induction of, desensitization. There is, however, evidence that GRK(s) contribute to desensitization based on the use of an inhibitory peptide in HEK293 cells and LC neurons (Johnson et al., 2006; Bailey et al., 2009b). There is also evidence that additional kinases contribute to ME-induced desensitization (Dang et al., 2009). Thus, although multiple mechanisms may induce opioid receptor desensitization, the present results indicate that GRK2 and β-arrestin-2 function specifically to control the recovery from desensitization.
Tolerance at the cellular and systems level
The relatively modest effects on opioid signaling measured at the single-cell level are surprising given the profound tolerance that can be observed in various physiological and behavioral assays in vivo after chronic treatment with opioids (Christie 2008; Gintzler and Chakrabarti 2008). These observations suggest that the mechanisms that underlie the development of tolerance to opioids involve cellular adaptive changes that may be subtle but lead to systems level adaptations that can vary considerably depending on the system as well as the agonist under study. There is no reason to think that cellular tolerance in the LC impacts analgesic tolerance. However, cellular tolerance of a similarly modest level has been reported in the periaqueductal gray after chronic morphine administration (Bagley et al., 2005). Conversely, there is an approximate fourfold shift in the concentration–response curve to normorphine in the guinea pig ileum from morphine-treated animals (Chavkin and Goldstein, 1984), yet the constipation induced by morphine is generally resistant to the development of tolerance (Ross et al., 2008). It thus appears that no single adaptive process, or cellular adaptation occurring in any single population of neurons, can account for the full range of behavioral measures that are found after chronic opioid treatment (Christie 2008, Gintzler and Chakrabarti 2008, Kieffer and Evans 2002).
The present study examined the sensitivity of LC neurons to opioids after the washout of the morphine that was initially present in the brain slices. Thus, the slices were in a state of acute withdrawal. Enhanced desensitization and reduced recovery persisted for hours after washout of morphine, but the state of withdrawal has been shown to alter normal cellular and synaptic physiology in two ways. First, there are numerous studies indicating that acute withdrawal results in a rebound increase in transmitter release (for review, see Williams et al., 2001). The increased transmitter release is sensitive to reapplication of opioids such that the opioid inhibition of transmitter release in withdrawn slices is larger than in slices from untreated animals. Second, when slices from morphine-treated animals were maintained in morphine, the sensitivity of LC neurons to morphine was significantly decreased (Bailey et al., 2009a). Placing slices from morphine-treated animals in morphine-free solutions resulted in a significant increase in the current induced by morphine. Thus, acute opioid withdrawal can have multiple receptor-dependent actions.
Summary
Agonist selective actions of acutely applied opioids have been extensively described. The present study identifies an agonist-selective regulation that occurs after chronic opioid administration. The adaptation found in the present study differs from previously described agonist selective actions because it is selectively induced by morphine and affects the recovery from desensitization rather than its induction. The present results also identify a previously unrecognized function of the GRK/arrestin system that may contribute to the complex sequence of events resulting from chronic morphine treatment.
Footnotes
This work was supported by National Institutes of Health Grants DA023793 (N.Q.), DA020972 (E.K.L.), DA08163 (J.T.W.), and DA012864 (M.v.Z.).
References
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, Williams JT. mu-Opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Quillinan N, Low MJ, von Zastrow M, Pintar J, Williams JT. Differential activation and trafficking of micro-opioid receptors in brain slices. Mol Pharmacol. 2008;74:972–979. doi: 10.1124/mol.108.048512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley EE, Chieng BC, Christie MJ, Connor M. Opioid tolerance in periaqueductal gray neurons isolated from mice chronically treated with morphine. Br J Pharmacol. 2005;146:68–76. doi: 10.1038/sj.bjp.0706315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G. μ-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci. 2003;23:10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, Henderson G. Role of protein kinase C and μ-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009a;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, McArdle CA, Smith FL, Dewey WL, Kelly E, Henderson G. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009b;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake AD, Bot G, Li S, Freeman JC, Reisine T. Differential agonist regulation of the human kappa-opioid receptor. J Neurochem. 1997;68:1846–1852. doi: 10.1046/j.1471-4159.1997.68051846.x. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- Chavkin C, Goldstein A. Opioid receptor reserve in normal and morphine-tolerance guinea pig ileum myenteric plexus. Proc Natl Acad Sci U S A. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addition. Br J Phamacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- Dang VC, Williams JT. Chronic morphine treatment reduces recovery from opioid desensitization. J Neurosci. 2004;24:7699–7706. doi: 10.1523/JNEUROSCI.2499-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Napier IA, Christie MJ. Two distinct mechanisms mediate acute mu-opioid receptor desensitization in native neurons. J Neurosci. 2009;29:3322–3327. doi: 10.1523/JNEUROSCI.4749-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gintzler AR, Chakrabarti S. The ambiguities of opioid tolerance mechanisms: barriers to pain therapeutics or new pain therapeutic possibilities. J Pharmacol Exp Ther. 2008;325:709–713. doi: 10.1124/jpet.107.135533. [DOI] [PubMed] [Google Scholar]
- Grecksch G, Bartzsch K, Widera A, Becker A, Höllt V, Koch T. Development of tolerance and sensitization to different opioid agonists in rats. Psychopharmacology. 2006;186:177–184. doi: 10.1007/s00213-006-0365-8. [DOI] [PubMed] [Google Scholar]
- Harris GC, Williams JT. Transient homologous mu-opioid receptor desensitization in rat locus coeruleus neurons. J Neurosci. 1991;11:2574–2581. doi: 10.1523/JNEUROSCI.11-08-02574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Jr, Lefkowitz RJ, Caron MG, Giros B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A. 1996;93:12974–12979. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Kenski DM, Zhang C, von Zastrow M, Shokat KM. Chemical genetic engineering of G protein-coupled receptor kinase 2. J Biol Chem. 2005;280:35051–35061. doi: 10.1074/jbc.M507594200. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ. Opioid tolerance: in search of the holy grail. Cell. 2002;108:587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- Koch T, Höllt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol Ther. 2008;117:199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Höllt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Osborne PB, Williams JT. Characterization of acute homologous desensitization of μ-opioid receptor-induced current in locus coeruleus neurones. Br J Pharmacol. 1995;115:925–932. doi: 10.1111/j.1476-5381.1995.tb15899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin I, Braksator E, Bailey CP, Goodchild S, Marrion NV, Kelly E, Henderson G. Methadone: does it really have low efficacy at μ-opioid receptors? Neuroreport. 2008;19:589–593. doi: 10.1097/WNR.0b013e3282f97b64. [DOI] [PubMed] [Google Scholar]
- Ross GR, Gabra BH, Dewey WL, Akbarali HI. Morphine tolerance in the mouse ileum and colon. J Pharmacol Exp Ther. 2008;327:561–572. doi: 10.1124/jpet.108.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Höllt V. Morphine induces terminal micro-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford K, Gomes AB, Shen J, Yoburn BC. mu-Opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol Biochem Behav. 2001;69:233–237. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- Torrecilla M, Quillinan N, Williams JT, Wickman K. Pre- and postsynaptic regulation of locus coeruleus neurons after chronic morphine treatment: a study of GIRK-knockout mice. Eur J Neurosci. 2008;28:618–624. doi: 10.1111/j.1460-9568.2008.06348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Young AM. Differential tolerance to antinociceptive effects of mu opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacology. 2001;154:131–142. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of μ opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]