

Abstract
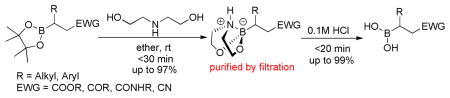
A two-step procedure for deprotection of alkylpinacolyl boronate esters via transesterification with diethanolamine followed by hydrolysis was successfully developed with the advantages of tolerance to various functional groups, short reaction time and ease of product isolation.
Boronic acids are important in organic synthesis due to their versatility as synthetic intermediates in the preparation of complex molecules. Their utility is exemplified by the powerful carbon-carbon bond forming Suzuki-Miyaura coupling reaction.1,2 As inhibitors of serine proteases, boronic acids inhibit therapeutically relevant proteases including chymotrypsin, thrombin, dipeptidyl peptidase, HCV NS3 protease, and the proteasome.3 Indeed, the first boron-containing drug, Bortezomib (Velcade), was approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma. Thus, methods for synthesizing boronic acids are vital in harnessing their full potential in a variety of applications.
Introduction of the boronyl moiety has been a subject of intense investigation. Traditionally, aryl boronic acids can be obtained from electrophilic borate trapping of arylmetal intermediates from aryl halides,4 transmetallation of arylsilanes and stananes,5 transition metal-catalyzed coupling between aryl halides/triflates and diboron reagents,6 and direct boration by transition metal-catalyzed aromatic C-H activation.7,8 Some boronic acids are unstable and incompatible to reaction conditions during functional group transformations; therefore they are converted to air- and chromatography stable boronate esters using diols such as pinanediol,9 cathecol,10,11 neopentyl glycol12–16 and pinacol.17,18 Although these groups are usually benign to various reaction conditions, their removal has been troublesome and unpredictable.19 Recently, trifluoroborate,20 N-methyliminodiacetic acid21–23 and 1,8-diaminonaphthalene24,25 units were introduced as alternative protecting groups for boronic acids. Organotrifluoroborate salts undergo hydrolysis in the presence of silica gel to afford the corresponding boronic acids26 and, more recently, a direct route to arylboronic acids was reported.27
Several deprotection methods for the removal of cyclic boronate esters are available. These include oxidative cleavage with sodium periodate,28 biphasic transesterification with other boronic acids,28,29 transborylation with boron trichloride, 30–32 and acidic hydrolysis.33,34 The yield and purity of these deprotection procedures vary greatly, notwithstanding issues that can arise from functional group incompatibilities and purification problems. Alternatively, a solid-phase process that utilizes polystyrene-boronic acid has been developed for arylpinacolyl boronic esters.35 However, this method requires acidic conditions, is time-consuming, and relies on the commercial availability of the resin. Recently, Hutton and co-workers36 reported a two-step deprotection of pinacol group via fluorinated intermediates37 that is followed by trimethylsilyl chloride38 or lithium hydroxide.
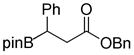
In the course of our work on the development of N-terminal peptidic boronic acids as protease inhibitors,39 we were faced with the difficulty of removing the pinacolyl group during the initial phase of our study. The boronyl moiety in all of these cases was directly attached to alkyl groups. For example, when β-boronopinacolyl-β-phenyl benzyl ester 1e was subjected to various deprotection conditions, either the starting material decomposed or no reaction occurred, and thus β-boronic acid 3e was not obtained (Scheme 1).
SCHEME 1.

Attempted deprotection of 1e.
In order to circumvent this issue, we investigated a transesterification procedure with diethanolamine (DEA) to form an sp3-hybridized boron·DEA adduct, which is then hydrolyzed to yield a boronic acid derivative. Indeed, a few examples of pinacol deprotection with DEA have been reported, but most of these are arylboronic esters.40–42 To date, only two examples of successful alkylpinacolyl boronate ester deprotection involving transesterification and hydrolysis to produce boron-containing amino acid analogs have been disclosed.43,44 In some cases, an acidic resin, long reaction time or distillation was necessary to effectively provide the boronic acid moiety. Because there has not been a comprehensive investigation of the boryl pinacolate deprotection with DEA, we explored the generality of a two-step protocol in the deprotection of alkylpinacolyl boronate esters. Herein, we report a simple and efficient deprotection method of pinacolyl boronate esters.
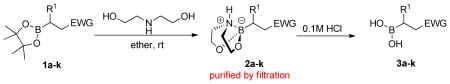
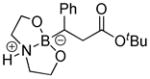
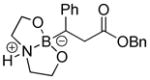
We initially employed an arylboronic ester as our test case (Scheme 2). Treatment of pinacolylphenylboronic ester with DEA in ether afforded the DEA boronate product, which was filtered and subsequently treated with 0.1M HCl for 20 minutes to provide phenylboronic acid in 99% yield. Having established the deprotection procedure, we investigated whether a one-pot deprotection method was possible with pinacolyl boronate ester substrates.45 However, none of these studies (changing solvents, biphasic solutions, sequential addition of reagents, etc.) was successful. Thus, we elected to pursue the two-step transesterification/hydrolysis using a variety of pinacolyl-β-alkylboronic esters. As shown in Table 1, transesterification of the pinacol group with DEA efficiently proceeded in up to 97% isolated yield. It should be noted that this reaction not only occurred in a short amount of time (<30 min) but the purification was a simple filtration as the DEA boronate product precipitated in the ethereal solution. For example, unhindered substrates containing benzyl or methyl esters provided compounds 2a-b in good yields (entries 1–2). Introduction of substituents on the β-position with a phenyl or methyl group was also effective (entries 3–7). In addition, cyclic and acyclic ketones as well as nitriles underwent the transesterification smoothly (entries 8–11). All the isolated compounds 2a-l were white solids, stable to atmospheric condition, and had long shelf lives. These compounds were characterized by 1H, 13C, 11B NMR and high resolution mass spectroscopy. Introduction of DEA produced desirable effects: the formation of a “boron-ate” species that is stable to oxidation and an adduct that is insoluble in ether. sp3-Hybridized boron complexes are reported to be stable and resistant to air oxidation.20,22 To further confirm the tetracoordination on boron, the single-crystal X-ray diffraction data for 2b was solved. As shown in Figure 1, boron adopts a tetrahedral geometry similar to what is observed in aryl·DEA boronates.
SCHEME 2.

Two-step protocol for the boronic ester deprotection.
TABLE 1.
Substrate scope of the two-step transesterification/hydrolysis of pinacolyl boronate esters.a
 | |||||
|---|---|---|---|---|---|
| entry | substrate | 2 | % yield of 2b | 3 | % yield of 3b |
| 1 |
1a
|
|
85 |
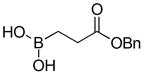
|
95 |
| 2 |
1b
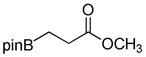
|
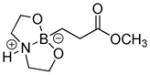
|
72 |
|
90 |
| 3 |
1c
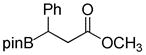
|
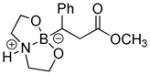
|
88 |
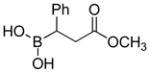
|
92c |
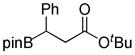
| 4 |
1d
|
|
75 |
|
96c |
| 5 |
1e
|
|
84 |
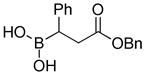
|
90c |
| 6 |
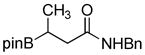
1f
|
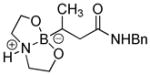
|
65 |
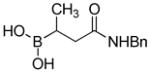
|
98 |
| 7 |
1g
|
|
68 |
|
69c |
| 8 |
1h
|
|
79 |
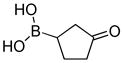
|
79c |
| 9 |
1i
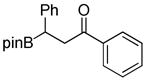
|
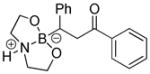
|
80 |
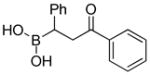
|
82 |
| 10 |
1j
|
|
76 |
|
31 |
| 11 |
1k
|
|
97 |
|
89c |
Each reaction was performed at least twice. Conditions: 1 (1 equiv), DEA (1.1 equiv), ether, rt, ~30 minutes followed by 0.1M HCl (20 min).
Isolated yield.
Product was isolated as the pinacol ester.
FIGURE 1.

X-ray crystal structure of 2b.
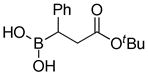
With the diethanolamine derivatives in hand, hydrolysis to the corresponding boronic acids was performed using a biphasic solution of 0.1M HCl and ether. As expected, the boronic acid products were readily formed within minutes. For example, esters 3a-3b, amides 3f-3g, ketone 3i and nitrile 3j were isolated as white solids in moderate to excellent yields (Table 1). However, in some cases, we experienced difficulty in isolating the desired alkylboronic acids. This was not surprising as some boronic acids are known to be unstable, undergoing facile decomposition in polar protic media via protodeboronation, oxidation, and/or polymerization.46–49 To test whether boronic acids were formed but decomposed during the reaction and/or work-up, the DEA boronates were subjected to a biphasic solution of 0.1M HCl and hexanes in the presence of 2 equiv. of pinacol. We hypothesized that DEA boronates 2 undergo rapid hydrolysis under acidic aqueous conditions as previously observed, and the unstable alkylboronic acids 3 subsequently trapped as the boron pinacolyl esters 1 (Scheme 3). Indeed, when the in situ hydrolysis-trapping protocol was performed, the pinacol boronate esters were achieved in excellent yields (Table 1).
SCHEME 3.

Rapid hydrolysis and subsequent trapping of 2.
In conclusion, a two-step deprotection protocol for alkyl pinacolyl boronic esters via DEA-protected boronates was successfully developed with the advantages of tolerance to various functional groups, short reaction time and ease of product isolation. Moderate to excellent yield was achieved for stable boronic acid products.
Experimental Section
General Procedure for the Synthesis of DEA Protected Boronic Esters 2a-l
To a solution of pinacolyl boronic ester 1 (1.7 mmol) in ether was added diethanolamine (0.199 g, 1.9 mmol). After a few minutes, a white precipitate formed and the reaction was allowed to continue until the starting material was completely consumed as monitored by TLC (~30 minutes). The precipitate was then filtered, washed with ether, and dried to afford the desired product 2.
8-(3-(Benzyloxy)-3-oxopropyl)hexahydro-[1,3,2] oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2a)
White solid (0.305 g, 85 % yield). mp 159.7–161.6 °C. 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.27 (m, 5H), 6.16 (s, 1H), 5.08 (s, 2H), 3.95 (m, 2H), 3.81 (m, 2H), 3.15 – 3.00 (m, 2H), 2.76 – 2.65 (m, 2H), 2.60 – 2.48 (m, 2H), 0.81 – 0.56 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 179.3, 136.1, 128.7, 128.3, 128.0, 66.3, 63.3, 51.3, 29.8. The signal for the carbon directly attached to boron was not observed due to quadrupolar relaxation. 11B NMR (160 MHz, CDCl3) δ 11.26. HRMS (ESI+) calculated for C14H21BO4 [M+H]+: 278.1558, found: 278.1532.
8-(3-Methoxy-3-oxopropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2b)
White solid, 72% yield; mp 147.3–148.4 °C; 1H NMR (500 MHz, CD3Cl) δ 6.39 (s, 1H), 4.00 (td, J = 5.3, 9.6, 2H), 3,92 – 3.81 (m, 2H), 3.65 (s, 3H), 3.26 – 3.14 (m, 2H), 2.86 – 2.73 (m, 2H), 2.53 – 2.45 (m, 2H), 0.77 – 0.60 (m, 2H).13C NMR (125 MHz, CD3Cl) δ 180.1, 63.3, 51.71, 51.3, 29.4. 11B NMR (160 MHz, CD3Cl) δ 11.25. HRMS (ESI+) Calculated for C8H17BNO4 [M+H]+: 202.1245, found: 202.1243.
8-(3-Methoxy-3-oxo-1-phenylpropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2c)
White solid, 88% yield; mp 188.7–189.9 °C; 1H NMR (500 MHz, CD3Cl) δ 7.29 – 6.98 (m, 5H), 5.65 (s, 1H), 4.01 – 3.88 (m, 2H), 3.87 – 3.78 (m, 2H), 3.62 (s, 3H), 3.12 – 3.01 (m, 1H), 3.00 – 2.89 (m, 2H), 2.76 – 2.64 (m, 3H), 2.32 (dd, J = 5.5, 8.1 Hz, 1H). 13C NMR (125 MHz, CD3Cl) δ 177.9, 147.34, 128.2, 127.8, 124.5, 63.4, 63.2, 51.7, 51.5, 51.4, 37.0. 11B NMR (160 MHz, CD3Cl) δ 11.25. HRMS (ESI+) Calculated for C14H21BNO4 [M+H]+: 278.1558, found: 278.1543.
8-(3-(tert-Butoxy)-3-oxo-1-phenylpropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2d)
White solid, 75% yield; mp 186.5–187.7 °C; 1H NMR (500 MHz, CD3Cl) δ 6.01 (s, 1H), 4.03 – 3.91 (m, 2H), 3.89 – 3.81 (m, 2H), 3.22 – 3.11 (m, 1H), 3.02 – 2.94 (m, 1H), 2.94 – 2.84 (m, 1H), 2.79 – 2.68 (m, 2H), 2.61 (dd, J = 4.8, 17.2 Hz, 1H), 2.27 (dd, J = 4.6, 8.9 Hz, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, CD3Cl) δ 155.7, 131.8, 128.0, 127.8, 124.2, 80.3, 77.3, 77.0, 76.7, 63.4, 63.2, 51.3, 51.20, 38.5, 28.0. 11B NMR (160 MHz, CD3Cl) δ 10.38. HRMS (ESI+) Calculated for C17H27BNO4 [M+H]+: 320.2028, found: 320.2006.
8-(3-Oxo-3-phenoxy-1-phenylpropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2e)
White solid, 84% yield; mp 173.9–174.8 °C; 1H NMR (400 MHz, CD3CN) δ 7.41 – 6.99 (m, 9H), 4.96 (s, 3H), 3.82 – 3.70 (m, 2H), 3.67 – 3.49 (m, 2H), 3.01 – 2.85 (m, 1H), 2.82 – 2.60 (m, 6H), 2.34 (dd, J = 5.8, 9.8 Hz, 1H). 13C NMR (100 MHz, CD3CN) δ 174.7, 128.3, 128.0, 127.7, 127.6, 124.0, 117.3, 105.0, 65.2, 62.5, 62.4, 51.2, 51.0, 36.7. 11B NMR (160 MHz, CD3Cl) δ 10.86. HRMS (ESI+) Calculated for C20H25BNO4 [M+H]+: 354.1871, found: 354.1878.
8-(4-(Benzylamino)-4-oxobutan-2-yl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2f)
White solid, 65% yield; mp 127.5–128.6 °C; 1H NMR (500 MHz, CD3Cl) δ 7.77 (s, 1H), 7.41 – 7.10 (m, 5H), 6.00 (s, 1H), 4.39 (d, J = 5.7 Hz, 2H), 4.04 – 3.77 (m, 4H), 3.35 – 3.01 (m, 2H), 2.82 – 2.68 (m, 2H), 2.49 – 2.10 (m, 2H), 0.99 (d, J = 6.1 Hz, 3H), 0.94 (dd, J = 9.1, 17.7 Hz, 1H). 13C NMR (125 MHz, CD3Cl) δ 177.3, 138.0, 128.8, 127.6, 127.6, 63.5, 63.4, 51.5, 51.3, 43.7, 40.8, 17.3. 11B NMR (160 MHz, CD3Cl) δ 11.37. HRMS (ESI+) Calculated for C15H24BN2O3 [M+H]+: 291.1874, found: 291.1890.
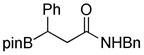
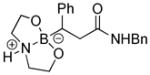
8-(3-(Benzylamino)-3-oxo-1-phenylpropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2g)
White solid, 68% yield; mp 197.3–198.2 °C; 1H NMR (500 MHz, CD3Cl) δ 7.34 – 7.19 (m, 10H), 7.08 – 7.01 (m, 1H), 6.04 (s, 1H), 4.39 (d, J = 5.7 Hz, 2H), 4.02 – 3.76 (m, 4H), 3.33 – 3.19 (m, 1H), 3.10 – 2.62 (m, 2H), 2.53 – 2.19 (m, 2H). 13C NMR (125 MHz, CD3Cl) δ 176.7, 148.1, 137.9, 128.8, 128.2, 127.9, 127.6, 124.4, 63.6, 63.5, 51.4, 43.8, 40.2. 11B NMR (160 MHz, CD3Cl) δ 11.03. HRMS (ESI+) Calculated for C20H26BN2O3 [M+H]+: 353.2031, found: 353.2034.
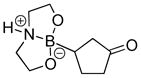
8-(3-Oxocyclopentyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2h)
White solid, 79% yield; mp 201.4–202.1 °C; 1H NMR (500 MHz, CD3CN) δ 5.13 (s, 1H), 3.88 – 3.77 (m, 3H), 3.73 – 3.63 (m, 3H), 3.16 – 3.03 (m, 3H), 2.82 – 2.73 (m, 3H), 2.13 – 2.07 (m, 1H), 2.06 – 1.97 (m, 2H), 1.92 – 1.77 (m, 3H), 1.67 – 1.55 (m, 1H), 1.24 – 1.11 (m, 1H). 13C NMR (125 MHz, CD3Cl) δ 171.2, 63.4, 63.3, 52.3, 42.2, 39.9, 31.1, 26.2. 11B NMR (160 MHz, CD3Cl) δ 11.56. HRMS (ESI+) Calculated for C9H17BNO3 [M+H]+: 198.1296, found: 198.1308.
8-(3-Oxo-1,3-diphenylpropyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2i)
White solid, 72% yield; mp 170.0–171.4 °C; 1H NMR (400 MHz, CD3CN) δ 7.98 – 6.96 (m, 8H), 3.87 – 3.71 (m, 2H), 3.67 – 3.52 (m, 2H), 3.46 – 3.28 (m, 2H), 3.03 – 2.89 (m, 1H), 2.88 – 2.74 (m, 1H), 2.75 – 2.64 (m, 2H), 2.56 – 2.45 (m, 1H). 13C NMR (125 MHz, CD3Cl) δ 199.4, 148.0, 137.2, 133.3, 128.6, 128.3, 128.3, 128.1, 124.5, 63.5, 63.3, 51.5, 51.3, 43.3, 24.8. 11B NMR (160 MHz, CD3Cl) δ 11.32. HRMS (ESI+) Calculated for C19H23BNO3 [M+H]+: 324.1766, found: 324.1777.
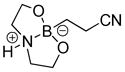
8-(2-Cyanoethyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2j)
White solid, 76% yield; mp 218.6–220.4 °C; 1H NMR (400 MHz, CD3CN) δ 3.81 (td, J = 9.3, 5.4 Hz, 1H), 3.74 – 3.63 (m, 1H), 3.23 – 3.04 (m, 1H), 2.84 – 2.69 (m, 1H), 2.20 (dd, J = 16.1, 7.8 Hz, 1H), 0.75 – 0.61 (m, 1H). 13C NMR (100 MHz, CD3CN) δ 117.3, 62.5, 51.2, 12.5. 11B NMR (160 MHz, CD3Cl) δ 11.28. HRMS (ESI+) Calculated for C7H14BN2O2 [M+H]: 169.1148, found: 169.1165.
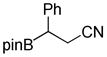
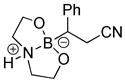
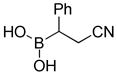
8-(2-Cyano-1-phenylethyl)hexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2k)
White solid, 97% yield; mp 147.5–148.9 °C; 1H NMR (500 MHz, CD3CN) δ 7.34 – 7.07 (m, 6H), 4.86 (s, 1H), 3.82 – 3.71 (m, 3H), 3.67 – 3.61 (m, 1H), 3.56 (dt, J = 4.8, 9.9 Hz, 1H), 3.04 – 2.94 (m, 1H), 2.74 – 2.61 (m, 7H), 2.18 (t, J = 8.3 Hz, 1H), 2.14 (s, 1H). 13C NMR (125 MHz, CD3CN) δ 145.3, 128.1, 128.0, 124.9, 117.4, 62.6, 62.5, 51.3, 51.2, 19.6. 11B NMR (160 MHz, CD3CN) δ 10.47. HRMS (ESI+) Calculated for C13H16BN2O2 [M+H]+: 243.1310, found: 243.1335.
8-Phenylhexahydro-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-4-ium-8-uide (2l)
White solid, 88% yield; mp 215.1–217.5 °C; 1H NMR (400 MHz, CD3CN) δ 7.55 – 7.15 (m, 2H), 3.97 (td, J = 9.3, 5.4 Hz, 1H), 3.93 – 3.81 (m, 1H), 3.28 – 3.14 (m, 1H), 2.90 – 2.78 (m, 1H). 13C NMR (100 MHz, CD3CN) δ 132.5, 126.9, 126.7, 63.1, 51.1. 11B NMR (160 MHz, CD3CN) δ 10.55. HRMS (ESI+) Calculated for C10H15BNO2 [M+H]+: 192.1196, found: 192.1204.
General Procedure for the Deprotection of DEA-Boronate Compound 2
To a solution of DEA-boronate 2 (1.1 mmol) in ether was added 0.1 M HCl. After about 20 minutes as judged by TLC, the reaction was extracted with ether (3x), washed with brine (1x), dried with sodium sulfate and the organic solvent was removed under reduced pressure. Evaporation of residual solvent provided the analytically pure product as white solid. Due to their facile dehydration, boronic acids tend to provide inconsistent melting points.50 Therefore the melting points for boronic acids were not taken.
(3-(Benzyloxy)-3-oxopropyl)boronic acid (3a)
White solid, 95% yield; 1H NMR (500 MHz, CD3Cl) δ 7.45 – 7.22 (m, 5H), 5.13 (s, 2H), 2.53 (t, J = 7.2 Hz, 2H), 1.12 (t, J = 7.0 Hz, 2H). 13C NMR (125 MHz, CD3Cl) δ 175.15, 136.02, 128.68, 128.35, 66.58, 28.69, 24.84. 11B NMR (160 MHz, CD3Cl) δ 31.59. HRMS (ESI+) Calculated for C10H13BO4Cl [M+Cl]+: 243.0601, found: 243.0619.
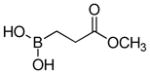
(3-Methoxy-3-oxopropyl)boronic acid (3b)
White solid, 90% yield; 1H NMR (500 MHz, CD3Cl) δ 3.66 (s, 3H), 2.48 (t, J = 7.2 Hz, 2H), 1.11 (t, J = 7.2 Hz, 2H). 13C NMR (125 MHz, CD3Cl) δ 175.9, 51.9, 28.4, 10.4. 11B NMR (160 MHz, CD3Cl) δ 31.49. HRMS (ESI+) Calculated for C4H10BO4[M+H]+: 133.0672, found: 133.0665.
(4-(Benzylamino)-4-oxobutan-2-yl)boronic acid (3f)
White solid, 98% yield; 1H NMR (500 MHz, CD3OD) δ 7.39 – 7.16 (m, 5H), 4.37 (q, J = 14.9 Hz, 2H), 2.32 (d, J = 8.3 Hz, 2H), 1.51 – 1.34 (m, 1H), 0.92 (d, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ 176.3, 138.4, 128.2, 127.3, 127.0, 43.2, 39.6, 14.3. 11B NMR (160 MHz, CD3OD) δ 26.96. HRMS (ESI+) Calculated for C11H20BN2O3[M+NH4]: 239.1561, found: 239.1568.
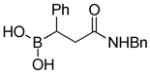
(3-(Benzylamino)-3-oxo-1-phenylpropyl)boronic acid (3g)
White solid, 69% yield; 1H NMR (500 MHz, CD3OD) δ 7.29 – 6.97 (m, 10H), 4.50 – 4.22 (m, 2H), 2.80 – 2.74 (m, 1H), 2.72 – 2.43 (m, 2H). 13C NMR (125 MHz, CD3OD) δ 176.7, 148.1, 137.9, 128.8, 128.2, 127.9, 127.7, 127.6, 124.4, 43.8, 40.2. 11B NMR (160 MHz, CD3OD) δ 25.39. HRMS (ESI+) Calculated for C16H19BNO3[M+H]+: 284.1453, found: 284.1478.
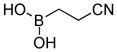
(2-Cyanoethyl)boronic acid (3j)
White solid, 31% yield; 1H NMR (500 MHz, (CD3)2CO) δ 2.42 (t, J = 5 Hz, 2H), 1.08 (t, J = 5 Hz, 2H). 13C NMR (125 MHz, CD3Cl) δ 120.8, 11.3. 11B NMR (160 MHz, (CD3)2CO) δ 18.89. HRMS (ESI+) Calculated for C3H6BNO2Cl[M+Cl]+: 134.0186, found: 134.0180.
Supplementary Material
Acknowledgments
We thank Dr. Carla Slebodnick for assistance with X-ray crystallography, and the Department of Chemistry at Virginia Tech and the National Institutes of Health (RO1 GM093834) for financial support.
Footnotes
Supporting Information Available: 1H, 13C and 11B NMR spectra of new compounds and CIF file for 2b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 2.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall D. Boronic acids: Preparation, applications in organic synthesis and medicine. Wiley-VCH GmbH & Co; Weinheim: 2005. [Google Scholar]
- 4.Brown HC, Cole TE. Organometallics. 2002;2:1316. [Google Scholar]
- 5.Sharp MJ, Cheng W, Snieckus V. Tetrahedron Lett. 1987;28:5093. [Google Scholar]
- 6.Ishiyama T, Murata M, Miyaura N. J Org Chem. 1995;60:7508. [Google Scholar]
- 7.Chen H, Hartwig JF. Angew Chem Int Ed Engl. 1999;38:3391. doi: 10.1002/(sici)1521-3773(19991115)38:22<3391::aid-anie3391>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- 9.Matteson DS. Tetrahedron. 1998;54:10555. [Google Scholar]
- 10.Kuivila HG, Keough AH, Soboczenski EJ. J Org Chem. 1954;19:780. [Google Scholar]
- 11.Pizer R, Babcock L. Inorg Chem. 1977;16:1677. [Google Scholar]
- 12.Nguyen P, Lesley G, Taylor NJ, Marder TB, Pickett NL, Clegg W, Elsegood MRJ, Norman NC. Inorg Chem. 1994;33:4623. [Google Scholar]
- 13.Lawlor FJ, Norman NC, Pickett NL, Robins EG, Nguyen P, Lesley G, Marder TB, Ashmore JA, Green JC. Inorg Chem. 1998;37:5282. [Google Scholar]
- 14.Clegg W, Elsegood MRJ, Lawlor FJ, Norman NC, Pickett NL, Robins EG, Scott AJ, Nguyen P, Taylor NJ, Marder TB. Inorg Chem. 1998;37:5289. [Google Scholar]
- 15.Fang H, Kaur G, Yan J, Wang B. Tetrahedron Lett. 2005;46:1671. [Google Scholar]
- 16.Moldoveanu C, Wilson DA, Wilson CJ, Leowanawat P, Resmerita AM, Liu C, Rosen BM, Percec V. J Org Chem. 2010;75:5438. doi: 10.1021/jo101023t. [DOI] [PubMed] [Google Scholar]
- 17.Männig D, Nöth H. J Chem Soc, Dalton Trans. 1985:1689. [Google Scholar]
- 18.Ishiyama T, Murata M, Ahiko TA, Miyaura N. Org Synth. 1999;77:176. [Google Scholar]
- 19.Brown HC, Rangaishenvi MV. J Organomet Chem. 1988;358:15. [Google Scholar]
- 20.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 21.Mancilla T, Contreras R, Wrackmeyer B. J Organomet Chem. 1986;307:1. [Google Scholar]
- 22.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 23.Gillis EP, Burke MD. J Am Chem Soc. 2008;130:14084. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noguchi H, Shioda T, Chou CM, Suginome M. Org Lett. 2008;10:377. doi: 10.1021/ol702420x. [DOI] [PubMed] [Google Scholar]
- 25.Noguchi H, Hojo K, Suginome M. J Am Chem Soc. 2007;129:758. doi: 10.1021/ja067975p. [DOI] [PubMed] [Google Scholar]
- 26.Molander GA, Cavalcanti LN, Canturk B, Pan PS, Kennedy LE. J Org Chem. 2009;74:7364. doi: 10.1021/jo901441u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coutts SJ, Adams J, Krolikowski D, Snow RJ. Tetrahedron Lett. 1994;35:5109. [Google Scholar]
- 29.Snow RJ, Bachovchin WW, Barton RW, Campbell SJ, Coutts SJ, Freeman DM, Gutheil WG, Kelly TA, Kennedy CA. J Am Chem Soc. 1994;116:10860. [Google Scholar]
- 30.Kinder DH, Katzenellenbogen JA. J Med Chem. 1985;28:1917. doi: 10.1021/jm00150a027. [DOI] [PubMed] [Google Scholar]
- 31.Matteson DS, Jesthi PK, Sadhu KM. Organometallics. 1984;3:1284. [Google Scholar]
- 32.Martichonok V, Jones JB. J Am Chem Soc. 1996;118:950. [Google Scholar]
- 33.Martin R, Jones JB. Tetrahedron Lett. 1995;36:8399. [Google Scholar]
- 34.Coutts SJ, Kelly TA, Snow RJ, Kennedy CA, Barton RW, Adams J, Krolikowski DA, Freeman DM, Campbell SJ, Ksiazek JF, Bachovchin WW. J Med Chem. 1996;39:2087. doi: 10.1021/jm950732f. [DOI] [PubMed] [Google Scholar]
- 35.Pennington TE, Kardiman C, Hutton CA. Tetrahedron Lett. 2004;45:6657. [Google Scholar]
- 36.Yuen AKL, Hutton CA. Tetrahedron Lett. 2005;46:7899. [Google Scholar]
- 37.Inglis SR, Woon ECY, Thompson AL, Schofield CJ. J Org Chem. 2009;75:468. doi: 10.1021/jo901930v. [DOI] [PubMed] [Google Scholar]
- 38.Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020. [Google Scholar]
- 39.Knott K, Fishovitz J, Thorpe SB, Lee I, Santos WL. Org Biomol Chem. 2010;8:3451. doi: 10.1039/c004247a. [DOI] [PubMed] [Google Scholar]
- 40.Jung ME, Lazarova TI. J Org Chem. 1999;64:2976. doi: 10.1021/jo9902751. [DOI] [PubMed] [Google Scholar]
- 41.Song YL, Morin C. Synlett. 2001:266. [Google Scholar]
- 42.Perttu EK, Arnold M, Iovine PM. Tetrahedron Lett. 2005;46:8753. [Google Scholar]
- 43.Kinder DH, Ames MM. J Org Chem. 1987;52:2452. [Google Scholar]
- 44.Arnold K, Batsanov AS, Davies B, Grosjean C, Schutz T, Whiting A, Zawatzky K. Chem Commun. 2008:3879. doi: 10.1039/b806779a. [DOI] [PubMed] [Google Scholar]
- 45.Gao M, Thorpe SB, Santos WL. Org Lett. 2009;11:3478. doi: 10.1021/ol901359n. [DOI] [PubMed] [Google Scholar]
- 46.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Todd RCJ, KVB, Gorr K, Priebe K, Gao P. Abstr Paps Am Chem Soc. 2007;233:ORGN780. [Google Scholar]
- 48.Johnson JR, Campen MGV, Grummitt O. J Am Chem Soc. 1938;60:111. [Google Scholar]
- 49.Lightfoot AP, Twiddle SJR, Whiting A. Org Biomol Chem. 2005;3:3167. doi: 10.1039/b507900d. [DOI] [PubMed] [Google Scholar]
- 50.Duggan P, Offerman D. Aust J Chem. 2007;60:829. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.