

Abstract
We demonstrated previously that human FEN1 endonuclease, an enzyme involved in excising single-stranded DNA flaps that arise during Okazaki fragment processing and base excision repair, cleaves model flap substrates assembled into nucleosomes. Here we explore the effect of flap orientation with respect to the surface of the histone octamer on nucleosome structure and FEN1 activity in vitro. We find that orienting the flap substrate toward the histone octamer does not significantly alter the rotational orientation of two different nucleosome positioning sequences on the surface of the histone octamer but does cause minor perturbation of nucleosome structure. Surprisingly, flaps oriented toward the nucleosome surface are accessible to FEN1 cleavage in nucleosomes containing the Xenopus 5S positioning sequence. In contrast, neither flaps oriented toward nor away from the nucleosome surface are cleaved by the enzyme in nucleosomes containing the high-affinity 601 nucleosome positioning sequence. The data are consistent with a model in which sequence-dependent motility of DNA on the nucleosome is a major determinant of FEN1 activity. The implications of these findings for the activity of FEN1 in vivo are discussed.
Keywords: Chromatin, DNA Repair, DNA Replication, Histones, Nucleosome, Flap Endonuclease 1, Nucleosome Positioning Sequence
Introduction
Flap endonuclease 1 (FEN1)2 plays critical roles in both DNA replication and repair. During processing of Okazaki fragments, FEN1 specifically recognizes a 5′-unannealed flap formed by the displacement synthesis activity of DNA polymerase δ. FEN1 binds and cleaves the phosphodiester backbone at the base of the flap, leaving a ligatable nick (1, 2). The significant role of FEN1 in DNA synthesis is highlighted by genetic studies demonstrating that RNase H is not essential for eliminating the RNA initiator (3) and that homozygous deletion of FEN1 results in a complete inactivation of DNA synthesis in mouse blastocysts (4).
FEN1 is also involved in the penultimate step of a long patch base excision repair pathway where the enzyme removes a flap containing the damaged base that was generated by DNA polymerase β displacement synthesis. In vivo studies have shown that deletion of both copies of the FEN1 gene results in hypersensitivity of cells to methylating agents and hydrogen peroxide (5), and a FEN1 null mutation has been found to render Saccharomyces cerevisiae and Schizosaccharomyces pombe sensitive to UV radiation (2). Further, mice with FEN1 haploinsufficiency show a mild predisposition phenotype which, in combination with a mutation in the adenomatous polyposis coli (Apc) gene, leads to rapid tumor progression and genomic instability. In addition, deletion of both copies of the FEN1 gene results in early embryonic lethality (6). Finally, blastocysts with both copies of the FEN1 gene deleted and exposed to γ radiation have been observed to undergo extensive apoptosis (4), likely because of the essential role of FEN1 in the repair of radiation-induced DNA damage in vivo.
A number of in vitro studies have addressed the kinetics and mechanism of FEN1 activity on DNA substrates. FEN1 specifically recognizes single-stranded flaps emanating from double-stranded replication and repair intermediates. Cleavage of the flap has been proposed to require threading of the single-stranded flap through a clamp-like structure in the protein (7, 8) because FEN1 is unable to cleave flaps containing a region of double-stranded DNA (9) or flaps tagged with biotin/streptavidin (8). A model whereby FEN1 threads the flap strand from its 5′ end is further supported from the crystal structure of Pyrococcus furiosus FEN1, which contains a clamp-like structure (7). FEN1 cleavage occurs primarily one nucleotide past the base of the 5′ flap structure and involves a one-nucleotide 3′ flap, leaving behind a simple nick that can be converted to a continuous strand by DNA ligase (10). Cleavage results in the release of intact 5′ flaps, ruling out an exonucleolytic mechanism whereby the enzyme digests the flap one nucleotide at a time from the 5′end until it reaches the junction (11).
While the activity of FEN1 on naked DNA substrates has been thoroughly investigated, much less is known regarding the details of how FEN1 processes chromatin substrates. Eukaryotic DNA is present as chromatin and gets replicated in conjunction with the DNA during S-phase. Indeed, evidence suggests that at least some immature Okazaki fragments are already associated with histones immediately after passage of the replication fork (12). Clearly, histone-DNA interactions pose a challenge for the enzymes involved in replication and repair. It has been shown that the enzymes involved in nucleotide excision repair, which results in replacement of a 25- to 30-nt stretch of DNA on the damaged strand, require ATP-dependent chromatin remodeling activities to gain access to the underlying DNA substrate (13, 14). However, base excision repair targets base damage that causes relatively small distortions in helical structure and results in replacement of only one (small patch) or two to 11 (large patch) nucleotide regions (15). Thus it is possible that the enzymes involved in this pathway do not always depend upon the ATP-dependent remodeling machinery to carry out their repair functions. Indeed, studies indicate that numerous DNA glycosylases and AP endonuclease 1 can efficiently process their targets when buried within nucleosomes (16–21). Likewise, DNA ligase I and FEN1 can cleave flap substrates assembled into nucleosomes, where a 5-nucleotide non-complementary flap was placed near the dyad and oriented away from the histone octamer (22, 23).
The robust activity of FEN1 previously detected on nucleosome substrates suggests that cleavage occurs without significant disruption of histone-DNA interactions (23). However, in this work, the flap was oriented maximally away from the core histone surface. In this study, we tested whether rotational orientation or DNA sequence affect FEN1 activity toward flaps in nucleosomes. We find that human FEN1 can recognize and efficiently process flaps oriented toward the histone octamer in nucleosomes reconstituted with the Xenopus 5 S rDNA nucleosome positioning element and that such flaps cause minor structural perturbations, which may facilitate access of the flap to FEN1 in these nucleosomes. However, flaps within nucleosomes containing the high-affinity 601 DNA sequence were completely resistant to FEN1 cleavage regardless of orientation. Thus, our data suggest that flaps within such high-affinity nucleosome binding sequences will require ATP-dependent remodeling activity for completion of base excision repair.
EXPERIMENTAL PROCEDURES
DNA Substrate Construction
Flap-containing FEN1 substrates were constructed using synthetic DNA sequences, including the nucleosome positioning elements derived from a Xenopus borealis somatic-type 5S ribosomal gene (23) and the 601 sequence (24). PAGE-purified oligonucleotides (purchased from IDT Corp., Coralville, IA) were annealed to form DNA substrates as described (23). Briefly, for both “flap-in” and “flap-out” 5 S templates (Fig. 1A), a 154-nt bottom strand was prepared by phosphorylating an 80-nt oligo (80-mer) with ATP and T4 polynucleotide kinase (Promega) and ligating to a 74-nt oligo (74-mer) after annealing both to a 40-nt splicer (Fig. 1A and supplemental Fig. S1). The resulting 154-nt product was gel-purified (23). For the flap-out substrates, a 57-mer was 5′ end-labeled with [γ-32P]ATP and polynucleotide kinase as described (23). The labeled 57-mer was ligated to a 35-mer after annealing to a 40-nt splint. The resulting 92-mer was PAGE-purified and annealed with a 66-mer and the 154-nt bottom strand to generate the double-stranded flap-out substrate. The final product was purified by native 5% polyacrylamide gel electrophoresis (1× Tris borate-EDTA). The 5 S flap-in substrate was constructed by ligating the labeled 57-mer with a 41-mer after annealing to a 40-nt splint (Fig. 1A and supplemental Fig. S1). The resulting 97-mer was PAGE-purified and annealed with a 61-mer and the 154-nt bottom strand. The final product was PAGE-purified as described above (Fig. 1A and supplemental Fig. S1). A fraction of the flap-out and flap-in naked DNA substrates were treated with EcoRV to cleave 44 base pairs off the downstream end to be used as an internal naked DNA control in the FEN1 reactions with nucleosomes. Substrates prepared for structural probing experiments were 5′ end-labeled with [γ-32P]ATP and polynucleotide kinase as indicated in the figure legends.
FIGURE 1.

Flap-out and flap-in nucleosome substrates. A, schematic of DNA oligonucleotides used to assemble the 154-bp flap-out and flap-in substrates. Substrates are based on the Xenopus 5S nucleosome positioning element. The length of the ligated oligos used to anneal each template are indicated in italics. The location of the flap, the EcoRV cleavage site, and predicted orientation of the flaps (right) are indicated. B, nucleosomes reconstituted on control (Con, no flap), flap-out (FO), and flap-in (FI) substrates were run on a 0.7% agarose nucleoprotein gel along with a 154-bp naked DNA control (DNA). C, flap-out and flap-in substrates labeled on either the top (flap) or bottom strands were run on a 5% polyacrylamide nucleoprotein gel. D, nucleosomes reconstituted on flap-out and flap-in templates were separated over sucrose gradients, and fractions were analyzed on 0.7% agarose nucleoprotein gels. Autoradiographs of the dried gels are shown.
Flap substrates based on the 601 nucleosome positioning element were constructed in a similar manner. A 151-nt bottom strand was formed by ligating a 70-mer and an 81-mer after annealing to a 40-nt splint (Fig. 7A and supplemental Fig. S2). The flap-out substrate was formed by ligating a labeled 39-mer and a 42-mer followed by annealing with a 76-mer and the bottom strand 151-mer. The flap-in substrate was in turn constructed by ligating the labeled 39-mer with a 48-mer followed by annealing with a 70-mer and the bottom strand 151-mer. A fraction of the flap-out and flap-in 601 substrates were treated with MspI to construct a shorter naked DNA substrate to be used as the control in FEN1 reactions.
FIGURE 7.

Flaps in 601 nucleosomes are refractory to FEN1, regardless of orientation. A, schematic of DNA oligonucleotides used to assemble the 601 151-bp flap-out (FO) and flap-in (FI) substrates. The length of the ligated oligos used to anneal each template are indicated in italics. The location of the flaps and the MspI cleavage site are indicated. B, translational gel analysis of 601 nucleosomes reconstituted with flap-out (lanes 2 and 4), flap-in (lanes 3 and 5), and control (no flap, lane 6) DNA fragments. Naked DNA is shown in lane 1. C, flap-containing 601 nucleosomes exhibit expected rotational orientation. FO and FI 601 nucleosomes were either mock-treated (lanes 1 and 3) or treated with hydroxyl radicals, and the cleavage patterns were analyzed by sequencing gel electrophoresis. The position of radioactive labels is on the 151-nt bottom strands, as indicated by the asterisk in A. The predicted location of the nucleosome dyad is indicated by the filled arrow. D, FEN1 cleavage of 601 DNA (lanes 1–4) and nucleosomes (lanes 5–10). FI and FO substrates were incubated in the absence (lanes 1, 3, 5, and 8) or presence (lanes 2, 4, 6, and 9) of FEN1 for 15 min. The samples in lanes 7 and 10 were incubated with FEN1 for 30 min. Cleavage was analyzed on 6% sequencing gels as described above. The full-length substrate and cleaved product are 86 and 81 nt, respectively. E, cleavage of 601 FI and FO nucleosomes and naked DNA substrates incubated in the same reaction. Naked 601 FI and FO DNA was truncated by cleavage with MspI (see A), mixed with an equivalent amount of FI and FO 601 nucleosomes, and then incubated together with FEN1 in the same reaction. Lanes 1 and 6 show undigested samples, whereas lanes 2–5 and 7-10 show samples digested with FEN1 for the times indicated above the gel. The positions of uncleaved nucleosome DNA and uncleaved and cleaved naked DNA are indicated.
Nucleosome Reconstitution
The four core histones (Xenopus) were expressed in Escherichia coli and purified as described (23). Equimolar amounts of the core histones were mixed with annealed flap substrates (∼2 × 105 cpm) and 5 μg of calf thymus DNA in 1× TE (10 mm Tris-HCl, pH 8.0, 1 mm EDTA) containing 2 m NaCl in 200 μl total volume. The nucleosomes were reconstituted using salt gradient dialysis and were purified using sucrose gradient fractionation as described (21).
Hydroxyl Radical Footprinting
For footprinting studies, substrates radiolabeled at the 5′ ends of the bottom or top strands were reconstituted and sucrose gradient purified. Sucrose was removed by spin column microfiltration (Millipore, YM-5 unit) with five 400-μl (1× TE) washes. Naked DNA and purified nucleosomes in TE were then treated with hydroxyl radicals as described (23). The DNA was recovered by ethanol precipitation and analyzed by 6% denaturing polyacrylamide sequencing gel electrophoresis. The gels were dried and analyzed by phosphorimagery. Alternatively, flap-out and flap-in nucleosomes were treated with hydroxyl radicals directly after reconstitution, followed by the separation of naked DNA and nucleosome components on preparative 0.7% agarose gels (0.5× Tris borate-EDTA). DNA was isolated from each band, and cleavage patterns were analyzed on sequencing gels as described above.
FEN1 Cleavage Reactions
Purified human FEN1 was a kind gift from Dr. Bob Bambara. Approximately 15 pmol (15,000 cpm) of sucrose gradient-purified nucleosome and truncated naked DNA substrates were treated with 1 pmol of FEN1 (1 μl) and 20 μl of aliquots removed at the times indicated in the figure legends. Aliquots were treated with 10× stop buffer (120 mm EDTA, 1.2% w/v SDS) and analyzed as described (23). In an alternative method, 100 fmol of reconstituted nucleosome substrates (without sucrose gradient purification) were treated with 2 pmol FEN1 for 10 min, 10 μl of stop buffer lacking SDS was added, and the nucleosome and naked DNA fractions were separated on preparative 0.7% agarose nucleoprotein gels (0.5× Tris borate-EDTA). The DNA was isolated from respective bands, and cleavage was analyzed on a sequencing gel as described above.
Cross-linking Reactions
H2A A45C/H2B dimers were prepared and subsequently modified with the cross-linking agent, 4-azidophenacyl bromide (APB) as described (25). Nucleosomes were reconstituted as described above with modified and unmodified dimers, irradiated at 365 nm for 40 s, and the cross-linked DNA was separated from non-cross-linked on 6% SDS-polyacrylamide gels (25). DNA purified from the polyacrylamide gel was treated with piperidine, precipitated, and loaded onto 6% denaturing polyacrylamide gel to determine sites of cross-linking within the DNA.
Exonuclease III Assays
Sucrose gradient-purified flap-out and flap-in nucleosomes and naked DNA were treated with 1 μl of ExoIII (100 units, New England Biolabs). Aliquots were collected at 2, 5, and 10 min and the reactions stopped by addition of 1/10th volume of stop buffer (120 mm EDTA, 1.2% w/v SDS). The DNA was precipitated by standard methods, and cleavage products were analyzed by sequencing gel electrophoresis as described above.
RESULTS
In previous work, we demonstrated that human FEN1 can efficiently cleave a conventional DNA flap substrate when assembled into a nucleosome and oriented away from the core histone octamer, based on the preferred rotational positioning of the DNA sequence (22, 23). To test whether DNA fragments containing a flap oriented in a direction opposite to the predicted rotational orientation of the DNA can be assembled into canonical nucleosomes and, if so, whether such structures are accessible to FEN1, we constructed DNA templates for nucleosome reconstitution based on the well characterized Xenopus 5S nucleosome positioning sequence (26, 27). These contained a double-flap configuration, with a complementary 1-nt 3′ flap and a non-complementary 5-nt 5′ flap, likely to be the physiologically substrate of FEN1 (10). Although flaps generated during replication and repair in vivo would have a sequence complementary to the underlying DNA template, we employed a non-complementary 5′ flap to prevent formation of alternative structures (10).
Nucleosomes were reconstituted in which the flap was predicted to be oriented away (flap-out) or toward (flap-in) the histone octamer, based on the known rotational preference of 5 S DNA (see Fig. 1A) (27, 28). Flap-containing nucleosomes and non-flap controls exhibited similar patterns of migration through agarose nucleoprotein gels, suggesting that the presence of the flaps had little or no effect on the overall structure or integrity of the nucleosome (Fig. 1B). In addition, the fraction of the radio-labeled DNA fragments assembled into nucleosomes when reconstituted with an excess of unlabeled competitor DNA fragments indicates that the histone octamer binds with similar affinity, regardless of presence of the flap or the predicted orientation with respect to the histone surface. The homogeneity of the nucleosomes assembled with flap-out and flap-in substrates was further examined by running samples on non-denaturing polyacrylamide gels, which separate nucleosomes based on translational position (Fig. 1C). We find that irrespective of the orientation of the flap, nucleosomes migrate as distinct bands, indicative of a single or set of closely related translational position(s). Each reconstituted fraction has some residual naked DNA that migrates slightly differently, depending on the orientation of the flap. However, the nucleosomes exhibit nearly identical electrophoretic migration rates, suggesting very similar conformations and translational positions regardless of the presence or position of the flap. Finally, both flap-in and flap-out nucleosomes sediment at equivalent rates through sucrose gradients, consistent with the native gel analyses (Fig. 1D).
We further characterized the structure of the 154-bp flap-in and flap-out nucleosomes using hydroxyl radical cleavage. Hydroxyl radical footprinting provides high-resolution information regarding the interaction of the DNA backbone with the core histone octamer surface and thus should report on any change in the rotational positioning of the DNA or alteration of nucleosome structure caused by the differing flap orientations. Flap-containing nucleosomes in which the bottom and top strands were individually radio-labeled at their 5′ ends were sucrose gradient-purified (Fig. 1D) and subjected to hydroxyl radical cleavage after removal of the sucrose. We find that patterns for the bottom strands from the flap-out and flap-in nucleosomes were nearly identical (Fig. 2A). Although the naked templates exhibited a fairly even cleavage profile throughout (Fig. 2A, lanes 3 and 5), the reconstituted templates exhibited the characteristic wave-like pattern (lanes 7 and 9) because of the alternate association of the phosphodiester backbone of the DNA helix with the surface of the core histone octamer (27, 28). Similar results were obtained when the top strands were radio-labeled at the 5′ end of the flap strand (Fig. 2B). Note that the patterns for flap-out and flap-in constructs are offset by 6 bp on these gels because of the different lengths of these constructs (see Fig. 2B, top). These results indicate that the orientation of the flap did not alter the general rotational positioning of the DNA with respect to the histone octamer. An identical result was obtained when nucleosomes were first subjected to hydroxyl radical cleavage and then separated from any non-nucleosomal components on a preparative agarose gel before sequencing gel analysis (data not shown).
FIGURE 2.

Hydroxyl radical cleavage analysis of flap-out and flap-in 5S nucleosomes. Flap-out (FO) and flap-in (FI) naked DNA and reconstituted nucleosomes were subjected to hydroxyl radical cleavage and the products analyzed by sequencing gel electrophoresis and phosphorimagery. A, cleavage pattern of the bottom strand of flap-out and flap-in substrates. The location of the label in each 154-bp substrate is indicated by the asterisk in the schematic at the top. Lane 1, G-rxn marker. Lanes 2–5 show naked FO (lanes 2 and 3) and FI (lanes 4 and 5) substrates either mock-treated (lanes 2 and 4) or digested with hydroxyl radicals (lanes 3 and 5), as indicated. Lanes 6–9 show nucleosomes containing FO (lanes 6 and 7) or FI (lanes 8 and 9) substrates either mock-treated (lanes 6 and 8) or digested with hydroxyl radicals (lanes 7 and 9), as indicated. Numbers and arrows indicate positions of cleavage peaks from nucleosome dyad at position 0. The positions of the flaps and dyad axis are indicated beside the gel. B, cleavage pattern of the top (flap) strand. As in A, except that the label is located at the 5′ end of the top strand (see schematics at top). Single arrows within the gel correspond to those shown on side but are displaced because of the gel slant and indicate peaks in cleavage as in A. Note that FI peaks are shifted 5 bp further up on the gel because of the location of the flap.
Interestingly, however, close inspection of the hydroxyl radical cleavage patterns reveals subtle differences between the flap-in and flap-out nucleosome. On the “bottom” strand, the flap-in substrate exhibits more cleavage in regions where the DNA backbone is presumably in contact with the histone proteins compared with the flap-out substrate, especially in regions close to the flap (Fig. 2A, small vertical lines). These differences can also be observed in scans of lanes 7 and 9 in Fig. 2A (supplemental Fig. S3A). This may be due to DNA exhibiting greater motility within the flap-in nucleosomes compared with the flap-out nucleosomes. Likewise, when the radioactive label is placed at the 5′ end of the top (flap) strand, a similar effect is observed (Fig. 2B, top and supplemental Fig. S3B), again suggesting that accommodation of the flap oriented toward the histone octamer increases conformational homogeneity or motility of the DNA on the histone surface. These results suggest that although on average the rotational orientation of the DNA is maintained in the presence of the inward-facing flap, the presence of this flap does cause some increased motility or conformational excursions of the DNA on the histone surface.
Exonuclease III (ExoIII) progressively degrades one strand of double-stranded DNA in a 3′ to 5′ fashion, with progress restricted by histone-DNA interactions at the edge of the nucleosome core region (29). We therefore used ExoIII to analyze the translational alignment of the upstream and downstream edges of the histone octamers within flap nucleosomes. Although naked DNA is completely digested by ExoIII, there is a significant portion of both the flap-in and flap-out nucleosome substrates remaining after 10 min of digestion (Fig. 3 and results not shown). Comparison of ExoIII digestion profiles of nucleosomes reconstituted with the flap-in and flap-out substrates reveals subtle differences, despite the fact that the double-stranded DNA sequence is identical in both nucleosomes. For example, digestion of flap-out nucleosomes results in pauses in digestion corresponding to the downstream edge of the core region, at +78, +70, +69, and +59 (label on the top strand) and at the upstream edge at −63, −53, and −44 (label on the bottom strand). However, with the flap-in nucleosomes, the pause at +70 on the top strand is lost, whereas a new pause at +48 is evident. Likewise, pauses on the bottom strand occur at −73, −63, and −54. These data are consistent with a slight difference in conformation or stability of DNA association at the nucleosome edges between the flap-out and flap-in substrates, wherein the top strand is more weakly protected at the downstream edge, consistent with the hydroxyl radical footprinting pattern. Moreover, the cleavage pattern suggests that the edge of the flap-in nucleosome is shifted slightly further upstream compared with that in the flap-out nucleosome.
FIGURE 3.

ExoIII analysis of flap-out and flap-in 154-bp 5 S nucleosome substrates. Nucleosomes were purified and digested with ExoIII as described in the text, and cleavage products were analyzed by sequencing gels and phosphorimagery. Shown are lanes corresponding to undigested and digested samples for each 3′ end of the flap-out (FO, top) and flap-in (FI, bottom) substrates. Numbers indicate the distance from the expected nucleosome dyad fixed at position 0.
The 154-bp templates used above provide only a few bp of dsDNA beyond that encompassed by the 147-bp nucleosome core region, which greatly restricts the choice of translational positions (25). To further investigate whether orientation of the flap can affect nucleosome translational position, we prepared templates that were extended ∼60 bp on the downstream edge of the 154-bp template to provide more opportunity for alternative translational positions, including positions in which the flaps would be located outside of the nucleosome core region. In addition, the extension of the templates increases the ability to resolve alternative translational positions on non-denaturing polyacrylamide gels. Flap-in and flap-out 215-bp templates were prepared containing a radio label at the 5′ end of the top strand oligonucleotide (Fig. 4A). Analysis on translational gels revealed that nucleosomes on either template adopt closely related translational positions, with three apparent positions resolved for the flap-out nucleosomes and two for the flap-in nucleosomes (Fig. 4B, lanes 3 and 4). We then performed restriction enzyme cleavage analysis to further characterize translational positioning. Cleavage of these nucleosomes with BamHI, which cuts the top strand at −63, 15 bp from the upstream end of the template, resulted in loss of the topmost bands in each sample (top two in the flap-out and one in the flap-in), whereas the most rapidly migrating bands were retained (Fig. 4B, lanes 5 and 6). (Note that the 15-bp radio-labeled cleavage product runs off the gel in this experiment.) This indicates that the translational positions corresponding to the fastest-migrating band in each sample include the BamH1 site, whereas the top bands correspond to positions downstream of this site. Cleavage with BbvI, which cuts the 5 S DNA at position −20, resulted in a loss of intensity in the topmost band for both the flap-out and flap-in nucleosomes, indicating that the middle band in the flap-out sample represents a translational position in which the upstream edge of the nucleosome core region is somewhere between the BamHI site at site −63 and the BbvI site at −23 (Fig. 4B, lanes 7 and 8). As expected, all translational positions are resistant to RsaI digestion, which cuts at +35 (Fig. 4B, lanes 11 and 12). These results are consistent with previous work (25, 30) showing nucleosomes on the 215-bp 5 S template adopt approximately two upstream translational positions at the 5′ end of the (non-flap) parent 5 S DNA fragment, with upstream edges at −75 and −65, and a downstream position with the upstream edge located approximately at −20 (see Fig. 4A). Therefore, the flaps do not drastically alter the distribution of translational positions observed on the 215-bp template, with only subtle differences detected between the flap-out and flap-in positions near the upstream edge, consistent with the ExoIII digestion results.
FIGURE 4.

Translational positions on 216-bp 5 S flap-out and flap-in nucleosomes. Nucleosomes were reconstituted on 215-bp templates as described in the text, and translational positions were analyzed on 5% native polyacrylamide gels. A, schematic of flap-out (FO) and flap-in (FI) nucleosome templates and translational positions of nucleosome cores as determined by restriction enzyme digestion analysis in B. B, restriction enzyme digestion analysis of FO and FI nucleosomes. Lane 1, naked DNA; lane 2, template reconstituted with H3/H4 tetramer only; lanes 3 and 4, 9 and 10, FO and FI nucleosomes, respectively; lanes 5–8 and 11 and 12, FO and FI nucleosomes digested with Bam H1, BbvI, or RsaI, as indicated. Filled arrowheads indicate the nucleosome translational positions in undigested nucleosomes, the open arrowhead indicates the position of tetramer-DNA complex, and the brackets indicate subnucleosomal complexes.
Interestingly, we note that incubation of the flap-in nucleosomes with BbvI did not result in efficient cleavage of the DNA, despite the loss in nucleosome bands on the gel (Fig. 4B, lane 8). This is most likely a result of the close apposition of the flap at −15 (top strand) and the BbvI cleavage site, which cuts the DNA backbone at positions −21/20 and −18/17 on the top and bottom strands, respectively. Surprisingly, despite the lack of cleavage, incubation of the flap-in nucleosomes with BbvI resulted in the loss of H2A/H2B dimers from the slowest migrating translational position to produce tetramer complexes (Fig. 4B, compare lanes 2, 4, and 8). Trial incubations with BbvI buffer alone show that the restriction enzyme is required for the displacement of the dimer from the nucleosomes (results not shown). These results suggest that the DNA binding activity of BbvI displaced H2A/H2B from the flap-in but not the flap-out nucleosomes, signifying that the H2A/H2B dimers are perhaps bound less tightly in a fraction of the flap-in nucleosomes.
The results above are consistent with the idea that accommodation of the flap structure in the flap-in nucleosomes causes some destabilization of histone-DNA interactions near the edge of the nucleosome, perhaps involving the association of the H2A/H2B dimers. To further probe possible changes in the nucleosome structure, we carried out histone → DNA cross-linking studies using an H2A site specifically modified with the photoactivatable cross-linking reagent APB (see “Experimental Procedures”). Nucleosomes were prepared with H2A A45C-APB, and cross-linking in 154-bp flap-out and flap-in templates was analyzed by base elimination and denaturing polyacrylamide gel electrophoresis, as described previously (25, 31). Flap-out and flap-in nucleosomes reconstituted with the modified histone showed no apparent difference in the migration pattern on agarose gels (not shown), suggesting that the modification did not affect the composition or integrity of the nucleosomes. Upon UV irradiation, ∼ 30% of the total radio-labeled DNA in the APB-modified nucleosomes migrated as cross-linked DNA-protein complexes (Fig. 5A). After isolation of DNA and base elimination, comparison of bands dependent on both UV irradiation and APB modification reveals a distinct cross-linking pattern in the flap-out and flap-in nucleosomes (Fig. 5B). Specifically, two major cross-links were observed in the flap-out nucleosome sample, at positions +30 and +40 (Fig. 5B, lane 6), consistent with nucleosome dyads located at −3 and −13 (25). However, these cross-links were somewhat diminished in intensity in the flap-in nucleosomes, and new bands were observed shifted further up the gel, including a rather robust cross-linking at position +25 (Fig. 5B, lane 7). This pattern of cross-linking suggests that the flap-in nucleosomes are equilibrating with an upstream position or are subtly altered in structure (such as altered H2A/H2B binding) because of the accommodation of the flap within the nucleosome, consistent with enzymatic mapping results.
FIGURE 5.
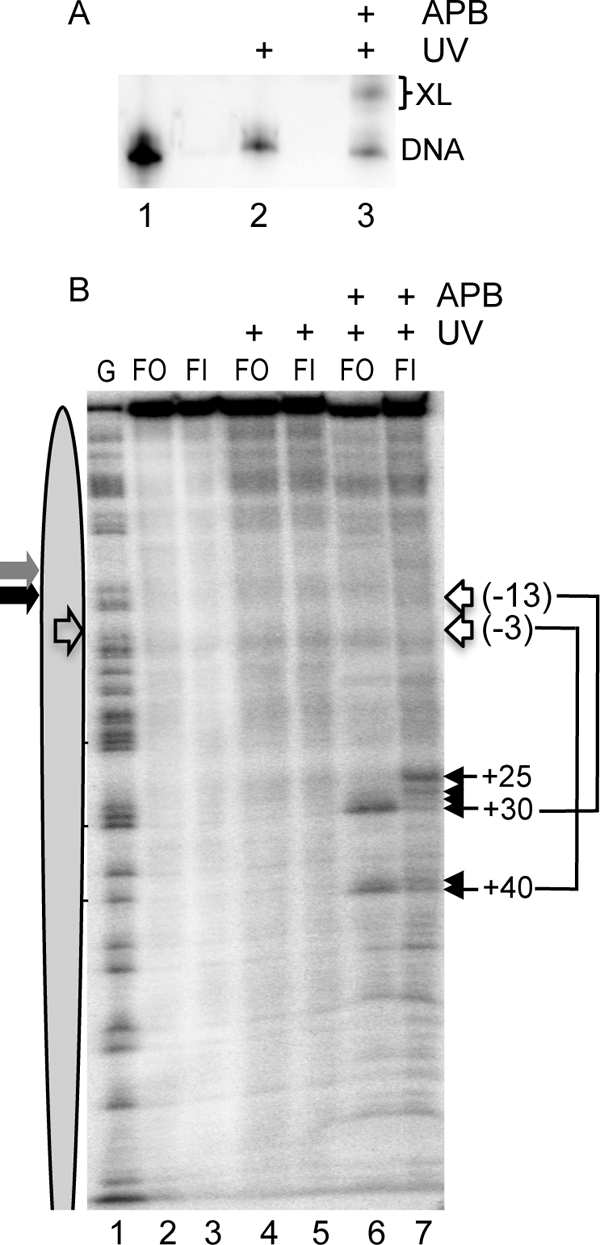
Site-directed protein-DNA cross-linking within flap-out and flap-in nucleosomes. Nucleosomes were reconstituted on flap-out (FO) and flap-in (FI) templates with H2A A45C-APB, and cross-linking was analyzed as described in the text. A, efficiency of cross-linking. Flap-out nucleosomes containing H2A A45C were irradiated with UV light for 40 s, and cross-linking was analyzed by SDS-PAGE. Lane 1 shows a naked DNA control. B, cross-link mapping in FO and FI nucleosomes. Nucleosomes were mock-treated (lanes 2 and 3), treated with UV only (lanes 4 and 5), or both APB-modified and UV-irradiated (lanes 6 and 7). Lane 1 shows a G-reaction marker. Positions of cross links are indicated with filled arrows. Lines and open arrows indicate positions of nucleosome dyads corresponding to specific cross-links according to the known cross-linking pattern of H2A A45C-APB (31). Gray and black arrows indicate the location of flaps in FO and FI nucleosomes, respectively. The open arrow in oval indicates the predicted location of the dyad in the 5 S nucleosome.
We next examined whether human FEN1 could catalyze cleavage of the 5-nt flap within the flap-in nucleosomes. We previously showed that FEN1 readily cleaves the flap-out nucleosomes when the flap was positioned near the nucleosome dyad. Consistent with the previous observation, FEN1 cleaved almost 50% of the flap-out nucleosomes during the course of the incubation, whereas the naked substrate in the reaction was nearly completely cleaved by FEN1 (Fig. 6B, lanes 1–4). Importantly, FEN1 also cleaved the flap-in nucleosome substrates with an efficiency similar to that of the flap-out substrate (Fig. 6B, lanes 5–8), indicating that FEN1 can access the flaps in both nucleosomal substrates irrespective of the rotational orientation.
FIGURE 6.

FEN1 cleaves both flap-out and flap-in substrates assembled into nucleosomes. A, schematics of the flap-out (FO) and flap-in (FI) nucleosomes and naked DNA substrates. Numbers in italics indicate the length of oligos. B, comparison of FEN1 cleavage of nucleosome and naked DNA substrates incubated in the same reaction. Lanes 1–4 and 5-8 show flap-out and flap-in substrates, respectively. Flap-out and flap-in nucleosome substrates and products are 99/93 nt and 104/98 nt, respectively. Substrates and products are 49/43 nt and 54/48 nt for the naked DNA flap-out and flap-in constructs, respectively. C, cleavage of nucleosome and naked DNA substrates in the presence of the restriction enzyme EcoRV. The restriction enzyme was added first to the sample to cleave any non-nucleosomal substrate present and then followed by FEN1 addition. The nucleosomes were incubated with both enzymes for an additional 10 min. Lanes 1–4, naked (nucleosome-length) DNA; lanes 5–10, nucleosomes. Flap-out and flap-in substrates are as indicated. Note EcoRV cleavage of the full-length flap-in and flap-out substrates generates products of 55 and 50 nt, respectively, whereas FEN1 cleavage of these products results in shorter bands on the gel (brackets). D, FEN1 cleaved gel isolated nucleosomes. Nucleosomes and naked DNA were incubated in the presence or absence of FEN1 as indicated and then isolated on preparative native agarose gels. DNA was isolated from each band and analyzed on the sequencing gel, as indicated. Lanes 1–4 and 5-8 show flap-in and flap-out samples, respectively.
Although the above FEN1 reactions were performed on sucrose gradient-purified nucleosomes, there was a possibility that some naked DNA was generated in our nucleosomal samples during the dialysis to remove sucrose. To eliminate this possibility, we treated the samples with EcoRV before subjecting the nucleosomes to FEN1 digestion. Treatment with EcoRV truncates any naked DNA present in the sample, whereas bona fide nucleosomes remain intact because of protection of the DNA by the histone octamer. Indeed, a small portion of the flap-in and flap-out nucleosomes are cleaved by EcoRV, whereas the vast majority of the nucleosome substrate is resistant to cleavage (Fig. 6C, lanes 6 and 9). Importantly, incubation of flap-in and flap-out nucleosomes with both EcoRV and FEN1 still resulted in the appearance of characteristic 98- and 93-nt FEN1 cleavage products, supporting that FEN1 can access flaps within bona fide flap-out and flap-in nucleosomes (Fig. 6C, lanes 5 and 7). We further confirmed the above result by using an alternate method whereby reconstituted nucleosomes were subjected to FEN1 digestion, after which the nucleosome and DNA fractions were isolated by preparative agarose gel electrophoresis. Analysis of cleavage in these samples indicates that FEN1 can indeed cleave a flap in 5 S nucleosomal substrates irrespective of orientation (Fig. 6C, lanes 1 and 5).
The above data indicate that any steric clash resulting from accommodation of a flap designed to be oriented toward the histone surface is insufficient to completely override the inherent propensity of the 5 S sequence to adopt a dominant rotational orientation on the nucleosomes surface. However, our solution structural analyses indicate that there are subtle structural differences between the flap-in and flap-out nucleosomes that likely contribute to accessibility of the flap observed in the FEN1 cleavage assays. It is possible that such structural perturbations in the flap-in nucleosome may be sequence-specific and that nucleosomes formed with a higher-affinity positioning element would be less likely to equilibrate to flap-accessible structures. To test this, we reconstituted nucleosomes using the 601 nucleosome positioning sequence identified by Lowary and Widom (24), known to have a ∼100-fold higher affinity for the histone octamer compared with the 5 S DNA sequence. Based on previous determinations of the rotational orientation of the 601 sequence on the nucleosome surface, we designed flap-out and flap-in substrates, both containing a 3′ 1-nt flap and 5′ non-complementary 5-nt flap located close to the dyad, as in the case of 5 S DNA substrates (Fig. 7A).
Nucleosomes reconstituted with the flap-containing 601-based substrates exhibited similar migration through agarose and acrylamide nucleoprotein gels (Fig. 7B and not shown). The absence of naked DNA in the reconstituted fractions is indicative of the higher affinity of the 601 sequence for the histone octamer. These nucleosomes were sucrose gradient-purified and subjected to hydroxyl radical cleavage. Both the flap-in and flap-out 601 nucleosomes exhibited identical hydroxyl radical cleavage patterns, indicating that the rotational setting of the DNA was not altered by the orientation of the flap (Fig. 7C). This is especially evident in a comparison of the cleavage pattern in the vicinity of the flaps.
Incubation of the naked 601 substrates with FEN1 resulted in quantitative cleavage of the flap (Fig. 7D, lanes 1–4). In contrast, no cleavage was observed with the nucleosomes reconstituted with either the flap-out or flap-in substrates (Fig. 7D, lanes 5–10). To ensure enzyme activity, we repeated the assay with a naked DNA control added directly to the nucleosome samples. Importantly, whereas the naked DNA was nearly completely cleaved by FEN1, the nucleosome-bound substrates were completely resistant to FEN1 digestion (Fig. 7E).
DISCUSSION
FEN1 is likely to encounter flap substrates buried within nucleosomes in vivo. Although the sequence dependence of processes that generate flaps is not well characterized, it is likely that flaps will not always occur in register with the natural nucleosome positioning preference of the underlying DNA sequence. Thus, we have investigated the effect of rotational positioning of the flap with respect to the histone octamer. We show that the ability of FEN1 to recognize and cleave flap substrates in chromatin is not dependent upon rotational orientation of the flap on the nucleosome surface, implying that if flap structures are prematurely assembled into histone-DNA complexes during replication or are inadvertently trapped in nucleosomes during DNA repair, the penultimate step of these processes can still proceed with reasonable efficiency (23). Coupled with our prior demonstration that human DNA ligase I can seal nicks buried in nucleosomes (22), our data suggest that Okazaki fragment processing or DNA repair can be completed in nucleosome substrates as long as DNA synthesis associated with these processes is completed.
We previously showed that flap cleavage within the 5 S nucleosome occurs at rates only a few-fold slower than naked DNA, suggesting that histone-DNA interactions are not significantly disrupted to accommodate the enzyme (23). Because we measured similar rates between flap-out and flap-in substrates, it is likely that the enzyme can access both substrates in an equivalent manner. It is possible that access to the flap requires equilibration to a similar “exposed” structure for both flap-in and flap-out nucleosomes, with similar conformational distortion required in both cases. X-ray crystallographic and nuclease digestion studies (32–34) indicate that nucleosomes are dynamic macromolecular assemblies. Moreover, nucleosomal DNA can access states in which the inner superhelical face of the DNA is exposed to allow binding of pyrrole-imidazole polyamides (34). Importantly, the binding of such drugs results in significant yet localized structural distortion in the DNA adjacent to the binding sites. Moreover, twist defects appear to be readily accommodated within nucleosomes (32, 35). Thus, intrinsic conformational flexibility within nucleosomes most likely facilitates access of the flap by FEN1 irrespective of its rotational orientation in the 5 S rDNA.
Our data suggest that the inward-facing flap structure can be easily accommodated within the nucleosome. Hydroxyl footprinting experiments of the flap-in nucleosomal substrates confirmed that the base of the flap is indeed oriented toward the histone octamer in these nucleosomes. However, we find no change in the efficiency of reconstitution of flap-in versus flap-out templates in the presence of a large excess of competitor DNA, indicating a similar overall affinity of each template for the histone octamer. Moreover, nucleosomes reconstituted with both templates exhibit a similar distribution of translational positions and no gross change in rotational orientation of the DNA on the histone surface. On the other hand, subtle differences in protection from ExoIII digestion, directed cross-linking, and protection from hydroxyl radicals are observed between flap-in and flap-out nucleosomes and indicate localized perturbations. This may lead to increases in conformational plasticity or motility of the DNA on the nucleosome surface, allowing FEN1 to recognize and cleave the flap regardless of rotational orientation. Interestingly, restriction enzyme accessibility assays indicate that flap orientation has no effect on the probability of DNA unwrapping from the edge of the nucleosome core region (unpublished results), suggesting that cleavage requires a similar distortion of both flap-in and flap-out structures.
Interestingly, we find that in the case of 601 nucleosomes, neither the “out” nor “in” orientations are accessible to the FEN1. The 601 nucleosome positioning element binds the core histone octamer 50–100-fold times more tightly than the 5 S DNA fragment used in these studies and >200 times more tightly than an average DNA sequence (24). It is likely that, given the extremely high binding affinity, conformational excursions or equilibrations necessary for FEN1 activity found in the 5 S nucleosome do not occur with significant frequency in the 601 nucleosome substrates. Indeed, this reduced conformational motility is evident in the hydroxyl radical footprinting pattern (Fig. 7C), whereas clear differences are observed between flap-out and flap-in substrates reconstituted with the 5 S template (Fig. 2). This idea is supported by recent work demonstrating that such high-affinity sequences can act as a barrier to transcriptional elongation because of increased stability of histone-DNA interactions (36). Given that high-affinity nucleosome-binding sequences occur relatively rarely in vivo (37), we assume that the bulk of nucleosomes will behave more like the 5 S nucleosome and that only rare sequences will be refractory to FEN1 activity upon nucleosome formation. A similar dependence may be the basis for the observation that DNA polymerase β exhibited activity in nucleosomal substrates reconstituted using 5 S rDNA, whereas complete inhibition of this enzyme activity on nucleosomes reconstituted with a higher-affinity sequence (19). It will be interesting in future work to test other factors within the base excision repair pathway on nucleosome substrates of varying composition.
Supplementary Material
Acknowledgment
We thank Dr. Robert Bambara for the kind gift of human FEN1.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1GM52426. This work was also supported by American Cancer Society Grant RPG GMC-99742.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- FEN1
- flap endonuclease 1
- nt
- nucleotide(s)
- APB
- 4-azidophenacyl bromide.
REFERENCES
- 1. Henricksen L. A., Bambara R. A. (1998) Leuk. Res. 22, 1–5 [DOI] [PubMed] [Google Scholar]
- 2. Liu Y., Kao H. I., Bambara R. A. (2004) Annu. Rev. Biochem. 73, 589–615 [DOI] [PubMed] [Google Scholar]
- 3. Qiu J., Qian Y., Chen V., Guan M. X., Shen B. (1999) J. Biol. Chem. 274, 17893–17900 [DOI] [PubMed] [Google Scholar]
- 4. Larsen E., Gran C., Saether B. E., Seeberg E., Klungland A. (2003) Mol. Cell. Biol. 23, 5346–5353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuzaki Y., Adachi N., Koyama H. (2002) Nucleic Acids Res. 30, 3273–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kucherlapati M., Yang K., Kuraguchi M., Zhao J., Lia M., Heyer J., Kane M. F., Fan K., Russell R., Brown A. M., Kneitz B., Edelmann W., Kolodner R. D., Lipkin M., Kucherlapati R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 9924–9929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hosfield D. J., Mol C. D., Shen B., Tainer J. A. (1998) Cell 95, 135–146 [DOI] [PubMed] [Google Scholar]
- 8. Murante R. S., Rust L., Bambara R. A. (1995) J. Biol. Chem. 270, 30377–30383 [DOI] [PubMed] [Google Scholar]
- 9. Robins P., Pappin D. J., Wood R. D., Lindahl T. (1994) J. Biol. Chem. 269, 28535–28538 [PubMed] [Google Scholar]
- 10. Kao H. I., Henricksen L. A., Liu Y., Bambara R. A. (2002) J. Biol. Chem. 277, 14379–14389 [DOI] [PubMed] [Google Scholar]
- 11. Bornarth C. J., Ranalli T. A., Henricksen L. A., Wahl A. F., Bambara R. A. (1999) Biochemistry 38, 13347–13354 [DOI] [PubMed] [Google Scholar]
- 12. Cusick M. E., Lee K. S., DePamphilis M. L., Wassarman P. M. (1983) Biochemistry 22, 3873–3884 [DOI] [PubMed] [Google Scholar]
- 13. Gong F., Fahy D., Liu H., Wang W., Smerdon M. J. (2008) Cell Cycle 7, 1067–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gong F., Fahy D., Smerdon M. J. (2006) Nat. Struct. Mol. Biol. 13, 902–907 [DOI] [PubMed] [Google Scholar]
- 15. Peterson C. L., Côté J. (2004) Genes Dev. 18, 602–616 [DOI] [PubMed] [Google Scholar]
- 16. Prasad A., Wallace S. S., Pederson D. S. (2007) Mol. Cell. Biol. 27, 8442–8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishibashi T., So K., Cupples C. G., Ausio J. (2008) Mol. Cell. Biol. 28, 4734–4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nilsen H., Lindahl T., Verreault A. (2002) EMBO J. 21, 5943–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beard B. C., Wilson S. H., Smerdon M. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beard B. C., Stevenson J. J., Wilson S. H., Smerdon M. J. (2005) DNA Repair 4, 203–209 [DOI] [PubMed] [Google Scholar]
- 21. Cole H. A., Tabor-Godwin J. M., Hayes J. J. (2010) J. Biol. Chem. 285, 2876–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chafin D. R., Vitolo J. M., Henricksen L. A., Bambara R. A., Hayes J. J. (2000) EMBO J. 19, 5492–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huggins C. F., Chafin D. R., Aoyagi S., Henricksen L. A., Bambara R. A., Hayes J. J. (2002) Mol. Cell 10, 1201–1211 [DOI] [PubMed] [Google Scholar]
- 24. Lowary P. T., Widom J. (1998) J. Mol. Biol. 276, 19–42 [DOI] [PubMed] [Google Scholar]
- 25. Yang Z., Zheng C., Hayes J. J. (2007) J. Biol. Chem. 282, 7930–7938 [DOI] [PubMed] [Google Scholar]
- 26. Rhodes D. (1985) EMBO J. 4, 3473–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thiriet C., Hayes J. J. (1998) J. Biol. Chem. 273, 21352–21358 [DOI] [PubMed] [Google Scholar]
- 28. Hayes J. J., Tullius T. D., Wolffe A. P. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 7405–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aoyagi S., Wade P. A., Hayes J. J. (2003) J. Biol. Chem. 278, 30562–30568 [DOI] [PubMed] [Google Scholar]
- 30. Aoyagi S., Narlikar G., Zheng C., Sif S., Kingston R. E., Hayes J. J. (2002) Mol. Cell. Biol. 22, 3653–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kassabov S. R., Henry N. M., Zofall M., Tsukiyama T., Bartholomew B. (2002) Mol. Cell. Biol. 22, 7524–7534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edayathumangalam R. S., Weyermann P., Dervan P. B., Gottesfeld J. M., Luger K. (2005) J. Mol. Biol. 345, 103–114 [DOI] [PubMed] [Google Scholar]
- 33. Polach K. J., Widom J. (1995) J. Mol. Biol. 254, 130–149 [DOI] [PubMed] [Google Scholar]
- 34. Suto R. K., Edayathumangalam R. S., White C. L., Melander C., Gottesfeld J. M., Dervan P. B., Luger K. (2003) J. Mol. Biol. 326, 371–380 [DOI] [PubMed] [Google Scholar]
- 35. Ong M. S., Richmond T. J., Davey C. A. (2007) J. Mol. Biol. 368, 1067–1074 [DOI] [PubMed] [Google Scholar]
- 36. Kulaeva O. I., Gaykalova D. A., Pestov N. A., Golovastov V. V., Vassylyev D. G., Artsimovitch I., Studitsky V. M. (2009) Nat. Struct. Mol. Biol. 16, 1272–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lowary P. T., Widom J. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 1183–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.