

Abstract
The cellular interferon regulatory factor-4 (IRF-4), which is a member of IRF family, is involved in the development of multiple myeloma and Epstein-Barr virus (EBV)-mediated transformation of B lymphocytes. However, the molecular mechanism of IRF-4 in cellular transformation is unknown. We have found that knockdown of IRF-4 leads to high expression of IRF-5, a pro-apoptotic member in the IRF family. Overexpression of IRF-4 represses IRF-5 expression. Reduction of IRF-4 leads to growth inhibition, and the restoration of IRF-4 by exogenous plasmids correlates with the growth recovery and reduces IRF-5 expression. In addition, IRF-4 negatively regulates IRF-5 promoter reporter activities and binds to IRF-5 promoters in vivo and in vitro. Knockdown of IRF-5 rescues IRF-4 knockdown-mediated growth inhibition, and IRF-5 overexpression alone is sufficient to induce cellular growth inhibition of EBV-transformed cells. Therefore, IRF-5 is one of the targets of IRF-4, and IRF-4 regulates the growth of EBV-transformed cells partially through IRF-5. This work provides insight on how IRFs interact with one another to participate in viral pathogenesis and transformation.
Keywords: Herpesvirus, Interferon, Lymphocyte, Transcription Factors, Transformation, EBV, IRF-4, IRF-5, Mum1, Lymphoma
Introduction
Interferon regulatory factors (IRFs)5 are a small family of transcription factors that have multiple functions. The hallmark of these factors is a conserved N-terminal DNA-binding domain, which mediates binding to consensus or similar DNA sequences such as interferon-stimulated response element (ISRE) and IRF-binding elements. The C-terminal portion is variable and defines biological functions (1–3). Currently, the IRF family has nine members with a variety of functions including, but not limited to, apoptosis, oncogenesis, host defense, viral latency, and immune response (2–4).
Epstein-Barr virus (EBV) is a human herpesvirus, belonging to the gamma herpesvirus family. EBV almost certainly triggers two fatal cancers without the necessity for cofactors as follows: AIDS-associated central nervous system lymphoma and post-transplantation lymphoproliferative disorder (5). In addition, EBV is associated with many different human malignancies, including Burkitt lymphoma, nasopharyngeal carcinoma, Hodgkin lymphoma, and several other diseases (6). EBV transforms adult primary B cells into continually growing lymphoblastoid cell lines and concomitantly establishes type III latency in vitro (5).
IRF-4 is involved in the pathogenesis of EBV-mediated transformation of B lymphocytes and plays an essential role in cell growth of multiple myeloma cells (7, 8). IRF-4 represses expression of certain genes implicated in the mitotic checkpoint, DNA repair, apoptosis, metastasis, and immune recognition and may provide important functions critical to the emergence of adult T cell leukemia (9–11). Interestingly, IRF-4 alone is apparently not sufficient for oncogenesis in transgenic mice overexpressing IRF-4 in lymphocytes (12), suggesting that additional factors are required for the oncogenic activity of IRF-4 in vivo. Moreover, the cellular environments may determine its specific functions. IRF-4 promotes cellular proliferation in EBV-transformed cells and in multiple myeloma, but in early B cell development, IRF-4 is considered as a tumor suppressor (13). IRF-4 interacts with many different cellular factors, which may determine its biological functions in various cellular environments. Other than oncogenesis, IRF-4 is essential for the function and homeostasis of both mature B and T lymphocytes (14). Furthermore, IRF-4 is critical for pre-B-to-B transition and development of certain dendritic cells (15, 16).
IRF-5 is involved in Toll-like receptor-mediated activation of innate and adaptive immune systems (17). Overexpression of IRF-5 modulates the expression of some genes in apoptotic pathways along with multiple cell cycle regulatory factors (18). Although WT p53 stimulates expression of IRF-5 (19), the pro-apoptotic and cell cycle regulatory effects of IRF-5 are completely independent of p53. The ectopic IRF-5 sensitizes p53-proficient and p53-deficient cancer cells to DNA damage-induced apoptosis (20). IRF-5 is selectively involved in apoptosis but not in cell cycle arrest in vivo (21, 22).
Previously, we have found that IRF-4 is a critical factor in the EBV-transformation process in vitro (7). To address this possible mechanism, we have identified IRF-5 as a cellular target of IRF-4; knockdown of IRF-4 leads to high expression of IRF-5, and overexpression of IRF-4 represses IRF-5. IRF-5 overexpression alone is sufficient to induce growth inhibition, and knockdown of IRF-5 rescues IRF-4 knockdown-mediated growth inhibition. Therefore, IRF-4 regulates cell growth partially through IRF-5 in EBV transformation.
MATERIALS AND METHODS
Plasmids and Antibodies
Expression plasmid of IRF-4, F-IRF-4, was made by PCR with pCEP-IRF-4 as a template and cloned into 3×FLAG-Myc-CMV expression vector (Sigma). pCEP-IRF-4 was a gift from Alessandra B. Pernis. IRF-5 PV1 and PV3 luciferase reporter constructs were gifts from Dr. Barnes. IRF-5 expression plasmid (v4) was a gift from Dr. Paula Pitha. The mPV3-luc has the same mutations in the ISRE region, as described previously (23). The mutations were introduced into the PV3 promoter reporter construct using the QuikChange® site-directed mutagenesis kit (Stratagene) following the recommended conditions. shLuc and shIRF-41, -42, and -43 were described previously (7). shIRF-4 was the mixture of shIRF-41, -42, and -43 in a 1:1:1 ratio. The shIRF4UTR5 (target sequence (5′-AGGGCGAGTGCAGAGCAGA-3′) and shIRF4UTR3 (5′-CAGATGAGCTTATTTCAAA-3′), shIRF-51 (5′-GGTCAACGGGGAAAAGAAA-3′), shIRF-52 (5′-AGGAAGAGCTGCAGAGGAT-3′), shIRF-53 (5′-GGCCAAGGAGACAGGGAAA-3′), and shIRF-54 (5′-CGAGAGAAGAAGCTCATTA-3′) were all cloned in the pHP vector, an shRNA expression plasmid (24). The shIRF4UTR was the mixture of shIRF4UTR5 and shIRF4UTR3 at a 1:1 ratio. The shIRF-5 could be shIRF-51 plus shIRF-53 or shIRF-52 plus shIRF-54 at a 1:1 ratio. In our system, we found the combination of two or more shRNAs was more effective in target gene reductions. IRF-5 antibody (10547-1-AP) was purchased from Proteintech Group. IRF-4 (28696), PARP (F-2), and GAPDH (0411) antibodies were purchased from Santa Cruz Biotechnology. Tubulin antibody was purchased from Sigma (T6557). Caspase 3 antibody was purchased from Imgenex (IMG-144K).
Cell Culture, Transient Transfection, and Isolation of Transfected Cells
DG75 is an EBV-negative Burkitt lymphoma cell line (25). IB4, Sav I, Sav III, and P2 are all EBV-transformed B cell lines (26–28). These cells were maintained in RPMI 1640 medium plus 10% fetal bovine serum (FBS). Electroporation (320 V; 925 microfarads) was used for transfection of IB4, DG75, and P2 cells and the isolation of CD4-positive cells with the use of Dynabeads CD4 (Dynal Inc) as described previously (29–31). 293T cells are a human fibroblast line and were maintained in DMEM plus 10% FBS. Effectene (Qiagen) was used for the transfection of these cells.
Transfection of IB4 Cells for Cell Growth Assays
Transfection of IB4 cells was achieved by using a nucleofector device from Amaxa. 1 × 106 cells were transfected with 5 μg of DNA in solution B and program U20. Transfected cells were immediately put into 12-well plates with RPMI 1640 medium plus 20% FBS. After transfection by using Amaxa transfection apparatus, ∼50% of cells were dead. The growth rates of cells after transfection were slower than untransfected cells, regardless of plasmid used. Approximately 70% of remaining live cells could be transfected with the protocol. One day later, live cells were isolated by Ficoll-Paque Plus (GE Healthcare). The live cells were counted and dispensed in a culture flask at 2 × 105 cells/ml in RPMI 1640 medium plus 10% FBS; this was counted as day 1 after transfection. Every day, a small portion of cells were stained with trypan blue, and live cells were counted using a hemocytometer.
DNA Fragmentation Assay and Apoptosis Inhibitors
On day 3, the same volumes of cells were pelleted, and DNA isolation was performed as described previously (32). The isolated DNA was separated on an agarose gel. N-Acetylcysteine was from Sigma (A7250). Z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)fluoromethyl ketone) was from R&D Systems (FMK001), and DMSO was purchased from Sigma (D2650).
Biotinylated Oligonucleotide Pulldown Assay
The assays were essentially following the published protocols with 600 μg of lysates (23, 33). Some samples also included 50 μg of nonlabeled competitors or poly(dI:dC) (Sigma, P4929). For PV3 promoter binding assays, biotinylated oligonucleotide sequences were IRF5ISRE-Bf, 5′-biotin-GGAGGCTGGGGCAGAAAGCGGAACTGAGCCCGC-3′, and IRF5ISRE-Br, 5′-biotin-GCGGGCTCAGTTCCGCTTTCTGCCCCAGCCTCC-3′. Biotinylated oligonucleotide sequences for mutated PV3 ISRE were IRF5-ISRE-MBf 5′-biotin-GGAGGCTGGGGCAGAAGCCGGAACTGAGCCCGC-3′, and IRF-5-ISRE-MBr, 5′-biotin-GCGGGCTCAGTTCCGGCTTCTGCCCCAGCCTCC-3′. Separation of proteins on SDS-PAGE and Western blot were carried out following standard protocol as described previously (31, 34).
Chromatin Immunoprecipitation Assay (ChIP)
EZ ChIPTM chromatin immunoprecipitation kit was used for ChIP assay following the manufacturer's recommendations (Millipore; catalog no. 17-371). 8 μg of IRF-4 antibody, normal rabbit serum, or no antibody was added. 50 ng of DNA were used for PCR using primers that flank the PV3 or PV1 IRF-binding sequence region within the two IRF-5 promoters as follows: IRF5PV3CHIPf, 5′-GGGGTCTACAGATACAACTATG-3′, and IRF5PV3CHIPr, 5′-GAGAGGTAAGGCCGGCCCTTGC-3′; IRF5PV1CHIPf, 5′-GGGTGACAGAGCAAGACTCC-3′, and IRF5PV1CHIPr, 5′-GTCAACAGGCAGCAGGTGTA-3′.
Detection of IRF-5 Promoter Usages
Primer sets that specifically recognize IRF-5 promoter regions (promoter V1 specific, 5′-CCTGGCGCAGCCACGCAGGCGCA-3′, and V3 specific, 5′-CTAGGCAGGTGCAACCCCAAAA-3′) and a common region in exon 4 of IRF-5 (5′-CCAAAAGAGTAATCCTCAGGG-3′) were used for detection of the promoter usages in target cells (23). Total RNA was isolated; cDNA was synthesized with random hexamers and subjected to PCR analysis with the promoter specific pairs. The PCR products were electrophoresed on an 8% polyacrylamide gel.
RESULTS
IRF-4 Negatively Regulates the Expression of IRF-5 in EBV-transformed Cells
IRF-5 is a pro-apoptotic gene that is highly expressed in EBV-transformed cells (35), and we suspect that IRF-4 might be required for the inhibition of the pro-apoptotic functions of IRF-5 in EBV transformation. We thus examined if the expression of IRF-5 is affected by the IRF-4 knockdown in EBV-transformed cells. IB4, a prototypical cell line transformed by EBV in vitro (28, 36–40), was used for our experiments as it has a relatively high transfection efficiency with Amaxa technology. As shown in Fig. 1, IRF-4 knockdown resulted in the increase of IRF-5 protein (lanes 1 and 2). To examine the role of IRF-4 further, we examined if ectopic expression of IRF-4 affected the expression of IRF-5. IB4 cells were transfected with IRF-4 expression plasmid. Overexpression of IRF-4 reduced the expression of IRF-5 (Fig. 1, lanes 3 and 4). Of note, the exposure times for the two panels were different. Minimum exposure was used to show the increase, and moderate exposure time was used for observation of inhibition. Collectively, those data suggest that IRF-4 negatively regulates IRF-5 expression.
FIGURE 1.
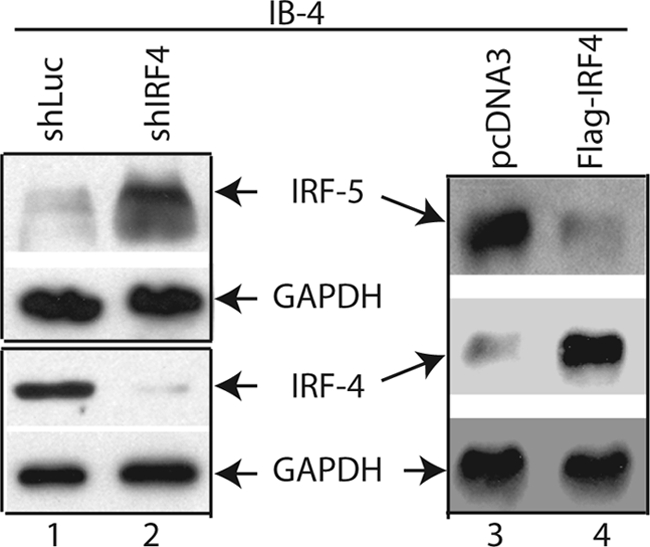
IRF-4 negatively regulates IRF-5 expression. IB4 cells were transfected with shLuc (5 μg), shIRF-4 (shIRF-41/shIRF-42/shIRF-43, 1:1:1 ratio, 5 μg), pcDNA3 (5 μg), or IRF-4 expression plasmid (1 μg) by use of a nucleofector device from Amaxa. Total DNA were normalized to 5 μg with vector DNA. Cells were collected 3 days later. The expression levels of IRF-4, IRF-5, and GAPDH were examined by Western blots. The identities of the proteins are shown. The images in the same box indicate that they are derived from the same membrane.
Restoration of IRF-4 Expression Rescues EBV-transformed Cells from Growth Inhibition
To further evaluate the role of IRF-4 in the cellular growth of EBV-transformed cells, we have established a system to specifically knockdown the endogenous IRF-4 but allow the expression of exogenously introduced IRF-4. We have chosen the 5′- and/or 3′-untranslated region (UTR) of IRF-4 mRNA as targets of shRNA (Fig. 2A). The shIRF4UTR expression plasmids (the combination of shIRF4UTR5 and shIRF4UTR3; Fig. 2A) were able to knock down IRF-4 expression in IB4 cells with the established transfection methods (Fig. 2B). The expression plasmid for IRF-4 was made only using the IRF-4 coding sequences with FLAG tag on the N terminus (Fig. 2A). Because there is no overlap between shIRF4UTR target sequences and IRF-4 coding sequences, the shIRF4UTR does not affect the expression of the IRF-4 derived from the expression vector. As expected, the IRF-4 expression was restored in shIRF4UTR + F-IRF-4 transfected cells (Fig. 2B). In addition, the cell growth of IRF-4-restored cells behaved similarly to the vector control cells (Fig. 2C). These data confirmed that IRF-4 is responsible for the growth inhibition in EBV-transformed cells. Furthermore, the expression of IRF-5 was induced when IRF-4 was knocked down (Fig. 2B, lanes 1 and 2) but reduced upon restoration of IRF-4 (lanes 3 and 4). Thus, the growth phenotype of IRF-4 knockdown cells was associated with IRF-5 regulation.
FIGURE 2.

Restoration of IRF-4 alleviates endogenous IRF-4 knockdown-mediated cell growth inhibition. A, schematic diagram of shIRF4UTR and expression plasmid. The target of the shIRF4UTR is shown. The expression plasmid was made with just the IRF-4 ORF region. B, restoration of IRF-4 in endogenous IRF-4 knockdown cells. IB4 cells were transfected with shLuc (4 μg), shIRF4UTR (shIRF4UTR5 + shIRF4UTR3; 4 μg), or shIRF4UTR plus F-IRF-4 (1 μg) by use of a nucleofector device from Amaxa. Total DNAs were normalized to 5 μg with vector DNA. The lanes 3 and 4 represent duplication. The expression levels of IRF-4, IRF-5, and GAPDH were examined by Western blots. The images in the same box indicate that they are derived from the same membrane. C, restoration of IRF-4 rescues cells from growth inhibition. One day (D) after transfection, live cells were isolated through Ficoll-Paque Plus and seeded onto fresh media. At the indicated days after transfection, surviving cells were enumerated by trypan blue exclusion. Each point represents the number of live cells (means ± S.D.) from three different counts. One representative from three independent experiments is shown.
IRF-4 Binds to IRF-5 Promoter
IRF-5 has at least two promoters (PV1 and PV3) (23). Both promoters are active in virally transformed cells (Fig. 3A and supplemental Fig. 1A), and both have putative IRF-binding sites. Because IRFs can bind to the same or similar DNA sequences through the conserved DNA-binding domain, we thus suspect that IRF-4 may bind to the IRF-5 promoter region. To test whether IRF-4 binds the IRF-5 promoter DNA in vivo, ChIP assays were performed with IB4 cell lysates, and the region around putative IRF-binding sites was used as target for amplifying DNA from the immunoprecipitates. As shown in Fig. 3B, the PV3 promoter region was amplified from immunoprecipitates when IRF-4 antibody was used (lane 2). However, PCR amplification failed to generate target DNA from the immunoprecipitates by normal rabbit antibody (Fig. 3B, lane 3) or without antibody addition (lane 4). Amplifications of a small fraction of input DNA before immunoprecipitations as well as positive control produced a target band at the expected size (Fig. 3B, lanes 1 and 5). The data suggest that IRF-4 binds to IRF-5 promoter regions in vivo.
FIGURE 3.

IRF-4 binds to IRF-5 promoter region. A, PV3 promoter is active in EBV-transformed cells. The RNAs were isolated from the EBV-transformed cells as shown at the top, and RT-PCR was used to determine the transcriptional activities with the target PV3 promoter-specific primers. The specific promoter usage, and specificity of the amplification, is indicated. B, IRF-4 binds to IRF-5 promoter in vivo. IRF-4 antibody (Ab) was used for ChIP analyses to detect in vivo DNA binding activities to the IRF-5 promoter region. Primers for IRF-5 PV3 promoter regions surrounding the putative IRF-4-binding site were used to amplify the DNA from immunoprecipitates. The identity of IRF-4-bound, PV3 promoter amplification is as shown. The plasmid, IRF-5 PV3-luc, was used as positive control. Input DNA represents the PCR amplification from 1/100th the amount of input lysates for immunoprecipitations. PCR amplification of immunoprecipitates from normal rabbit serum is also shown as control. C, IRF-4 binds to the IRF-5 promoter in vitro. Cell extracts were made from IB4 cells and incubated with biotinylated oligonucleotides containing the IRF-5 PV3 ISRE sequences. The un-biotinylated IRF-5 PV3 ISRE, PV3 mISRE, Zta-response element oligonucleotides, or poly(dI:dC) were used as competitors as shown at the top. 1/20th of input cell lysates for pulldown experiments was used as a control. Western blots were carried out for the detection of IRF-4 and GAPDH.
We further tested whether IRF-4 binds to the IRF-5 promoter by DNA affinity binding assays with extracts from IB4 cells. The biotinylated IRF-5 PV3 ISRE was used to pull down the binding proteins, and various combinations of other annealed oligonucleotides were used as competitors. As shown in Fig. 3C, IRF-4 did bind to DNA (lane 1). The IRF-4 binding was competed out by incubating with nonbiotinylated wild type PV3 ISRE (Fig. 3C, lane 2). In contrast, mutated PV3-ISRE, nonrelevant Zta-response element, and poly(dI:dC) did not compete for IRF-4 binding (Fig. 3C, lanes 3–5). The residual carry-over GAPDH was used as an internal control for affinity binding assays. A small fraction of input IB4 cell lysates was also shown (Fig. 3C, lane 6). The mutated ISRE seemed to bind to IRF-4 also in a less efficient manner in this assay, possibly due to the specificity of IRF-4 and the IRF-5 PV3 ISRE, and/or the oligonucleotide pulldown assay itself (supplemental Fig. 1D). IRF-4 is also able to bind to PV1 promoter (supplemental Fig. 1, B and C). In summary, these results suggest that IRF-4 is able to bind IRF-5 promoter.
IRF-4 Represses IRF-5 Promoter Activity
Once the IRF-4 binding to IRF-5 PV3 promoter was established, we examined if IRF-4 could repress the IRF-5 promoter reporter constructs as IRF-4 is predominantly a transcriptional repressor (10, 11). Both PV1 and PV3 promoter luciferase reporter constructs were used. The promoter reporter construct and IRF-4 expression plasmid were co-transfected into 293T cells, and the reporter activities were measured. As shown in Fig. 4A, IRF-4 was able to repress the IRF-5 PV3 promoter reporter construct. However, IRF-4 failed to repress the reporter construct with ISRE mutations, which correlates with that data that IRF-4 binds to mutated ISRE less efficiently. Interestingly, IRF-4 failed to repress the PV1 reporter construct in this assay (supplemental Fig. 1E).
FIGURE 4.

IRF-4 negatively regulates IRF-5 promoter activity. A, IRF-4 represses IRF-5 promoter reporter activity. 293T cells were transfected with various reporter constructs and expression plasmid (0.01, 0.05, and 0.1 μg) as shown at the top. The mPV3-luc has the IRF-5 ISRE sequences mutated in the ISRE in the promoter reporter construct. Cell lysates were used for the luciferase and β-galactosidase assays. Relative promoter reporter activities (luciferase/β-galactosidase) are shown. Standard error bars are shown. B, endogenous IRF-4 regulates IRF-5 promoter activity. IB4 cells were transfected with the indicated plasmid as shown at the top. shIRF4UTR and F-IRF-4 usages were the same as in Fig. 2. Two days later, luciferase and β-galactosidase activities were measured. Relative promoter reporter activities are shown.
Furthermore, we tested if the endogenous IRFs affected IRF-5 PV3 promoter activities. The shIRF-4 and other plasmids were transfected into IB4 cells, along with the promoter reporter construct. As shown in Fig. 4B, shIRF4UTR up-regulated the IRF-5 PV3 promoter reporter activities and co-transfection with IRF-4 repressed the activity. In addition, mutated ISRE PV3 promoter (mPV3-luc) barely responded to shIRF4UTR expression. The IRF-5 PV1 promoter reporter construct had hardly any detectable activities in IB4 cells (data not shown). We used the conditional medium from shLuc- or shIRF4UTR-transfected IB4 cells and the treated 293T cells transfected with IRF-5 promoter construct (PV3-Luc). The conditional medium did not have an impact on the promoter reporter activity of PV3 (supplemental Fig. 2A). In addition, similar results to Fig. 4B can be obtained in P2 cells, a STAT-1-null EBV-transformed cell line (supplemental Fig. 2B) (26). Therefore, the potential inflammatory cytokines induced by shRNAs were probably not responsible for the activation of PV3 promoter reporter in IB4 cells. All these results collectively suggest that IRF-4 represses IRF-5 PV3 promoter reporter construct.
Knockdown of IRF-5 Rescues IRF-4 Knockdown-mediated Growth Inhibition
Next, we examined the role of IRF-5 in EBV-transformed cells. IRF-5 is considered as a tumor suppressor. The shIRF-5 plasmids were transfected into IB4 cells, and the knockdown of IRF-5 greatly enhanced the growth of EBV-transformed cells (Fig. 5A). Reduction of IRF-5 was confirmed by Western blot (Fig. 5B). Thus, the physiological levels of IRF-5 were able to repress cellular growth in EBV-transformed cells.
FIGURE 5.

Knockdown of IRF-5 rescues IRF-4 knockdown-mediated growth inhibition. A, IB4 cells were transfected with shLuc (5 μg; line +), shIRF-4 (shIRF-41 + shIRF-42 + shIRF-43, 1:1:1 ratio, 2.5 μg; line *), shIRF5 (shIRF-51 + shIRF-53, 1:1 ratio, 2.5 μg; solid square), shIRF4 plus shIRF-5 (shIRF-51 + shIRF-53; open square), shIRF-5 (shIRF-52 + shIRF-54, 1:1 ratio, 2.5 μg; solid circle), or shIRF-4 plus shIRF-5 (shIRF-52 + shIRF-54; open circle) by Amaxa nucleofector technology. Total DNA was normalized to 5 μg with shLuc DNA. One day (D) after the transfection, live cells were isolated. At the indicated days after transfection, surviving cells were enumerated. Each point represents the number of live cells (means ± S.D.). The use of shIRF52 plus shIRF-54 also produced curative data. Results from one representative experiment are as shown. B, expression levels of the various proteins. IB4 cells were transfected with various plasmids as shown at the top. The expression levels of IRF-4, IRF-5, and GAPDH were examined by Western blots 2 days after transfection. The identities of the proteins are shown.
Whether the knockdown of IRF-5 alleviates the IRF-4, knockdown-mediated growth inhibition was examined. As shown in Fig. 5A, shIRF-4 represses the cell growth as shown before. However, when shIRF-5 and shIRF-4 were co-transfected, the growth inhibition of shIRF-4 was partially alleviated (open circle and square). This suggests that IRF-5 is one of the targets for IRF-4 in the regulation of cellular growth in EBV-transformed cells.
Caspase Inhibitor Rescues IRF-4 Knockdown-mediated Cell Growth Inhibition
To evaluate the role of apoptosis in IRF-4 knockdown-mediated growth inhibition, we used caspase inhibitors to block the caspase dependent apoptosis. IB4 cells were transfected with shLuc or shIRF-4. One day after the transfection, live cells were isolated, seeded, and treated with DMSO, Z-VAD-FMK (20 μm), or NAC (10 mm), respectively. Z-VAD-FMK is a pan-caspase inhibitor that irreversibly binds to the catalytic site of caspase proteases and can inhibit induction of apoptosis. At lower concentrations, NAC is nontoxic to cultured cells and can protect against apoptosis induced by intracellular reactive oxygen species. As shown in Fig. 6A, Z-VAD-FMK almost completely recovered the IRF-4 knockdown-mediated cell growth inhibition but NAC did not (Fig. 6A). As expected, DMSO has no effect on cellular growth, and no drugs have an effect on shLuc-transfected cells (Fig. 6A, right panel). It is known that IRF-4 knockdown leads to caspase 3 activation (data not shown) (7). Thus, it is likely that caspase-dependent apoptosis is the major factor affecting IRF-4 knockdown-mediated growth inhibition.
FIGURE 6.

Caspase inhibition prevents IRF-4 knockdown-mediated growth inhibition. A, caspase inhibition and cell growth. IB4 cells were transfected with 5 μg of shLuc (right panel) or shIR-F4 (shIRF-41 + hIRF-42 + shIRF-43, 1:1:1 ratio) (left panel). One day (D) after the transfection, live cells were isolated through Ficoll-Paque Plus. The cells were seeded at desired concentrations in different wells with no treatment or with treatment of DMSO, Z-VAD (20 μm), or NAC (10 mm). At the indicated days after treatment, surviving cells were enumerated. Each point represents the number of live cells (mean ± S.D.). B, overexpression of IRF-5 inhibits cell growth of EBV-transformed cells. One day after the transfection, live cells were isolated. At the indicated days after transfection, surviving cells were enumerated. Each point represents the number of live cells (mean ± S.D.). C, protein cleavage in IRF-5-overexpressing cells. Lysates from transfected cells at day 3 were used for Western blot analysis with PARP, IRF-5, caspase 3, and GAPDH antibodies. The molecular masses (kDa) of PARP fragments are indicated in parentheses. The images in the same box are derived from the same membrane. The identities of the proteins are as shown. D, DNA fragmentation in IRF-5-transfected cells. Total cellular DNAs were isolated on day 3. The sizes of the DNA markers are as shown on the left in base pairs.
Overexpression of IRF-5 Inhibits Cellular Growth in EBV-transformed Cells
Because of the facts that IRF-4 knockdown inhibited the cellular growth and induced IRF-5 expression, whether IRF-5 overexpression itself is able to cause inhibition of cell growth was examined. IRF-5 expression plasmid (splicing variant 4) was transfected into IB4 cells, and the cell growth was monitored on a daily basis. As shown in Fig. 6B, the IRF-5-transfected cells had a much slower growth rate. The reason to use the IRF-5 splicing variant 4 in this experiment is that the variant has a similar property to other splicing variants as a full-length protein, and the variant was the first to be cloned and the most extensively characterized splicing variant of IRF-5. Furthermore, hallmarks of apoptosis were examined. We found that IRF-5 induced the cleavage of PARP and caspase 3, as well as the DNA fragmentation; those markers are indicative of apoptosis (Fig. 6, C and D). Of note, IRF-5 is predominantly localized in the cytoplasm in EBV-transformed cells (supplemental Fig. 3), but IRF-5 seems to be functional as endogenous levels of IRF-5 inhibit cell growth (Fig. 5). Therefore, IRF-5 overexpression inhibits cellular growth, partially through induction of apoptosis.
DISCUSSION
The involvement of IRFs in oncogenesis has been extensively studied. Previously, we have established that IRF-4 is involved in the EBV-mediated transformation process in vitro and possibly in vivo for lymphoma in the central nervous system (7). In this study, we have uncovered a novel mechanism for IRF-4 to regulate EBV-mediated cellular growth, i.e. by repressing the expression of IRF-5, a pro-apoptotic member of IRFs.
We have demonstrated the following. First, IRF-5 is induced once IRF-4 is down-regulated (Figs. 1 and 2B), and the restoration of IRF-4 by exogenous plasmids reduces IRF-5 expression (Fig. 2B). Moreover, overexpression of IRF-4 represses IRF-5 expression (Fig. 1). Second, IRF-4 binds to IRF-5 promoter regions. IRF-5 has at least two promoters (PV1 and PV3) that are active in EBV-transformed cells. Both promoters have putative IRF-binding sites, and IRF-4 binds to the PV3 promoter in vivo and in vitro (Fig. 3 and supplemental Fig. 1). Third, IRF-4 represses the IRF-5 PV3 promoter reporter construct (Fig. 4A), and the PV3 promoter reporter construct behaves just like the IRF-5 protein in EBV-transformed cells (Fig. 4B). Interestingly, IRF-4 did not repress the IRF-5 PV1 reporter construct in our experimental settings (supplemental Fig. 1E). Therefore, IRF-5 is apparently a target of IRF-4 at the PV3 promoter level in EBV-transformed cells. It is of note that IRF-4 itself has marginal effects on the IRF-5 promoter reporter (Fig. 4A), but in the context of EBV latency, IRF-4 seems to have a stronger effect (compare Fig. 4, A and B). The large increase of IRF-5 luciferase activity in IB4 cells might be related to the fact that IRF-4 has multiple targets, and some of the targets may also be involved in the regulation of IRF-5 expression.
The regulation of IRF-5 by IRF-4 may be related to the biological function of IRF-4 in the regulation of cell growth in EBV transformation. First, knockdown of IRF-4 inhibits the growth properties of transformed cells (Figs. 2C and 5A), and the inhibition of cell growth is positively associated with IRF-5 up-regulation (Figs. 1 and 2). Second, supplement of endogenous IRF-4 with plasmid-derived IRF-4 restores the growth of transformed cells and the down-regulation of IRF-5 (Fig. 2). Third, IRF-5 itself is a pro-apoptotic factor. IRF-5 seems to inhibit cell growth at the physiological levels (Fig. 5A), and overexpression of IRF-5 in EBV-transformed cells inhibits cell growth, at least partially through apoptosis induction (Fig. 6, B–D). Fourth, knockdown of IRF-5 rescues IRF-4 knockdown-mediated growth inhibition (Fig. 5A). Fifth, IRF-4 knockdown-mediated growth inhibition is mainly through the caspase-dependent apoptotic pathway (Fig. 6A), which correlates IRF-5 induction and the induction of apoptosis by IRF-5 (Figs. 1, 2, and 6, C and D). Sixth, and finally, the inhibition of cell growth by IRF-4 is not restricted to IB4 cells, other EBV-transformed cells were also inhibited by shIRF-4 transfection (supplemental Fig. 4). Interestingly, the EBV-negative Burkitt lymphoma line, DG75, IRF-4 has limited effects, suggesting IRF-4 might be preferentially playing a critical role in EBV-transformed cells (supplemental Fig. 4).
Of note, two sets of shIRF-5 (shIRF-51 plus shIRF-53 or shIRF-52 plus shIRF-54) generated similar results (Fig. 5A), suggesting the off-target effects of shIRF-5 are minimal. In addition, the off-targeting and other nonspecific effects of the shIRF-4 may be minimal (7). We also show that the other sets of shIRF-4 are able to reduce the expression of endogenous IRF-4. Furthermore, the restoration of IRF-4 by exogenous plasmids restored the growth properties of EBV-transformed cells (Fig. 2). IRF-4 is therefore a critical factor in EBV transformation by blocking the IRF-5-mediated apoptosis processes.
EBV activates many pro-apoptotic (such as p53) and anti-apoptotic (such as Bcl-2) factors during transformation processes (41, 42). EBV also encodes anti-apoptotic genes, such as latent membrane protein 1 and two viral Bcl-2 homologous genes (6, 43–45). IRF-5 is one of the pro-apoptotic gene products induced by EBV. It is apparent that the physiological levels of IRF-5 are involved in the growth of EBV-transformed cells (Fig. 5). Thus, EBV must use another gene(s) to block such pro-apoptotic action of IRF-5. IRF-4 is clearly a candidate for this purpose. Other than repressing the expression of IRF-5, IRF-4 may regulate IRF-5 targets directly. It is known that IRF-4 binds to MyD88, competes with IRF-5, and negatively regulates IRF-5-mediated activation of proinflammatory cytokines (17). In addition, IRF-4 may inhibit IRF-5-mediated activation of certain genes by competing for binding to promoter regions as IRF-binding sites are similar.
It is interesting that both IRF-4 and IRF-5 are highly expressed in EBV-transformed cells (35). Therefore, it is apparent that EBV uses IRF-4 to prevent a super-activation of IRF-5 during EBV transformation. The reciprocal inhibition and/or activation between the two factors may contribute to apoptosis or proliferation in infected cells. EBV may use this dynamic equilibrium for its own benefit in various microenvironments for the survival of the virus in vivo. It is suspected that the ratio of IRF-1 (a tumor suppressor)/IRF-2 (an oncogene) is important in certain tumor development (46–48). Here, we have provided another possible scenario that two IRFs may regulate cell growth in a different setting.
Apparently oncogenic herpesviruses and IRFs have intimate connections. EBV has been known to have an intimate relation with IRF-7 (29, 34, 49, 50). The genome of Kaposi sarcoma-associated herpesvirus contains several homologous IRFs (vIRFs) (51–53). Kaposi sarcoma-associated herpesvirus is implicated in the pathogenesis of Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease (51, 54, 55). Rhesus rhadinovirus, another oncogenic herpesvirus, has eight vIRFs in the genome (56, 57). In this study, we have identified a novel mechanism that IRF-4 regulates cellular growth by targeting pro-apoptotic IRF-5 during EBV transformation. The modulation of IRF pathway is apparently a common theme used by oncogenic herpesviruses.
Supplementary Material
Acknowledgments
We thank Drs. Betsy Barnes, Paula Pitha, Jean-Laurent Casanova, Jeffery Sample, and Alessandra Pernis for providing various reagents for the study and Dr. Lingjun Zhao for valuable advice on shRNA target selection.
This work was supported, in whole or in part, by National Institutes of Health Grants CA138213 and CA108951 and Grant RR15635 from NCRR (to L. Z.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
- IRF
- interferon regulatory factor
- EBV
- Epstein-Barr virus
- ISRE
- interferon-stimulated response element
- Z-VAD-FMK
- carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)fluoromethyl ketone
- PARP
- poly(ADP-ribose) polymerase
- NAC
- N-acetylcysteine.
REFERENCES
- 1. Zhang L., Pagano J. S. (2001) Semin. Cancer Biol. 11, 445–453 [DOI] [PubMed] [Google Scholar]
- 2. Nguyen H., Hiscott J., Pitha P. M. (1997) Cytokine Growth Factor Rev. 8, 293–312 [DOI] [PubMed] [Google Scholar]
- 3. Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. (2001) Annu. Rev. Immunol. 19, 623–655 [DOI] [PubMed] [Google Scholar]
- 4. Tanaka N., Taniguchi T. (2000) Semin. Cancer Biol. 10, 73–81 [DOI] [PubMed] [Google Scholar]
- 5. Pagano J. S. (1999) Proc. Assoc. Am. Physicians 111, 573–580 [DOI] [PubMed] [Google Scholar]
- 6. Kieff E. (1996) in Virology (Fields B. N., Knipe D. M., Howley P. M. eds) 3rd Ed., pp. 2343–2396, Lippinscott-Raven Publishers, Philadelphia [Google Scholar]
- 7. Xu D., Zhao L., Del Valle L., Miklossy J., Zhang L. (2008) J. Virol. 82, 6251–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaffer A. L., Emre N. C., Lamy L., Ngo V. N., Wright G., Xiao W., Powell J., Dave S., Yu X., Zhao H., Zeng Y., Chen B., Epstein J., Staudt L. M. (2008) Nature 454, 226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamagata T., Nishida J., Tanaka S., Sakai R., Mitani K., Yoshida M., Taniguchi T., Yazaki Y., Hirai H. (1996) Mol. Cell. Biol. 16, 1283–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mamane Y., Grandvaux N., Hernandez E., Sharma S., Innocente S. A., Lee J. M., Azimi N., Lin R., Hiscott J. (2002) Oncogene 21, 6751–6765 [DOI] [PubMed] [Google Scholar]
- 11. Mamane Y., Loignon M., Palmer J., Hernandez E., Césaire R., Alaoui-Jamali M., Hiscott J. (2005) J. Interferon Cytokine Res. 25, 43–51 [DOI] [PubMed] [Google Scholar]
- 12. Saito T., Yamagata T., Takahashi T., Honda H., Hirai H. (1999) Biochem. Biophys. Res. Commun. 260, 329–331 [DOI] [PubMed] [Google Scholar]
- 13. Acquaviva J., Chen X., Ren R. (2008) Blood 112, 3798–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mittrücker H. W., Matsuyama T., Grossman A., Kündig T. M., Potter J., Shahinian A., Wakeham A., Patterson B., Ohashi P. S., Mak T. W. (1997) Science 275, 540–543 [DOI] [PubMed] [Google Scholar]
- 15. Lu R., Medina K. L., Lancki D. W., Singh H. (2003) Genes Dev. 17, 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tamura T., Tailor P., Yamaoka K., Kong H. J., Tsujimura H., O'Shea J. J., Singh H., Ozato K. (2005) J. Immunol. 174, 2573–2581 [DOI] [PubMed] [Google Scholar]
- 17. Negishi H., Ohba Y., Yanai H., Takaoka A., Honma K., Yui K., Matsuyama T., Taniguchi T., Honda K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15989–15994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barnes B. J., Kellum M. J., Pinder K. E., Frisancho J. A., Pitha P. M. (2003) Cancer Res. 63, 6424–6431 [PubMed] [Google Scholar]
- 19. Mori T., Anazawa Y., Iiizumi M., Fukuda S., Nakamura Y., Arakawa H. (2002) Oncogene 21, 2914–2918 [DOI] [PubMed] [Google Scholar]
- 20. Hu G., Mancl M. E., Barnes B. J. (2005) Cancer Res. 65, 7403–7412 [DOI] [PubMed] [Google Scholar]
- 21. Couzinet A., Tamura K., Chen H. M., Nishimura K., Wang Z., Morishita Y., Takeda K., Yagita H., Yanai H., Taniguchi T., Tamura T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2556–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yanai H., Chen H. M., Inuzuka T., Kondo S., Mak T. W., Takaoka A., Honda K., Taniguchi T. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mancl M. E., Hu G., Sangster-Guity N., Olshalsky S. L., Hoops K., Fitzgerald-Bocarsly P., Pitha P. M., Pinder K., Barnes B. J. (2005) J. Biol. Chem. 280, 21078–21090 [DOI] [PubMed] [Google Scholar]
- 24. Xu D., Coleman T., Zhang J., Fagot A., Kotalik C., Zhao L., Trivedi P., Jones C., Zhang L. (2007) J. Virol. 81, 6068–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ben-Bassat H., Goldblum N., Mitrani S., Goldblum T., Yoffey J. M., Cohen M. M., Bentwich Z., Ramot B., Klein E., Klein G. (1977) Int. J. Cancer 19, 27–33 [DOI] [PubMed] [Google Scholar]
- 26. Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I. Z., Al-Jumaah S., Yang K., Chapgier A., Eidenschenk C., Eid P., Al Ghonaium A., Tufenkeji H., Frayha H., Al-Gazlan S., Al-Rayes H., Schreiber R. D., Gresser I., Casanova J. L. (2003) Nat. Genet. 33, 388–391 [DOI] [PubMed] [Google Scholar]
- 27. Nonkwelo C., Skinner J., Bell A., Rickinson A., Sample J. (1996) J. Virol. 70, 623–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Henderson A., Ripley S., Heller M., Kieff E. (1983) Proc. Natl. Acad. Sci. U.S.A. 80, 1987–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang L., Pagano J. S. (2001) J. Virol. 75, 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang L., Wu L., Hong K., Pagano J. S. (2001) J. Virol. 75, 12393–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang L., Pagano J. S. (2000) J. Virol. 74, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin L., Perng G. C., Brick D. J., Naito J., Nesburn A. B., Jones C., Wechsler S. L. (2004) J. Virol. Methods 118, 9–13 [DOI] [PubMed] [Google Scholar]
- 33. Ragione F. D., Cucciolla V., Criniti V., Indaco S., Borriello A., Zappia V. (2003) J. Biol. Chem. 278, 23360–23368 [DOI] [PubMed] [Google Scholar]
- 34. Zhang L., Zhang J., Lambert Q., Der C. J., Del Valle L., Miklossy J., Khalili K., Zhou Y., Pagano J. S. (2004) J. Virol. 78, 12987–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin H. J., Lee J. M., Walls D., Hayward S. D. (2007) J. Virol. 81, 9748–9758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carter K. L., Cahir-McFarland E., Kieff E. (2002) J. Virol. 76, 10427–10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hurley E. A., Klaman L. D., Agger S., Lawrence J. B., Thorley-Lawson D. A. (1991) J. Virol. 65, 3958–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cahir McFarland E. D., Izumi K. M., Mosialos G. (1999) Oncogene 18, 6959–6964 [DOI] [PubMed] [Google Scholar]
- 39. Cahir-McFarland E. D., Davidson D. M., Schauer S. L., Duong J., Kieff E. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6055–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frost V., Delikat S., Al-Mehairi S., Sinclair A. J. (2001) J. Gen. Virol. 82, 3057–3066 [DOI] [PubMed] [Google Scholar]
- 41. Allday M. J., Sinclair A., Parker G., Crawford D. H., Farrell P. J. (1995) EMBO J. 14, 1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henderson S., Huen D., Rowe M., Dawson C., Johnson G., Rickinson A. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8479–8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altmann M., Hammerschmidt W. (2005) PLoS Biol. 3, e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bellows D. S., Howell M., Pearson C., Hazlewood S. A., Hardwick J. M. (2002) J. Virol. 76, 2469–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marshall W. L., Yim C., Gustafson E., Graf T., Sage D. R., Hanify K., Williams L., Fingeroth J., Finberg R. W. (1999) J. Virol. 73, 5181–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willman C. L., Sever C. E., Pallavicini M. G., Harada H., Tanaka N., Slovak M. L., Yamamoto H., Harada K., Meeker T. C., List A. F. (1993) Science 259, 968–971 [DOI] [PubMed] [Google Scholar]
- 47. Taniguchi T., Harada H., Lamphier M. (1995) J. Cancer Res. Clin. Oncol. 121, 516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nguyen H., Mustafa A., Hiscott J., Lin R. (1995) Oncogene 11, 537–544 [PubMed] [Google Scholar]
- 49. Zhang L., Pagano J. S. (1997) Mol. Cell. Biol. 17, 5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ning S., Hahn A. M., Huye L. E., Pagano J. S. (2003) J. Virol. 77, 9359–9368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moore P., Chang Y. (2001) in Virology (Knipe D. M. ed) pp. 2803–2833, Lippincott Williams and Wilkins, Philadelphia [Google Scholar]
- 52. Chang Y., Cesarman E., Pessin M. S., Lee F., Culpepper J., Knowles D. M., Moore P. S. (1994) Science 266, 1865–1869 [DOI] [PubMed] [Google Scholar]
- 53. Chang Y., Moore P. S. (1996) Infect. Agents Dis. 5, 215–222 [PubMed] [Google Scholar]
- 54. West J. T., Wood C. (2003) Oncogene 22, 5150–5163 [DOI] [PubMed] [Google Scholar]
- 55. Dourmishev L. A., Dourmishev A. L., Palmeri D., Schwartz R. A., Lukac D. M. (2003) Microbiol. Mol. Biol. Rev. 67, 175–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pitha P. M., Au W. C., Lowther W., Juang Y. T., Schafer S. L., Burysek L., Hiscott J., Moore P. A. (1998) Biochimie 80, 651–658 [DOI] [PubMed] [Google Scholar]
- 57. Alexander L., Denekamp L., Knapp A., Auerbach M. R., Damania B., Desrosiers R. C. (2000) J. Virol. 74, 3388–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.