

Abstract
A highly regio- and stereoselective asymmetric synthesis of various C5′-alkyl side chains of rhamnosyl- and amicetosyl-digitoxigenin analogues has been established via palladium-catalyzed glycosylation with postglycosylated dihydroxylation or diimide reduction. The C5′-methyl group in both α-l-rhamnose and α-l-amicetose digitoxin analogues displayed a steric directed apoptosis induction and tumor growth inhibition against nonsmall cell human lung cancer cells (NCI-H460). The antitumor activity is significantly reduced when the steric hindrance is increased at the C5′-stereocenter.
Keywords: Digitoxin, rhamnose, amicetose, apoptosis, cancer, NCI-H460
For centuries, cardiac glycosides have been used for the treatment of congestive heart failure. The mechanism leading to this cardiotonic activity is the result of plasma membrane Na+/K+ ATPase inhibition.1,2 The accumulated intracellular Na+ concentration increases Ca2+ sequestration by the sarcoplasmic reticulum and enhances myocardial cell contractility via excitation−contraction coupling.3 Digitoxin (3; Figure 1), a commonly used cardiac glycoside originally isolated from Digitalis purpurea, has been recognized for its positive inotropic activity, and more recently for its potential application in oncology.4,5
Figure 1.
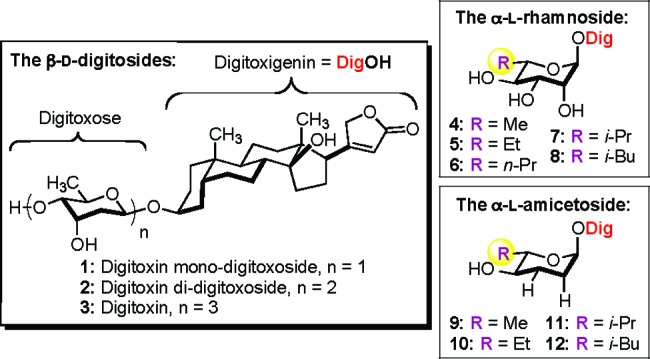
Digitoxin (3) and related carbohydrate analogues.
The cause of digitoxin cancer cell cytotoxicity has inevitably been associated with Na+/K+ ATPase inhibition and the resultant change in Ca2+ ion concentration. However, many research groups have found digitoxin induces apoptotic cell death at a subcardiotoxic concentration in plasma.6−10 These findings suggest the possible role of digitoxin binding to other biological targets as an apoptotic inducing event. In fact, direct evidence correlating Na+/K+ ATPase inhibition anticancer effects is still lacking.
Digitoxin consists of digitoxigenin (pharmacophore) and the trisaccharide moiety, which are known to play a crucial role in both its cardiotoxicity and anticancer activity.11 Because digitoxin’s cardiotonic activity has been known for a much longer time, most structure activity relationship (SAR) studies have focused on the Na+/K+ ATPase inhibition and examined the effect of modifying the carbohydrate portion of the cardiac glycosides.12−15 More recently, Karlish demonstrated that inhibition of the α2-isoform over the α1-Na+/K+ ATPase isoform could induce cardiac contraction with minimal Ca2+ overload and less cardiotoxicity.16,17 These studies also suggest that an α2-selective cardiac glycoside could result from sugar modification, as the structural differences in these isoforms lie primarily in the extracellular carbohydrate binding loops. Thus, sugar modification could provide the potential for the discovery of new and safer digitoxin analogues with improved anticancer activity.
The first anticancer SAR study of digitoxin was conducted by Thorson in 2005, when they screened a neoglycoside library of digitoxin monosaccharide. Several β-l-sugar analogues were found to have improved cytotoxicity over digitoxin.18 More recently, we have had success in performing a series of systematic SAR studies on the carbohydrate portion of digitoxin for apoptotic cytotoxicity in the human lung cancer (NCI-H460) cell line. These studies include a comparison of digitoxin MeNO-neo- and O-glycosides (with O- better than MeNO-neo-glycosides),8 a study of sugar-chain length (with mono- better than di- or trisaccharides),10 and a stereochemical survey of digitoxin monosaccharides (with β-d-digitoxo-, α-l-amiceto-, and α-l-rhamno-glycosides being the most active).9 From these studies, we concluded that O-glycosides were better than MeNO-neoglycosides8 and that sugars with β-d-digitoxo-, α-l-amiceto-, and α-l-rhamno-stereochemistry were the most active.9 Regardless of the glycosidic linkage (O-/neo- and/or α-/β-), the sugar stereochemistry of the monosaccharides was more effective than those of di- and trisaccharides.10
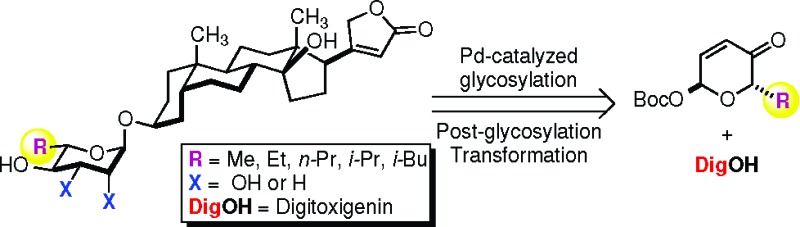
It is worth noting that these observations were stereospecific (i.e., α-d-rhamno- and α-d-amiceto-glycosides were significantly less active than their α-l-sugar diastereomers) and, thus, not attributable to nonspecific solubility and/or membrane transport properties. To further probe the specificity of the sugar portion of the α-l-digitoxin glycosides, we decided to test the SAR-importance of the C5′-alkyl position. Herein, we report our successful synthesis and cytotoxicity study of four digitoxigenin α-l-rhamno-glycosides (5−8) and three digitoxigenin α-l-amiceto-glycosides (10−12) using our de novo asymmetric approach to glycoside assembly (Figure 1).
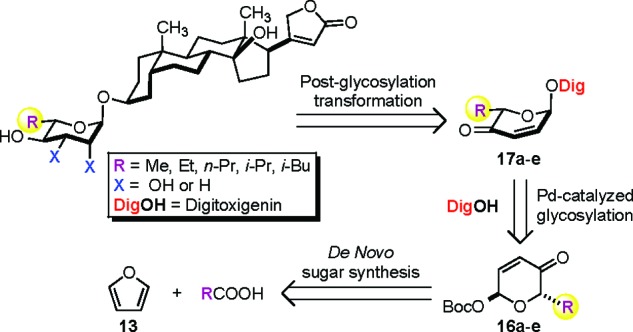
Outlined in Scheme 1 is our retrosynthetic analysis for the preparation of various C5′-alkyl substituted digitoxin analogues. We envisioned that the desired rhamno- and amiceto-stereochemistries and functionalities could be installed by a diastereoselective ketone reduction and a dihydroxylation, or alternatively a diimide reduction, of the digitoxin pyranone precursors 17a−e. These structurally diverse pyranones, in turn, could be prepared by a stereospecific palladium-catalyzed glycosylation to couple digitoxigenin (DigOH) with the corresponding α-l-Boc pyranones 16a−e. The preparation of these desired glycosyl donors could be simply amended by the use of our de novo asymmetric carbohydrate synthesis from an achiral furan starting material with different α-substituted alkyl carboxylic acids.
Scheme 1. Retrosynthetic Analysis of Digitoxin Analogues.
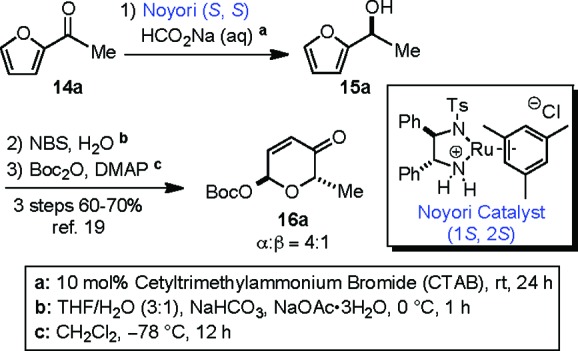
As we have previously described, the synthesis of C6′-deoxy-α-l-Boc-pyranone 16a was successfully achieved by a practical three-step sequence from acetylfuran 14a (Scheme 2).19 The absolute l-sugar stereochemistry was installed via an enantioselective Noyori asymmetric reduction.20 The resulting furfural alcohol 15a (>99% ee) was converted to the corresponding pyranone via an Achmatowicz oxidative rearrangement and a stereodivergent Boc-protection to yield an easily separable 4 to 1 ratio of α-/β-mixtures of Boc-pyranones in good overall yield (60−70%).21
Scheme 2. De Novo Approach to α-l-Boc-pyranone 16a.
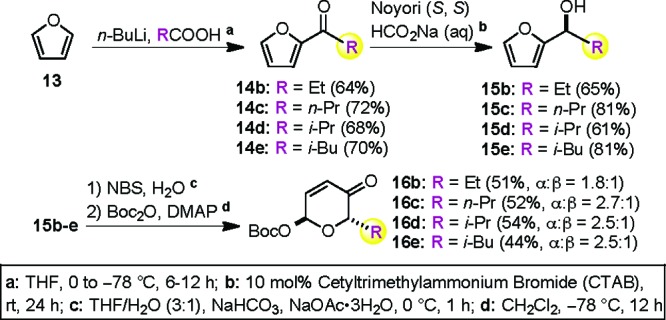
The flexibility of this de novo Achmatowicz approach allowed us to further expand the diversity of C5′-alkyl substituted Boc-pyranone sugar building blocks (16b−e, Scheme 3). The required acetyl furans (14b−e) with various α-alkyl substitutions (i.e., ethyl, n-propyl, isopropyl, and isobutyl) could be prepared by a one-pot, two-step procedure.22−24 Thus, the addition of furan (13) to a hexane solution of n-BuLi generated a solution of 2-lithiofuran, to which was added a THF solution of the appropriate substituted carboxylic acid (<0.5 equiv) to provide 14b−e in good yields. Exposure of the acylfurans to our preferred Noyori asymmetric reduction phase transfer conditions (Noyori (S, S), HCO2Na (aq), 10 mol % CTAB) provided good yields of the furan alcohols 15b−e with excellent enantiomeric purity (>98% ee). The furan alcohols 15b−e were then oxidized under the Achmatowicz conditions (NBS/H2O), and the resulting pyranone intermediates were protected with Boc2O to give the desired Boc-pyranones 16b−e in good overall yields with modest α-selectivity.
Scheme 3. De Novo Approach to the Pyranone Building Blocks 16b−e.
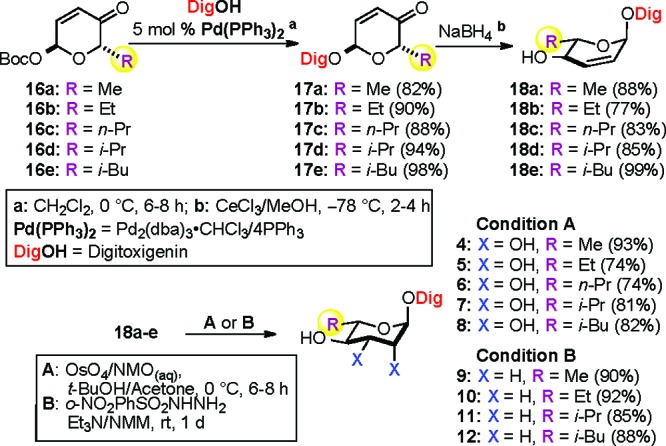
With the desired C5′-alkyl substituted α-l-Boc-pyranone sugar building blocks 16a−e in hand, we next employed them in a palladium-catalyzed glycosylation with digitoxigenin (DigOH) to provide α-l-digitoxin pyranones 17a−e (82−98%) as a single diastereomer with complete stereocontrol at the anomeric center (Scheme 4). The desired C4′-hydroxyl group was stereoselectively installed via Luche reduction to give equatorial allylic alcohols 18a−e (77−99%). The resulting C2′−C3′ double bonds in 18a−e were readily dihydroxylated under Upjohn conditions to provide C5′-alkyl substituted rhamnosyl digitoxin analogues 4−8 (Condition A).25 Alternatively, the double bonds were reduced to provide C5′-alkyl substituted amicetosyl digitoxin analogues 9−12 via a diimide reduction (Condition B).26
Scheme 4. Systematic Approach to α-l-Digitoxin C5′-Alkyl Analogues (4−12).
With the desired rhamno- and amiceto-glycosides in hand, we decided to first evaluate their apoptotic cytotoxicity against NCI-H460 human lung cancer cells, with α-l-rhamnose (4) and α-l-amicetose (9) serving as controls. Using our standard conditions, the nine compounds were tested at a single concentration (50 nM, 12 h), with the degree and the mechanism of cell death (apoptosis or necrosis) quantified using the Hoechst 33342 nuclear stain/propidium iodide method (Figure 2).27 The blue Hoechst nuclear stained cells with condensed and fragmented nuclei are indicative of apoptosis, whereas the completely ruptured cells (indicative of necrosis) were stained red with propidium iodide (Figure 2C/D). While all the digitoxin analogues 4−12 displayed varying degrees of cytotoxicity, the C5′-Me analogues rhamno-4 and amiceto-9 were the most cytotoxic (Figure 2A and B). Regardless of potency, apoptosis was the predominant mechanism of cell death for all the analogues. Interestingly, the activity trend of apoptosis significantly decreased when the size of the C5′-alkyl substituent increased.
Figure 2.

Apoptotic cell death and modification of C5′ bulk. (A and B) Apoptotic cell death (%) was compared for each C5′-alkyl substituted digitoxin rhamnoside and amicetoside at 50 nM concentration (one-way ANOVA; N = 6; ***, P < 0.001). (C and D) Hoechst 33342 stained apoptotic cells appear in blue and propidium iodide stained necrotic cells in red at 50 nM drug concentration.
The cytotoxicity (apoptosis and necrosis) was further evaluated in a 12 h treatment of increasing concentration (10 nM to 10 μM) of drugs. These results were plotted in Figure 3, which showed that all the analogues induced apoptotic cell death in a dose-dependent manner. The degree of cytotoxicity resulting in α-l-rhamno-C5′-Me (4) and α-l-amiceto-C5′-Me (9) concur with the literature data from the previous study, where both analogues 4 and 9 showed at least 4-fold stronger potency than natural digitoxin.9 The IC50 values were determined by a nonlinear regression analysis and recorded in Table 1. As in the single dose experiments, the IC50 values increased commensurately with the size of the C5′-substituent. This trend could be seen in both the rhamnosides (4−8, Figure 3A) and amicetosides (9−12, Figure 3B). Although demonstrably different, the potency of the C5′-Et-rhamno-analogue (5; IC50 57 nM) was only slightly weaker than that of the C5′-Me-rhamno-analogue (4; IC50 52 nM). The sensitivity to this negative steric effect greatly increased as subsequent methylene groups were added (e.g., C5′-n-Pr (6; IC50 116 nM), C5′-i-Pr (7; IC50 130 nM), and C5′-i-Bu (8; IC50 180 nM)). Interestingly, this effect of the C5′-alkyl substitution appeared to be even more significant on the amiceto-series (9−12), where there was a more significant increase in the IC50 values with the addition of methylene groups (cf., C5′-Me (9; IC50 52 nM), C5′-Et (10; IC50 87 nM), C5′-i-Pr (11; IC50 533 nM), and C5′-i-Bu (12; IC50 458 nM)). It is worth noting that α-l-amiceto-C5′-Me (9) has shown consistently stronger potency in growth inhibition against the NCI-panel of 60 human cancer cell lines than amiceto-C5′-i-Pr (11) and C5′-i-Bu (12) (see Supporting Information).
Figure 3.

Cytotoxicity as a function of drug concentration in the comparison of C5′-alkyl substitution. Dose response curve of total cell death (apoptosis/necrosis) mediated by digitoxin analogues in a 12 h treatment at increasing concentrations (10 nM to 10 μM). All the data were analyzed by two-way ANOVA (N = 6; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
Table 1. Cytotoxicity Evaluation of Digitoxin Analogues on NCI-H460 Human Lung Cancer Epithelial Cells.
| compd | IC50a | GI50b | compd | IC50a | GI50b |
|---|---|---|---|---|---|
| α-l-rhamnoside | α-l-amicetoside | ||||
| C5′-Me-4 | 52 ± 1 | 2 ± 1 | C5′-Me-9 | 52 ± 1 | 2 ± 1 |
| C5′-Et-5 | 57 ± 1 | 2 ± 1 | C5′-Et-10 | 87 ± 1 | 2 ± 1 |
| C5′-n-Pr-6 | 116 ± 1 | 5 ± 1 | C5′-i-Pr-11 | 533 ± 1 | 8 ± 1 |
| C5′-i-Pr-7 | 130 ± 1 | 6 ± 1 | C5′-i-Bu-12 | 458 ± 1 | 8 ± 1 |
| C5′-i-Bu-8 | 180 ± 1 | 13 ± 1 | |||
The IC50 value was measured by a 12 h treatment in Hoechst and propidium iodide stain assays.
The GI50 value was measured by a 48 h treatment in an MTT assay. All values represent the standard error of the mean value of three independent experiments with duplicate determinations.
To demonstrate that this steric-demand trend was not specific to apoptosis, we decided to measure the cytotoxicity of these digitoxin analogues with a different assay. We chose to use the MTT assay for this comparison, which evaluated the cell viability based on the measurement of mitochondrial enzymatic activity after 48 h of exposure.28,27 These results (Table 1 and Figure 4) showed a dose-dependent cytotoxic activity for each analogue that was similar to that of the apoptosis assay. As seen earlier, a commensurate drop in cytotoxicity was observed with the increasing size of the C5′-substituent. This trend was consistent across both rhamnoside (Figure 4A) and amicetoside (Figure 4B) series of digitoxin analogues. It should be noted, however, that the greater steric sensitivity of the amiceto-series in the apoptosis assay was not observed in the MTT assay.
Figure 4.

Cell viability as a function of drug concentration in the comparison of C5′-alkyl substitution. The dose response curve of cell viability in a 48 h treatment at increasing concentrations (1 nM to 10 μM). All the data were analyzed by two-way ANOVA (N = 9; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
In summary, we have demonstrated the SAR-effects of C5′-alkyl substitution on digitoxin α-l-rhamnoside (4) and α-l-amicetoside (9). Our results suggest that both the rhamno- and amiceto-analogues occupy a similar binding site and orientation in its target with a small hydrophobic pocket for the C5′-Me. While these cytotoxicity trends are not inconsistent with the models for Na+/K+ ATPase inhibition, further study is needed. The efficiency with which this systematic SAR-study was conducted was enabled by the flexibility of this unique de novo approach to carbohydrate synthesis. Further efforts aimed at optimizing cytotoxicity and elucidation of mechanism are ongoing and will be reported in due course.
Acknowledgments
We thank Anand Krishnan Iyer (Hampton University), Todd A. Stueckle, and Yongju Lu (West Virginia University) for their advice on cell culturing and cytotoxicity assays. We also thank Jonathan Boyd (West Virginia University) for his help with data analysis. We are grateful to the NIH (GM088839) and NSF (CHE-0749451) for the support of our research.
Supporting Information Available
Assay protocols, statistical analysis data, synthetic procedures, characterization data, and NMR spectra. This information is available free of charge via the Internet at http://pubs.acs.org.
Notes
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Greeff K.Cardiac Glycosides, Part 1: Experimental Pharmacology. Handbook of Experimental Pharmacology; Springer-Verlag: Berlin, New York, 1981; Vol. 56. [Google Scholar]
- Forbush B. Cardiotonic Steroid Binding to Na,K-ATPase. Curr. Top. Membr. Transp. 1983, 19, 167–201. [Google Scholar]
- Bers D. M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Stenkvist B.; Bengtsson E.; Eriksson O.; Holmquist J.; Nordin B.; Westman-Naeser S. Cardiac glycosides and breast cancer. Lancet 1979, 1, 563. [DOI] [PubMed] [Google Scholar]
- Stenkvist B.; Bengtsson E.; Dahlqvist B.; Eriksson O.; Jarkrans T.; Nordin B. N. Cardiac glycosides and breast cancer, revisited. N. Engl. J. Med. 1982, 306, 484. [PubMed] [Google Scholar]
- Haux J.; Solheim O.; Isaksen T.; Angelsen A. Digitoxin, in non-toxic concentrations, inhibits proliferation and induces cell death in prostate cancer cell lines. Z. Onkol. 2000, 32, 11–16. [Google Scholar]
- Newman R. A.; Yang P.; Pawlus A. D.; Block K. I. Cardiac Glycosides as Novel Cancer Therapeutic Agents. Mol. Interventions 2008, 8, 36–49. [DOI] [PubMed] [Google Scholar]
- Iyer A. K. V.; Zhou M.; Azad N.; Elbaz H.; Wang L.; Rogalsky D. K.; Rojanasakul Y.; O’Doherty G. A.; Langenhan J. M. Direct Comparison of the Anticancer Activities of Digitoxin MeON-Neoglycosides and O-Glycosides: Oligosaccharide Chain Length-Dependent Induction of Caspase-9-Mediated Apoptosis. ACS Med. Chem. Lett. 2010, 1, 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y. L.; Xin W.; Zhou M.; Stueckle T. A.; Rojanasakul Y.; O’Doherty G. A. Stereochemical Survey of Digitoxin Monosaccharides. ACS Med. Chem. Lett. 2011, 21pp 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y. L.; Rojanasakul Y.; O’Doherty G. A.. Synthesis and Evaluation of the α-d-/α-l-Rhamnosyl and Amicetosyl Digitoxigenin Oligomers as Anti-tumor Agents. ACS Med. Chem. Lett. 2011, (see DOI 10.1021/ml100290d). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lazaro M.; Pastor N.; Azrak S. S.; Ayuso M. J.; Austin C. A.; Cortes F. Digitoxin Inhibits the Growth of Cancer Cell Lines at Concentrations Commonly Found in Cardiac Patients. J. Nat. Prod. 2005, 68, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Yoda A. Structure−Activity Relationships of Cardiotonic Steroids for the Inhibition of Sodium- and Potassium-Dependent Adenosine Triphosphatase. Mol. Pharmacol. 1973, 9, 51–60. [PubMed] [Google Scholar]
- Brown L.; Erdmann E.; Thomas R. Digitalis structure−activity relationship analyses: Conclusions from indirect binding studies with cardiac (Na+ + K+)-ATPase. Biochem. Pharmacol. 1983, 32, 2767–2774. [DOI] [PubMed] [Google Scholar]
- Chiu F. C. K.; Watson T. R. Conformational Factors in Cardiac Glycoside Activity. J. Med. Chem. 1985, 28, 509–515. [DOI] [PubMed] [Google Scholar]
- Rathore H.; From A. H. L.; Ahmed K.; Fullerton D. S. Cardiac Glycosides. 7. Sugar Stereochemistry and Cardiac Glycoside Activity. J. Med. Chem. 1986, 29, 1945–1952. [DOI] [PubMed] [Google Scholar]
- Katz A.; Lifshitz Y.; Bab-Dinitz E.; Kapri-Pardes E.; Goldshleger R.; Tal D. M.; Karlish S. J. D. Selectivity of Digitalis Glycosides for Isoforms of Human Na,K-ATPase. J. Biol. Chem. 2010, 285, 19582–19592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansapura A. N.; Lasko V.; Xie Z.; Fedorova O. V.; Bagrov A. Y.; Lingrel J. B.; Lorenz J. N. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive α1 Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 1833–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenhan L. M.; Peters N. R.; Guzei I. A.; Hoffmann M.; Thorson J. S. Enhancing the anticancer properties of cardiac glycosides by neoglycorandomization. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12305–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the synthesis of α/β-L-Boc pyranones, see:Guo H.; O’Doherty G. A. De Novo Asymmetric Synthesis of Anthrax Tetrasaccharide and Related Tetrasaccharide. J. Org. Chem. 2008, 73, 5211–5220. [DOI] [PubMed] [Google Scholar]
- Fujii A.; Hashiguchi S.; Uematsu N.; Ikariya T.; Noyori R. Ruthenium (II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid-Triethylamine Mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar]
- Achmatowicz O.; Bukowski P.; Szechner B.; Zwierzchowska Z.; Zamojski A. Synthesis of methyl 2,3-dideoxy-DL-alk-2-enopyranosides from furan compounds: A general approach to the total synthesis of monosaccharides. Tetrahedron 1971, 27, 1973–1996. [Google Scholar]
- Li M.; O’Doherty G. A. An enantioselective synthesis of phomopsolide D. Tetrahedron Lett. 2004, 45, 6407–6411. [Google Scholar]
- Guo H.; O’Doherty G. A. De Novo Asymmetric Synthesis of D- and L-Swainsonine. Org. Lett. 2006, 8, 1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coral J. A.; Guo H.; Shan M.; O’Doherty G. A. A De Novo Asymmetric Approach to 8a-epi-Swainsonine. Heterocycles 2009, 79, 521–529. [Google Scholar]
- VanRheenen V.; Kelly R. C.; Cha D. Y. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17, 1973–1976. [Google Scholar]
- Haukaas M. H.; O’Doherty G. A. Enantioselective Synthesis of 2-Deoxy- and 2,3-Dideoxyhexoses. Org. Lett. 2002, 4, 1771–1774. [DOI] [PubMed] [Google Scholar]
- Chanvorachote P.; Nimmannit U.; Stehlik C.; Wang L.; Jiang B.-H.; Ongpipatanakul B.; Rojanasakul Y. Nitric Oxide Regulates Cell Sensitivity to Cisplatin-Induced Apoptosis through S-Nitrosylation and Inhibition of Bcl-2 Ubiquitination. Cancer Res. 2006, 66, 6353–6360. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.