

Abstract
We report the development of a microfabricated electrophoretic device for assembling high-density arrays of antibody-conjugated microbeads for chip-based protein detection. The device consists of a flow cell formed between a gold-coated silicon chip with an array of microwells etched in a silicon dioxide film and a glass coverslip with a series of thin gold counter electrode lines. We have demonstrated that 0.4 and 1 μm beads conjugated with antibodies can be rapidly assembled into the microwells by applying a pulsed electric field across the chamber. By assembling step-wise a mixture of fluorescently labeled antibody-conjugated microbeads, we incorporated both spatial and fluorescence encoding strategies to demonstrate significant multiplexing capabilities. We have shown that these antibody-conjugated microbead arrays can be used to perform on-chip sandwich immunoassays to detect test antigens at concentrations as low as 40 pM (6 ng/mL). A finite element model was also developed to examine the electric field distribution within the device for different counter electrode configurations over a range of line pitches and chamber heights. This device will be useful for assembling high-density, encoded antibody arrays for multiplexed detection of proteins and other types of protein-conjugated microbeads for applications such as the analysis of protein-protein interactions.
1. Introduction
The ability to interrogate proteins in a sensitive, quantitative, multiplexed and high-throughput manner has many applications in proteomic analysis,1-3 cancer research,4,5 diagnostics6 and drug discovery.7 Although established methods such as western blots8 and enzyme-linked immunosorbent assays9 (ELISA) can be used for sensitive and reliable protein detection and quantification, they are labor-intensive and require large sample volumes. Furthermore, they allow for the analysis of only a small number of samples and proteins at a time. Alternatively, the use of spotted protein and antibody microarrays enable greater multiplexing and significantly reduced sample volumes.10-18 Other groups have demonstrated the potential advantages of assays that employ protein- and antibody-conjugated microbeads, which allow for even greater multiplexing and scalability than those performed in microtiter plates or on spotted arrays.19-26 The majority of these microbead-based immunoassays are typically performed in solution19,20 or on-chip.21-26 The solution-based formats are fast and sensitive but they require specialized flow cytometry equipment for sample analysis. In contrast, chip-based formats are well suited for analysis via epifluorescence microscopy and allow for the integration of additional lab-on-a-chip processes such as nucleic acid extraction and genetic profiling from single cells or whole blood.27,28
Methods for assembling or capturing antibody-conjugated microbeads on chip-based platforms include micromanipulation,21 microfluidic trapping,22,23 evaporation of microbead suspensions on etched silicon24 or fiber-optic bundles,25 and electrostatic self-assembly on chemically-modified substrates.26 Many of these platforms enable multiplexed analysis by using a mixed population of encoded microbeads or by physically isolating each population in separate microfluidic channels. In this work, we report the development of a new approach for fabricating and assembling microbead arrays. We utilize an electric field to direct the assembly of antibody-conjugated microbeads onto a microfabricated array of wells. The process takes place within a microfluidic device and arrays of micron to sub-micron beads can be assembled in 15–45 s. Moreover, we have demonstrated that antibody-conjugated microbead arrays can be assembled and used for sensitive, multiplexed protein detection in many samples in parallel. In contrast to previously reported methods, our approach enables much faster and more scalable array assembly. The array format provides the order and spatial separation necessary for packing a large number of microbeads into an extremely small footprint. For instance, nearly 7000 sub-micron beads can be rapidly assembled on an array just 100 μm × 100 μm in size. This small footprint may enable the analysis of entire proteomes at the single cell level.29,30 In addition, our device may provide a means for electrophoretically accelerating the transport of antigens to decrease assay times and to enhance sensitivity.31-33
Another key advantage of our approach is the ability to assemble arrays in a controlled, stepwise fashion. By introducing and assembling a small number of microbeads from a single population at a time, we can record their physical locations on the array.34 This spatial encoding method enables a large range of multiplexing capabilities without the need for fluorescence encoding35,36 or other more complex strategies.37,38 In this study, we demonstrate the feasibility of a combined encoding approach by assembling two different fluorescence microbead populations per round. This combination of both fluorescence and spatial encoding schemes gives us even greater multiplexing potential.
The method and device described here also encompass significant improvements over those previously reported for rapid electric-field directed assembly of streptavidin-conjugated microbead arrays.39 In our previous work, the high-density array of wells was patterned in an epoxy-based photoresist on a gold-coated silicon wafer. The gold served as the working electrode, whereas the counter electrode consisted of an indium-tin oxide (ITO) film on a glass coverslip. In this study, the microwells are fabricated in a silicon dioxide film and the counter electrode consists of a series of thin gold lines on a glass coverslip. The use of a silicon dioxide layer results in a more robust platform and enables more precise control of the microwell geometry. The use of the gold lines as counter electrodes results in better light transmission and eliminates the problems associated with the degradation of ITO from the by-products of electrolysis.40
2. Materials and methods
Fabrication of arrays of microwells in an oxide on gold
Fig. 1A illustrates the general procedure for the fabrication of an array of microwells in a silicon dioxide film on a gold-coated wafer. First, 150 mm diameter silicon wafers were cleaned and then coated with silicon dioxide as described previously.39 Films of titanium, gold and titanium were sequentially deposited on the oxide-coated wafer using a Denton Discovery 18 sputter system. The deposition chamber was typically evacuated to a base pressure of 9 × 10−7 Torr or less and the films were deposited at 150–200 W DC in 3.0 × 10−3 Torr Ar flowing at a rate of 36 sccm. The two titanium layers, which serve as adhesion layers between the oxide and gold films, were approximately 10 nm thick. The thickness of the gold film was ~300 nm. Following metallization, another 100–300 nm of silicon dioxide was deposited via PECVD as described previously.39 A Filmetrics F20 measurement system was used to determine the oxide film thickness.
Fig. 1.

Fabrication and assembly of the electrophoretic device. (A). Fabrication of an array of microwells in silicon dioxide on a titanium-gold-titanium stack on a silicon wafer. (B) Fabrication of gold counter electrode lines on a glass coverslip via a lift-off process. The gold lines serve as counter electrodes in the assembled chamber and are only 25 μm wide with a pitch of 320 μm to allow for imaging of the microbead arrays. (C) Assembly of the device and mounting to a custom-built aluminium plate with tapped ports for fluidic connections. Drawings are not to scale.
To fabricate the microwell arrays, the wafers were first coated with a bottom anti-reflective coating (BARC) (ARC 29A-8, Brewer Science) by spin-coating at 2250 rpm for 30 s. After baking the BARC at 220 °C for 60 s, a 250 nm-thick film of deep UV photoresist (ARF AR1682J-15, JSR Micro) was applied via spin-coating at 1000 rpm for 40 s. Edge bead was removed from the front and back sides of the wafers with propylene glycol monomethyl ether acetate (Baker BTS-220, J. T. Baker). All coating, baking and edge bead removal steps were performed on a SVG 90-SE coat track. The resist was baked at 110 °C for 90 s and then exposed on a PAS 5500/950B Step and Scan System (ASML) equipped with a 10 W, 193 nm ArF excimer laser (ELS-6610A, Cymer). Arrays of posts were patterned on the substrates using a quartz reticle containing chrome contacts on a clear background. Typical doses ranged from 12–24 mJ/cm2. The exposed wafers were baked at 110 °C for 60 s, developed in MF-319 (Rohm & Haas Electronic Materials) for 60 s and rinsed with dH2O in a quick dump rinser. The wafers were then rinsed and dried in a spin-rinse-dry tool (PSC-101, Semitool). To remove the BARC, the wafers were exposed to an oxygen-based plasma for 75 s at 50 W RF at 8 sccm and 4.0 × 10−2 Torr in a RIE system (System VII, Plasma-Therm).
Next, the patterned wafers were coated with 30 nm of nickel via a Temescal BJD 1800 electron-beam evaporation system. The chambers were typically evacuated to base pressures of 7 × 10−7 Torr or less and nickel films were deposited at 1.0–2.0 Å/s. The resist and unwanted metal was removed using Shipley Microposit Remover 1165 (Rohm & Haas Electronic Materials) at 70 °C with ultrasonic agitation for ~6 h. After rinsing in dH2O and drying with nitrogen, the exposed oxide was etched in a Panasonic FP-EA01A ICP etcher using 40 sccm CHF3 at 0.5 Pa with 900 W forward RF power and 200 W reverse RF power. The substrates were cooled via backside helium flow at 15 sccm and 700 mTorr. Under these conditions, the average etch rate for PECVD-grown silicon dioxide was ~0.20 μm/min. However, etch times were extended by as much as 50% to ensure that the upper layer of titanium was also completely removed to fully expose the underlying gold film.
Following the etching process, the wafers were coated with a thick layer of Shipley Megaposit SPR220-3.0 (Rohm & Haas Electronic Materials) photoresist by spin-coating at 2000 rpm for 30 s and then baking at ~100 °C for 5 min. Holes for fluidic connections were then drilled in the wafers using a 1.0 mm diamond-coated drill bit (Cat. # MD16, C. R. Laurence Co.) and a high speed rotary tool (38481 IB/E, Proxxon) mounted to a CNC milling machine (PCNC-1100, Tormach). The wafers were secured in a custom-built jig and flooded with a dilute coolant solution (Formula #77, Kool Mist) while drilling. The wafers were then diced with a dicing saw (DAD3220, Disco). The resist was stripped by soaking in acetone for 3 min and then in isopropanol for 1 min. After drying with nitrogen, the nickel layer was stripped for 10 min at room temperature in a nickel etchant (Type TFB, Transene Co.). The wafers were then rinsed with dH2O and dried with nitrogen.
Fabrication of counter electrode lines on glass coverslips
Fig. 1B illustrates the general procedure for the fabrication of the counter electrode lines on a glass coverslip. Using a custom-built PTFE rack, 50 mm × 75 mm × 0.170 mm (Erie Scientific Co.) glass coverslips were cleaned as described previously.39 Prior to fabrication, the coverslips were blown dry with nitrogen and then baked on a hotplate at ~200 °C for 5 min to remove any remaining moisture. After cooling, HMDS was applied and allowed to sit for 30 s before spin-drying at 4000 rpm for 30 s. A layer of Shipley Megaposit SPR220-3.0 was then applied by spin-coating at 3500 rpm for 30 s. The resist was baked at 115 °C for 90 s and then exposed to 365–405 nm light on a Quintel contact aligner using a photomask printed on a transparency film. An exposure time of 18 s at ~10 mW/cm2 was typically used to print 25 μm wide lines at a pitch of 320 μm. After baking the exposed coverslip at 115 °C for 90 s, the resist was developed for 90 s in MF-24A developer (Rohm & Haas Electronic Materials), rinsed with dH2O, and dried with nitrogen.
After a 3 min oxygen plasma treatment in a Technics PEII-B plasma system at 100 W RF and 3.0 × 10−1 Torr O2, a Denton Discovery 18 sputter system was used to deposit a 10 nm-thick titanium film followed by a 300 nm-thick Au film. Sputtering was performed at 150 W with 3.0 × 10−3 Torr Ar at 36–38 sccm. The resist and unwanted metal was then removed by soaking the substrate in Shipley Microposit Remover 1165 at 70 °C in an ultrasonic bath for up to 1 h. The coverslip was then washed with acetone, rinsed with dH2O and dried with nitrogen. Line height measurements were obtained with a Dektak 150 surface profiler (Veeco Instruments).
Device assembly
An exploded view of the electrophoretic device is shown in Fig. 1C. The general method for assembly of this device has been described elsewhere.39 Briefly, each chip was cleaned by exposing it to oxygen-based plasma at 100 W RF and 3 × 10−1 Torr O2 for 3 min in a Technics PEII-B plasma system. After rinsing with dH2O and drying with compressed air, the chip was mounted to a custom-built aluminium plate using a double-coated adhesive tape. The flow cell was then formed by attaching the coverslip with the counter electrodes to the chip via a second double-coated adhesive tape containing cutouts of the fluidic channels. Channel dimensions were 2 mm wide by 10 mm long with a height of ~110 μm. Electrical connections were made to the gold film on the chip and the counter electrode lines using copper tape. Fluidic connections were made via ports in the aluminium plate (Fig. 2).
Fig. 2.

Photographs of the assembled electrophoretic fluidic device. (A) Top view of the custom-built aluminium plate through which the fluidic connections are made to each channel in the device. (B) Bottom view of the plate and an assembled device containing 14 separate channels. The bottom side of the aluminium plate has been coated with titanium and silicon dioxide films to aid in the adhesion of the double-coated tape.
Antibody conjugation to microbeads
Biotinylated antibodies (biotin-XX goat anti-mouse IgG (H + L), Cat. # B2763 and biotin-XX F(ab’)2 fragment of goat anti-rabbit IgG (H + L), Cat. # B21078, Invitrogen) were conjugated to 0.4 or 1 μm streptavidin-coated green (ex. 480/em. 520) and red (ex. 660/em. 690) fluorescent polystyrene beads (Cat. # CP01F/8682, CP01F/7678 and CP01F/8963, Bangs Laboratories) by adding drop-wise a 0.2% microbead suspension to a solution containing one of the biotinylated antibody species. Each conjugation was performed in phosphate buffered saline (PBS, 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM sodium phosphate dibasic and 2 mM potassium phosphate monobasic, pH 7.4) at an antibody concentration corresponding to five to ten times the amount required to cover the surface of all microbeads in the suspension. A 4 μL drop of the microbead suspension was delivered to the antibody solution every 5 s using a syringe pump (Cavro XR Rocket Pump, Tecan Group) and the mixture was vortexed throughout the mixing process. After the addition of the entire microbead suspension, the mixture was shaken at room temperature for 1 h. The microbeads were then washed four times with PBS and stored at 4 °C until use.
Microbead array assembly
Antibody-conjugated microbeads were assembled using optimized parameters as described elsewhere.39 Briefly, the microbeads were exchanged into a low conductance buffer (LCB, 4.5 mM tris(hydroxymethyl) aminomethane, 4.5 mM boric acid and 0.02% Triton X-100, pH 8.6, 60 μS/cm conductivity) and introduced into the flow cell at a concentration of 0.02% to 0.2% solids. The microbeads were assembled by applying 3.0 V DC pulses at 1 Hz and a 10% duty cycle for 15–45 s in 15 s intervals with a 1–2 min pause between each interval. The microbeads were pulled into the wells via electrophoresis and were permanently bound to the gold surface through electrochemically-induced interactions. Excess microbeads were then washed away using a syringe pump.
Spatial and fluorescence encoding of microbeads
A mixture of two populations of antibody-conjugated microbeads, one with green fluorescence and one with red fluorescence, were introduced into the flow cell and then subjected to exactly four electrical pulses. Microbeads that were not captured were washed away and then the array was imaged using a DM LFSA epifluorescence microscope (Leica Microsystems) equipped with a 40×/0.55 NA objective, a Cascade 650 CCD camera (Photometrics) and a fast wavelength-switching light source with a 300 W xenon arc lamp (Lambda DG-5, Sutter Instrument Co.). Array scanning was achieved via a BioPrecision 2 XY microscope stage and a MAC 5000 controller system (Ludl Electronic Products). This process was repeated a total of ten times to demonstrate the principle of combining both fluorescence and spatial encoding schemes to record the positions of 20 different microbead populations. The images were aligned and combined using custom macros in ImageJ41 to produce a spatial map of the array.
Sandwich immunoassays
Two antibody-conjugated microbead populations (goat anti-mouse IgG microbeads and goat anti-rabbit IgG microbeads) were combined at a 1 : 1 ratio and diluted to ~0.02% for each species. The microbeads were assembled into an array using 5–15 pulses then washed with PBS with 0.05% Tween-20 (PBS-5T). A blocking solution (SuperBlock blocking buffer in Tris-buffered saline, pH 7.4, Cat. # 37545, Pierce Biotechnology) with 0.05% Tween-20 (SB-5T) was then introduced into the chamber. After a 30 min incubation, the chamber was washed with PBS-5T. A solution containing the antigens diluted to specified concentrations using PBS-5T with 10% SB-5T was then loaded. After a 90 min incubation, the chamber was washed with PBS-5T and a solution containing both detection antibodies was introduced into the chamber. After a 30 min incubation, the chamber was washed with PBS-5T and the array was imaged on a fluorescence microscope. The test antigens were mouse and rabbit immunoglobulin proteins (Mouse IgG, Cat. # 2-6502; Rabbit IgG, Cat. # 2-6102, Invitrogen) and the detection antibodies were fluorescently labeled antibodies (Alexa Fluor 568 goat anti-rabbit IgG (H + L), Cat. # A11036; Alexa Fluor 568 goat anti-mouse IgG (H + L), Cat. # A11031, Invitrogen). To generate standard curves, 10 μL of the antigen solutions were introduced at concentrations ranging from 6 to 625 pM (100 ng/mL). Experiments were repeated 3 times at each antigen concentration. Negative controls were also conducted using an identical procedure but without antigen in the solution. After a 90 min incubation, the chamber was washed with PBS-5T. A solution containing a mixture of the detection antibodies diluted with PBS-5T to a concentration of 4 μg/mL each was then introduced into the chamber. After a 30 min incubation, the chamber was washed with PBS-5T and then imaged with an automated epifluorescence microscopy system. The microbead assembly and sandwich immunoassay process is illustrated in Fig. 3.
Fig. 3.
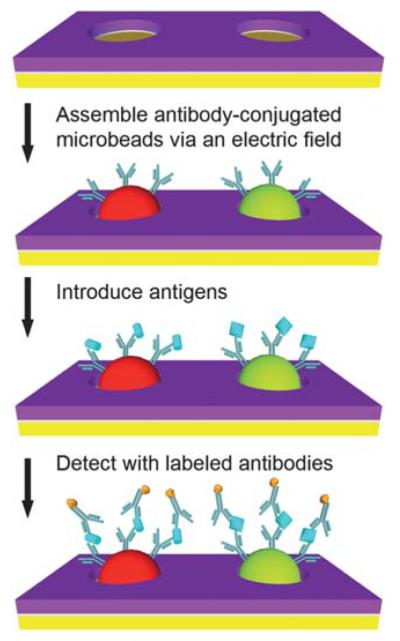
Sandwich immunoassay on antibody-conjugated microbeads assembled via electrophoretic deposition. Antibody-conjugated microbeads are assembled on the microfabricated arrays via the application of a pulsed electric field. Multiplexed protein detection is then performed within the same microfluidic channel using a sandwich immunoassay. Drawings are not to scale.
Fluorescence imaging
Quantitative fluorescence imaging of the antibody arrays was performed on an Axio Observer.Z1 epifluorescence microscope (Carl Zeiss) equipped with a 40×/1.3 NA oil objective, a 1-megapixel EMCCD camera (iXon + 885, Andor Technology) and a Lambda DG-5 light source. Imaging on the Axio Observer.Z1 was fully-automated using custom software. Auto focusing was performed with a Definite Focus System (Carl Zeiss) and array scanning was controlled via a BioPrecision 2 XY microscope stage and a MAC 5000 controller system. For each antibody array, multiple fields of view were acquired along the length of the channel. For each field of view, images were taken in three fluorescence channels using the appropriate filter cubes (FITC-3540B, TXRED-4040B, and Cy5.5-A, Semrock). To image the microbeads in the FITC channel, a neutral density filter (ND 2.0 A, Chroma Technology) was used to reduce the output from the light source.
Image analysis
The data from the sandwich immunoassays was analyzed in ImageJ using a custom macro. Briefly, the program generated a mask of the anti-rabbit and anti-mouse microbeads for each field of view using the images taken in the FITC and Cy5.5 channels, respectively. After locating the center of each microbead, the program identified the pixels associated with the microbead and calculated the mean pixel intensity in the corresponding Alexa 568 detection channel. The mean background pixel intensity for each microbead type was obtained from negative control experiments in which no antigen was added. A microbead was considered to have detected a given antigen if its mean fluorescence intensity in the corresponding channel was greater than three times the standard deviation of the mean background intensity of the microbeads in the same channel.42 A standard curve was then generated for each microbead type by plotting their mean background subtracted intensities as a function of antigen concentration. Results were gathered from the analysis of multiple images and each image contained ~150 microbeads.
SEM imaging
Scanning electron microscopy (SEM) was performed on a Phillips XL30 ESEM or FEI Sirion operating in high-vacuum mode. Prior to imaging with the XL30, the samples were coated with iridium using an Emitech K575X desktop sputtering system.
ITO transmission and resistivity measurements
The effects of electrophoretic assembly conditions on the optical transparency and resistivity of ITO films were studied using a modified chamber design. To enable easy disassembly, each ITO-coated glass slide (Cat. # CG–511N–S115, Delta Technologies) was positioned over a gold-coated slide outfitted with a PEEK gasket (Cat. # 5804K42, McMaster-Carr Co.) and held in place with binder clips. The gap between the two substrates was filled with LCB via capillary action. The chip was then subjected to a 3 V DC potential in 1 min intervals. The device was disassembled after each interval and the optical transmittance of the ITO film was recorded with a Lambda 20 UV-Vis spectrometer (Perkin Elmer). The resistance across the ITO film was also measured after each interval using a digital multimeter (Model # 2010, Keithley Instruments).
Finite element analysis
The variation in the electric field strength within the device was modeled using COMSOL Multiphysics v3.4 (COMSOL AB) and MATLAB 7.7.0 (Mathworks). A cross-section of the chamber, which included ~830 microwells across a span of 2 mm, was drawn to scale and the conductive media DC application was used to plot the electric field strength in the media using different counter electrode configurations. In the first study, the chamber height was fixed at 110 μm and the pitch between the counter electrode lines was varied from 160 μm to 640 μm. In a second study, the counter electrode line pitch was fixed at 320 μm and the chamber height was varied from 55 μm to 220 μm. In both studies, the height of the counter electrode lines was 0.3 μm and the wells were 0.25 μm deep by 1.2 μm wide at a 2.4 μm pitch. The media was assigned a conductivity of 60 μS/cm, which corresponds to the conductivity of the LCB used during microbead assembly. The bottom of each well was set to 3.0 V DC while the counter electrode lines were set to ground. All other entities were electrically insulating. Horizontal line plots were generated for each counter electrode line configuration at a height of 5 μm above the surface of the array and spanned between the centers of two adjacent counter electrode lines.
3. Results and discussion
We have developed a microfabricated electrophoretic device comprised of a high-density array of wells in silicon dioxide on a gold-coated silicon chip and a glass coverslip containing a series of thin gold lines. A typical SEM image of an array of microwells fabricated in silicon dioxide on a gold film is shown in Fig. 4A. By applying a pulsed electric field across the device, we have demonstrated that 0.4 and 1 μm antibody-conjugated microbeads can be rapidly assembled into high-density arrays with excellent filling efficiencies and near perfect order. A sample fluorescence micrograph and SEM image of a small portion of an array of antibody-conjugated microbeads assembled within the oxide wells is shown in Fig. 4B and 4C, respectively.
Fig. 4.

Arrays of microwells and antibody-conjugated microbeads. (A) SEM image of a small portion of an array of ~0.5 μm wells at a 1.2 μm pitch etched in a silicon dioxide film that was deposited on a gold-coated wafer. (B) Raw fluorescence image of a small portion of an assembled array of 0.4 μm antibody-conjugated beads at a 1.2 μm pitch. (C) SEM image of a small portion of an assembled array of 0.4 μm antibody-conjugated beads at a 1.2 μm pitch. The scale bars in (A) and (C) are 1 μm. The scale bar in (B) is 24 μm.
Spatial and fluorescence microbead encoding
To perform multiplexed immunoassays, we utilize both fluorescence and spatial encoding schemes to enable the identification of each microbead after it has been assembled on the array. An example of a combined encoding scheme is shown in Fig. 5. In this instance, a mixture of two populations of microbeads with either red or green fluorescence were introduced into the flow cell and then subjected to exactly four electrical pulses. Microbeads that were not captured were then washed away and the array was imaged via epifluorescence microscopy, thus recording the exact location of each microbead. This process, which only takes a few seconds per round, was repeated multiple times to demonstrate the gradual filling of the array and our ability to map each microbead type following each assembly round. The number of microbeads assembled in each round can be controlled by varying the microbead concentrations as well as the number of pulses applied during each round. Additionally, if more fluorescent barcodes are used, many more populations of antibody-conjugated microbeads can be assembled simultaneously during each round, resulting in much greater multiplexing capabilities.
Fig. 5.

Spatial and fluorescence encoding of antibody-conjugated microbead arrays. Fluorescent micrographs of a small portion of an antibody-conjugated microbead array assembled in a stepwise fashion over the course of ten rounds using two different fluorescent microbeads (Panels 1–10). In panels 2–10, the brightness of the previously assembled microbeads has been reduced to emphasize the newly assembled microbeads.
Immunoassays
Sandwich immunoassays were conducted to demonstrate that our platform could support sensitive, quantitative and multiplexed protein detection. For each immunoassay, a mixture of two populations of antibody-conjugated microbeads were assembled onto an array via an electric-field and then treated with a blocking solution prior to being exposed to a solution containing the antigens. The microbeads were then probed with fluorescently labeled detection antibodies and subsequently imaged via automated epifluorescence microscopy. With each type of capture antibody conjugated to a different fluorescently-labeled microbead, we were able to detect simultaneously two different antigens. The fluorescent signal from each microbead was measured in the detection channel (Alexa 568) and an antigen was considered present if the mean intensity in its corresponding channel was at least three times the standard deviation of the mean microbead background intensity in the same channel.42 Standard curves were generated using antigen concentrations ranging from 0 to 625 pM and our detection limit was determined to be ~40 pM (6 ng/mL) (Fig. 6), which is similar to the sensitivities of other microbead-based immunoassays.25,26 The variation in our mean intensities can be attributed to nonuniformity in the microbead populations and slight focusing inconsistencies with fluorescence imaging.
Fig. 6.
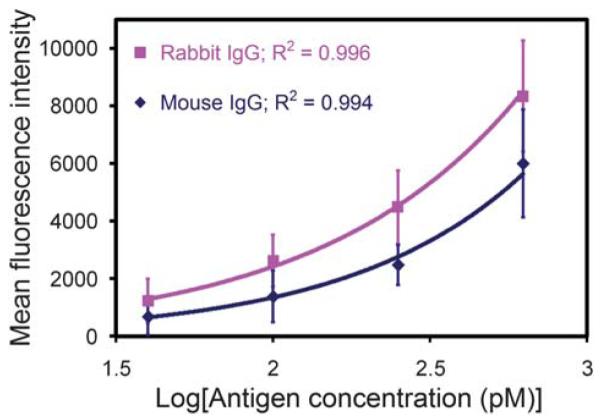
Standard curves for the sandwich immunoassays performed on antibody-conjugated microbead arrays. Graph of the mean fluorescence intensity from 1 μm antibody-conjugated microbeads as a function of the log10 of the antigen concentration. Our detection limit was determined to be ~40 pM (6 ng/mL) for both antigens.
Optimization of parameters such as surface chemistry,43,44 incubation times and the number of a given type of antibody-conjugated microbead assembled on the array may help bring the sensitivity of this approach closer to that of ELISA but without enzymatic signal amplification. The latter of these factors may be of importance at low antigen concentrations due to the small number of antigens available per microbead. Therefore, it may be beneficial to limit the number of microbeads of a given type to the minimum necessary to give statistically significant data. Other potential improvements include fluid oscillation, optimization of the incubation temperature and the application of an electric-field31-33 to direct the antigens toward the electrode-bound microbeads. These approaches may enhance the diffusion-limited process of capturing antigens, thereby increasing the sensitivity of the assay and reducing the total assay time. Furthermore, the sensitivity of our immunoassays may also be improved through the use of quantum dot-labeled detection antibodies45 or an immunoRCA strategy.46
Silicon dioxide wells
The use of silicon dioxide as a dielectric for the electrophoretic assembly of colloidal crystals has been demonstrated elsewhere.47 We have utilized this material in our device because it offers numerous advantages over the epoxy-based photoresist used in previous work.39 Although the fabrication process is simpler when using epoxy-based photoresist, the wells are partially destroyed during the plasma cleaning of the gold electrodes. In addition, the photoresist is more susceptible to chemical damage from the by-products of electrolysis during the microbead assembly process. In contrast, silicon dioxide can withstand harsh environments and processes and may enable the use of various chip bonding techniques such as anodic and thermal bonding. It is also well-suited for direct bonding to PDMS. The use of silicon dioxide also enables more control over the geometric properties of the wells. For instance, precise depths and vertical wall profiles are easily produced. In addition, the use of a silicon dioxide layer facilitates the use of higher resolution microfabrication processes such as DUV lithography and RIE/ICP. Once the array of microwells have been fabricated, the surface properties of the oxide can be modified using silane-based chemistry, which would allow, for example, the passivation of the oxide with polyethylene glycol (PEG) to prevent non-specific binding of microbeads and biomolecules.43
Counter electrode lines
The use of a counter electrode that consists of a series of gold lines fabricated on a glass coverslip offers several advantages over the use of an ITO-coated coverslip. First, there is no loss of light when imaging between the gold lines. In contrast, ITO films are typically only 80–90% transparent to light in the visible spectrum, which could effectively reduce the sensitivity of epifluorescence-based assays. Second, the gold lines can be fabricated using a relatively simple lift-off process whereas the deposition of high-quality ITO films requires the optimization of multiple parameters. Third, the gold lines are also very durable compared to ITO films, which tend to degrade when subjected to high electrical currents or the by-products of electrolysis that are produced under electrophoretic conditions. While a small number of 3.0 V DC electrical pulses may not have a significant effect, continuous exposure to these electrophoretic conditions for just one minute can result in a significant reduction in the transmission of visible light through the film. To demonstrate this phenomenon, we measured the transmittance of an ITO film after subjecting it to a continuous 3.0 V DC potential (Fig. 7). The experimental results indicate that the drop in transmittance is wavelength dependent, but is greater than 90% in the lower end of the visible spectrum after just three minutes. This decay would significantly affect the imaging sensitivity of a system using ITO as a counter electrode. The electrical properties of the ITO film were also affected to some degree as evidenced by a doubling of the sheet resistance after three minutes.
Fig. 7.
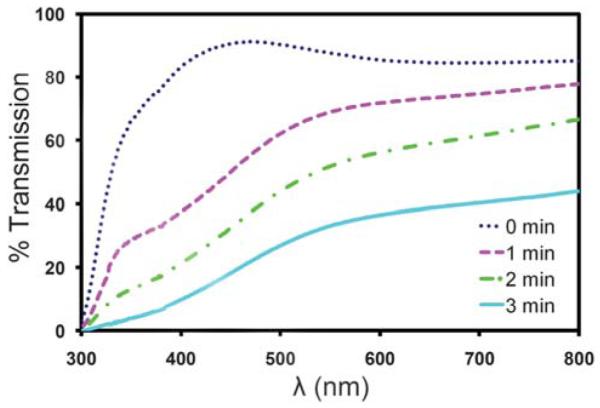
Light transmission through an ITO film subjected to electrophoretic conditions. The percent transmittance of light through an ITO-based electrophoretic device is plotted as a function of total exposure time to electrophoretic conditions.
Although the use of gold lines as counter electrodes may produce non-uniform electric fields within the chamber, microbead assembly was still rapid and uniform across the entire array when 25 μm counter electrode lines at a pitch of 320 μm were used. However, our attempts to assemble similar arrays using lines at a 640 μm pitch were unsuccessful. We developed a 2-D finite element model of the device to examine the electric field distribution within the chamber for varying counter electrode line pitches and chamber heights. A cross-sectional view of a section of the chamber is illustrated in Fig. 8A. A screenshot of a portion of a solved model is shown in Fig. 8B. For each device configuration studied, a horizontal line plot between adjacent counter electrode line centers was generated at a height of 5 μm above the surface of the array. In Fig. 8C, the chamber height was held at 110 μm while the counter electrode line pitch was varied from 160 μm to 640 μm. At this chamber height, the drop in field strength from a position directly underneath the center of an electrode line to the midpoint between adjacent electrode lines varied from 10% at a pitch of 160 μm to 96% at a pitch of 640 μm. At a pitch of 320 μm, this drop was 60%, and yet we were still able to achieve uniform assembly with this configuration. However, observations made using electrode lines at a 640 μm pitch confirmed that there is a threshold below which assembly cannot be performed. In Fig. 8D, the pitch was held at 320 μm while the chamber height was varied from 55 μm to 220 μm. At this pitch, the drop in field strength from a position directly underneath the center of an electrode line to the midpoint between adjacent electrode lines varied from 10% at a height of 220 μm to 95% at a height of 55 μm. This indicates that a uniform electric field could be attained by simply changing the height of the chamber. However, a significant increase in height will prohibit the use of microscope objectives with short working distances and may require a higher voltage for assembly. In our particular model, we do not take into account electrohydrodynamic and convective flow,48,49 but future models that include these factors may allow us to optimize further the electrode configurations, device geometry, and electrophoretic conditions.
Fig. 8.

Finite element analysis of the electric-field distribution within the microfabricated electrophoretic device. (A) An illustrative cross-section of a portion of the device. (B) Surface and contour plot of the y-component of the electric-field strength, Ey, in a scaled COMSOL model of the device. In this particular model, the counter electrode lines are 25 μm wide at a pitch of 320 μm. The chamber is 110 μm high by 2 mm wide and the wells are 0.25 μm deep by 1.2 μm wide with a pitch of 2.4 μm. The inset shows the electric field distribution within a single well. The scale bar is 50 μm. (C) A plot of Ey as a function of the horizontal position between neighboring counter electrode line centers for varying counter electrode line pitches. The channel height was held at 110 μm. (D) A plot of Ey as a function of the horizontal position between neighboring counter electrode line centers for varying chamber heights. The counter electrode line pitch was held at 320 μm. The line plots in (C) and (D) were generated at a fixed height of 5 μm above the surface of the array and the applied potential across the chamber was 3.0 V DC.
We found that 25 μm-wide lines at a pitch of 320 μm allow for excellent microbead assembly as well as unobstructed imaging between them when imaged with a 40× objective and an EMCCD camera with 1004 × 1002 pixels (8 μm × 8 μm pixels). Lines that were too close together lead to the presence of shadows in the images. As shown in Fig. 9, these shadows diminish the signal from the microbeads up to a distance of ~50 μm from the line. The image was acquired in the FITC channel in a region of the chip where 1 μm antibody-conjugated beads were located directly underneath a 25 μm-wide counter electrode line. Even though the counter electrode lines need to be spaced such that imaging can be performed far enough from the lines to avoid their shadows, this approach offers greater durability and better light transmission than ITO while still providing the means for uniform, efficient assembly of the antibody-conjugated microbeads.
Fig. 9.

Imaging of microbeads under and near a gold counter electrode line. (A) Fluorescent micrograph of 1 μm antibody-conjugated, fluorescent polystyrene beads assembled near and under a 25 μm gold counter electrode line. The approximate location of the electrode line is given by the dashed lines and is 110 μm above the focal plane in this image. (B) A corresponding profile of the signal to background ratio across the entire 200 μm wide image. The scale bar in (A) is 24 μm.
4. Summary
We have demonstrated that high-density, antibody-conjugated microbead arrays can be assembled via electrophoretic deposition on microfabricated arrays of wells in silicon dioxide on gold-coated silicon chips. In addition, thin gold lines fabricated on glass coverslips were used as counter electrodes to provide a more robust platform for assembly and enable greater imaging sensitivity than possible with ITO-coated coverslips. Assembly of the antibody-conjugated microbeads was rapid and resulted in high-density arrays with minimal defects. We have demonstrated the feasibility of a spatial encoding scheme and have also shown that the assembled antibody arrays could be used to detect test antigens at concentrations as low as 40 pM (6 ng/mL) using sandwich immunoassays. Our microfabricated electrophoretic device and methods will be useful for rapid assembly of encoded antibody arrays for multiplexed detection of proteins. Furthermore, our ability to assemble antibody-conjugated microbeads may be extended to a multitude of other types of protein-conjugated microbeads for other applications such as the analysis of protein-protein interactions.
Acknowledgements
This work was supported in part by grants from the NIH/NHGRI (R21HG004130 and R01HG004804) and the NSF under a CAREER award to X. H. (BES-0547193). A portion of this work was conducted at the Triangle National Lithography Center at North Carolina State University (NCSU). We thank David Vellenga and Marcio Cerullo at NCSU for their training, technical support and services, which included DUV photolithography and BARC etching. A portion of this work was done in the nanofabrication facility at the University of California, Santa Barbara (UCSB), part of the NSF funded NNIN network. We thank Dr Brian Thibeault for training and technical support at UCSB. Part of this work was also performed in the Nano3 facility at CalIT2 at the University of California, San Diego (UCSD). We thank Michael Clark for dicing our wafers and Larry Grissom and Ryan Anderson for training and technical support at UCSD. We also thank Nora Theilacker for advice and assistance with antibodies and image analysis routines.
References
- 1.Pandey A, Mann M. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 2.MacBeath G. Nat. Genet. 2002;32(Suppl):526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 3.Phizicky E, Bastiaens PIH, Zhu H, Snyder M, Fields S. Nature. 2003;422:208–215. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- 4.Madoz-Gúrpide J, Wang H, Misek DE, Brichory F, Hanash SM. Proteomics. 2001;1:1279–1287. doi: 10.1002/1615-9861(200110)1:10<1279::AID-PROT1279>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 5.Knezevic V, Leethanakul C, Bichsel VE, Worth JE, Prabhu VV, Gutkind JS, Liotta LA, Munson PJ, Petricoin EF, III, Krizman DB. Proteomics. 2001;1:1271–1278. doi: 10.1002/1615-9861(200110)1:10<1271::AID-PROT1271>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Bellisario R, Colinas RJ, Pass KA. Early Hum. Dev. 2001;64:21–25. doi: 10.1016/s0378-3782(01)00167-0. [DOI] [PubMed] [Google Scholar]
- 7.Burbaum J, Tobal GM. Curr. Opin. Chem. Biol. 2002;6:427–433. doi: 10.1016/s1367-5931(02)00337-x. [DOI] [PubMed] [Google Scholar]
- 8.Renart J, Reiser J, Stark GR. Proc. Natl. Acad. Sci. U. S. A. 1979;76:3116–3120. doi: 10.1073/pnas.76.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engvall E, Perlmann P. Immunochemistry. 1971;8:871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- 10.Silzel JW, Cercek B, Dodson C, Tsay T, Obremski RJ. Clin. Chem. 1998;44:2036–2043. [PubMed] [Google Scholar]
- 11.Arenkov P, Kukhtin A, Gemmell A, Voloshchuk S, Chupeeva V, Mirzabekov A. Anal. Biochem. 2000;278:123–131. doi: 10.1006/abio.1999.4363. [DOI] [PubMed] [Google Scholar]
- 12.MacBeath G, Schreiber SL. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 13.de Wildt RMT, Mundy CR, Gorick BD, Tomlinson IM. Nat. Biotechnol. 2000;18:989–994. doi: 10.1038/79494. [DOI] [PubMed] [Google Scholar]
- 14.Holt LJ, Bussow K, Walter G, Tomlinson IM. Nucleic Acids Res. 2000;28:72e. doi: 10.1093/nar/28.15.e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 16.Moody MD, Van Arsdell SW, Murphy KP, Orencole SF, Burns C. Biotechniques. 2001;31:186–194. doi: 10.2144/01311dd03. [DOI] [PubMed] [Google Scholar]
- 17.Haab BB, Dunham MJ, Brown PO. GenomeBiology. 2001;2:0004.1–0004.13. [Google Scholar]
- 18.Huang R-P, Huang R, Fan Y, Lin Y. Anal. Biochem. 2001;294:55–62. doi: 10.1006/abio.2001.5156. [DOI] [PubMed] [Google Scholar]
- 19.Dasso J, Lee J, Bach H, Mage RG. J. Immunol. Methods. 2002;263:23–33. doi: 10.1016/s0022-1759(02)00028-5. [DOI] [PubMed] [Google Scholar]
- 20.Kellar KL, Iannone MA. Exp. Hematol. 2002;30:1227–1237. doi: 10.1016/s0301-472x(02)00922-0. [DOI] [PubMed] [Google Scholar]
- 21.Goodey A, Lavigne JJ, Savoy SM, Rodriguez MD, Curey T, Tsao A, Simmons G, Wright J, Yoo S-J, Sohn Y, Anslyn EV, Shear JB, Neikirk DP, McDevitt JT. J. Am. Chem. Soc. 2001;123:2559–2570. doi: 10.1021/ja003341l. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann M, Roy E, Veres T, Tabrizian M. Lab Chip. 2007;7:1546–1552. doi: 10.1039/b707883h. [DOI] [PubMed] [Google Scholar]
- 23.Diercks AH, Ozinsky A, Hansen CL, Spotts JM, Rodriguez DJ, Aderem A. Anal. Biochem. 2009;386:30–35. doi: 10.1016/j.ab.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu X, Thompson JA, Chen Z, Liu C, Chen D, Ramprasad S, Mauk M, Ongagna S, Barber C, Abrams WR, Malamud D, Corstjens PLAM, Bau HH. Biomed. Microdevices. 2009;11:1175–1186. doi: 10.1007/s10544-009-9334-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blicharz TM, Siqueira WL, Helmerhorst EJ, Oppenheim FG, Wexler PJ, Little FF, Walt DR. Anal. Chem. 2009;81:2106–2114. doi: 10.1021/ac802181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivagnanam V, Song B, Vandevyver C, Gijs MAM. Anal. Chem. 2009;81:6509–6515. doi: 10.1021/ac9009319. [DOI] [PubMed] [Google Scholar]
- 27.Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, Hughes MA, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19272–19277. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong JF, Chen Y, Marcus JS, Scherer A, Quake SR, Taylor CR, Weiner LP. Lab Chip. 2008;8:68–74. doi: 10.1039/b712116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai L, Friedman N, Xie XS. Nature. 2006;440:358–362. doi: 10.1038/nature04599. [DOI] [PubMed] [Google Scholar]
- 30.Newman JRS, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 31.Ewalt KL, Haigis RW, Rooney R, Ackley D, Krihak M. Anal. Biochem. 2001;289:162–172. doi: 10.1006/abio.2000.4927. [DOI] [PubMed] [Google Scholar]
- 32.Morozov VN, Groves S, Turell MJ, Bailey C. J. Am. Chem. Soc. 2007;129:12628–12629. doi: 10.1021/ja075069m. [DOI] [PubMed] [Google Scholar]
- 33.Wu J, Yan Y, Yan F, Ju H. Anal. Chem. 2008;80:6072–6077. doi: 10.1021/ac800905k. [DOI] [PubMed] [Google Scholar]
- 34.Ng JK, Selamat ES, Liu W-T. Biosens. Bioelectron. 2008;23:803–810. doi: 10.1016/j.bios.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 35.Michael KL, Taylor LC, Schultz SL, Walt DR. Anal. Chem. 1998;70:1242–1248. doi: 10.1021/ac971343r. [DOI] [PubMed] [Google Scholar]
- 36.Han M, Gao X, Su JZ, Nie S. Nat. Biotechnol. 2001;19:631–635. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 37.Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, Doucet D, Milewski M, Yang R, Siegmund C, Haas J, Zhou L, Oliphant A, Fan J-B, Barnard S, Chee MS. Genome Res. 2004;14:870–877. doi: 10.1101/gr.2255804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birtwell S, Morgan H. Integr. Biol. 2009;1:345–362. doi: 10.1039/b905502a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barbee KD, Hsiao AP, Heller MJ, Huang X. Lab Chip. 2009;9:3268–3274. doi: 10.1039/b912876j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Folcher G, Cachet H, Froment M, Bruneaux J. Thin Solid Films. 1997;301:242–248. [Google Scholar]
- 41.Abramoff MD, Magelhaes PJ, Ram SJ. Biophotonics Intl. 2004;11:36–42. [Google Scholar]
- 42.Anderson DJ. Clin. Chem. 1989;35:2152–2153. [PubMed] [Google Scholar]
- 43.Hucknall A, Kim D-H, Rangarajan S, Hill RT, Reichert WM, Chilkoti A. Adv. Mater. 2009;21:1968–1971. doi: 10.1002/adma.200803125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain P, Baker GL, Bruening ML. Annu. Rev. Anal. Chem. 2009;2:387–408. doi: 10.1146/annurev-anchem-060908-155153. [DOI] [PubMed] [Google Scholar]
- 45.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat. Mater. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 46.Schweitzer B, Wiltshire S, Lambert J, O’Malley S, Kukanskis K, Zhu Z, Kingsmore SF, Lizardi PM, Ward DC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:10113–10119. doi: 10.1073/pnas.170237197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dziomkina NV, Hempenius MA, Vancso GJ. Adv. Mater. 2005;17:237–240. [Google Scholar]
- 48.Trau M, Saville DA, Aksay IA. Langmuir. 1997;13:6375–6381. [Google Scholar]
- 49.Cordovez B, Psaltis D, Erickson D. Appl. Phys. Lett. 2007;90:024102. [Google Scholar]