

Abstract
Resistance to multiple anticancer agents is a major obstacle in the successful treatment of cancers. Overexpression of some ATP-binding cassette (ABC) membrane transporters such as ABCC1 has been shown to be a major contributor of multidrug resistance (MDR) in both laboratory cell line models and the clinical setting. ABCC1 has been thought to function as a homodimer with a putative dimerization domain located in the first 281 amino acid residues, including MSD0 and L0 domains. In this study, we further mapped in detail the dimerization site and placed it in TM5 and ECL3 in MSD0 using co-expression and co-immunoprecipitation of a series of deletion constructs. TM5 and ECL3 in one subunit appear to interact with TM5 and ECL3 in the opposing subunit in a sequence-independent manner, but their physical location together with the hydrophobicity of TM5 and the length of ECL3 appears to be important contributors to the dimerization ability of ABCC1.
Multidrug resistance (MDR)1 is a major obstacle in the successful treatment of human cancers. Overexpression of some ATP-binding cassette (ABC) membrane transporters, such as P-glycoprotein (Pgp/MDR1/ABCB1), breast cancer resistance protein (BCRP/ABCG2), and multidrug resistance-associated protein 1 (MRP1/ABCC1, termed ABCC1 hereafter), has been demonstrated to be a major cause of MDR in both laboratory cell models and the clinical setting (1–3). These ABC transporters actively efflux anticancer drugs from cells, resulting in a decreased level of intracellular accumulation and the decreased cytotoxicity of these drugs.
Human ABCC1 is one of the 13 members of the human ABCC subfamily (4). ABCC1 has been demonstrated to mediate ATP-dependent cellular efflux of a wide variety of anticancer drugs, xenobiotics, and a broad spectrum of organic anions, including oxidized and reduced glutathione (GSSG and GSH, respectively) as well as anionic conjugates of GSH, glucuronide, and sulfate (4). Endogenous organic anion substrates of ABCC1 include but are not limited to cysteinyl leukotriene C4 (LTC4) and conjugated estrogens E217βG, E13SO4, and dehydroepiandrosterone sulfate (5). Unlike most other human ABC family members, such as ABCB1, which contain two membrane-spanning domains (MSD) and two nucleotide-binding domains (NBD) with an MSD1–NBD1–MSD2–NBD2 domain structure, ABCC1 as well as some other members of the ABCC subfamily, including ABCC2, -3, -6, and -7, contains an additional membrane-spanning domain (MSD0) consisting of five predicted transmembrane segments with a putative extracellular amino-terminal end.
The functional role of the additional MSD0 of ABCC1 is yet to be fully elucidated. Removal of up to 204 amino acids of MSD0 resulted in a functional ABCC1 transporter, suggesting that MSD0 may not be essential for ABCC1 function (6). However, further deletion of up to 281 amino acid residues resulted in a functionally defective protein. Most recently, it was reported that MSD0 may play roles in processing and trafficking of human ABCC1 (7). It has also been reported that the amino terminus is functionally important (8) and may form a U-shaped structure and function as a gating mechanism for drug transport activity (9, 10).
Previously, using multiple approaches, including nondenaturing polyacrylamide gel electrophoresis (PAGE), gel filtration chromatography, sucrose density gradient sedimentation, chemical cross-linking, and co-immunoprecipitation, we found that human ABCC1 is a homodimer and that MSD0 is essential and sufficient for homodimerization of human ABCC1 (11). In this study, we further mapped the dimerization site to transmembrane segment 5 (TM5) and extracellular loop 3 (ECL3) in MSD0, and it appears that the hydrophobicity of TM5 and the length of ECL3 but not their amino acid sequences contribute to ABCC1 dimerization.
Materials and Methods
Materials
Monoclonal antibody MRPr1 and antibody anti-HA were from Kamiya and Covance, respectively. Anti-FLAG antibody M2, HRP-conjugated goat anti-mouse IgG, and rabbit anti-rat IgG were from Sigma. Polyvinylidene difluoride (PVDF) membranes and concentrated protein assay dye reagents were from Bio-Rad. LipofectAMINE, G418, and cell culture media and reagents were obtained from Invitrogen. Pfu Turbo DNA polymerase and the enhanced chemiluminescence (ECL) system were purchased from Agilent Technologies and Amersham/Pharmacia, respectively. Protein G-agarose was from Santa Cruz Biotechnology. All other reagents of molecular biology grade were purchased from Sigma or Fisher Scientific.
Engineering Human ABCC1 Constructs
pcDNA3.1(+)-MRP1WT encoding human wild-type ABCC1, pcDNA3.1(+)-ABCC1F-FLAG encoding FLAG-tagged ABCC1, and pcDNA3.1-(+)-ABCC1F-HA encoding HA-tagged ABCC1 were constructed in a previous study (11).
To generate the NH2-terminally truncated ABCC1281N constructs (11), polymerase chain reaction (PCR) using pcDNA3.1-(+)-MRP1WT as a template was performed with a specific forward primer containing an NheI site followed by the Kozak initiation sequence and a universal reverse primer MRP1–281RP from nucleotide 1270 to 1287 of human ABCC1 cDNA (Table 1 of the Supporting Information). The PCR products were then digested with NheI and BamHI and subsequently cloned into pcDNA3.1(+)-ABCC1F-FLAG digested with NheI and BamHI. The resulting constructs were then digested with BamHI and NotI and blunted by Klenow followed by self-ligation, resulting in ABCC11–281N, ABCC133–281N, ABCC162–281N, ABCC195–281N, ABCC1120–281N, ABCC1155–281N, ABCC1159–281N, ABCC1163–281N, ABCC1167–281N, ABCC1169–281N, and ABCC1171–281N. Translation of these constructs terminates at a stop codon in the vector, as previously described (6, 12).
To generate COOH-terminally truncated ABCC1281N, PCR using pcDNA3.1(+)-ABCC11–281N (11) as a template was performed with a universal forward primer MRP1-N-FP from nucleotide 1 to 21 of human ABCC1 cDNA with a NheI site followed by the Kozak translation initiation sequence and a specific reverse primer with an HA epitope, a stop codon, and a NotI site (Table 2 of the Supporting Information). The PCR products were then digested with NheI and subsequently cloned into pcDNA3.1(+)-ABCC1F-HA digested with NheI, resulting in ABCC11–154N-HA, ABCC11–170N-HA, ABCC11–191N-HA, and ABCC11–281N-HA.
Two-step PCR was used to generate the TM5 and ECL3 replacement and polyalanine mutagenesis constructs. First, the NH2-terminal half of the first 281 amino acids was amplified using the universal forward primer MRP1-N-FP described above and a specific reverse primer carrying the replacement sequences (mutagenic primer) (Table 3 of the Supporting Information); the COOH half was also amplified using the specific forward primer carrying the replacement sequences (mutagenic primer) and the universal reverse primer MRP1–281-RP as described above (Table 3 of the Supporting Information). The secondary PCR was performed using primers MRP1-N-FP and MRP1–281RP and the amplified NH2-half and COOH-half PCR product mixture described above as a template. The PCR products were then digested with NheI and BamHI and subsequently cloned into pcDNA3.1(+)-ABCC1F-FLAG digested with NheI and BamHI. The resulting constructs were then digested with BamHI and NotI and blunted with Klenow followed by self-ligation, resulting in ABCC11–281N-TM5→3, ABCC11–281N-TM5→9, ABCC11–281N-TM5A1, ABCC11–281N-TM5A2, ABCC11–281N-TM5A3, ABCC11–281N-ECL3→7A, and ABCC11–281N-ECL3→7B. All constructs described above were confirmed by double-strand DNA sequencing.
Cell Culture and Transfection
HEK293 cells with or without stable expression of ABCC1F-FLAG or ABCC1F-HA were established previously (11) and maintained in DMEM supplemented with 10% fetal bovine serum in the presence of 100 units/mL penicillin and 100 μg/mL streptomycin. For transient transfection, 2.5–15 μg of plasmid DNA was transfected into HEK293 cells with or without stable expression of ABCC1F-FLAG or ABCC1F-HA in 150 mm dishes using LipofectAMINE according to the manufacturer's instructions. Forty-eight hours after transfection, cells were harvested for preparation of cell lysates as described previously (8).
Immunoprecipitation
Immunoprecipitation was performed as previously described (13, 14). Briefly, cell lysates (1.5 mg) were first precleared by incubation with 1 μg of normal mouse IgG at 4 °C for 1 h. Approximately 50 μL of protein G-agarose beads (50% slurry) was added to the reaction mixture, and it was incubated at 4 °C for an additional 1 h with rotation followed by centrifugation at 500g for 5 min to remove nonspecifically bound materials. The cleared supernatants were then incubated with 10 μg of anti-FLAG or anti-HA antibodies at 4 °C with rotation for 4 h before being mixed with 50 μL of a 50% protein G-agarose slurry. The mixtures were further incubated at 4 °C overnight with rotation followed by centrifugation to collect precipitates that were then washed five times with lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100] followed by Western blot analysis.
Confocal Microscope Imaging
Confocal microscope imaging was performed as previously described (8). Briefly, 48 h following transient transfection of HEK293 cells cultured on coverslips with various constructs, the cells were washed with phosphate-buffered saline, fixed with acetone and methanol, and blocked with bovine serum albumin. The cells were then probed with primary antibody MRPr1 or antibody anti-HA at room temperature for 1 h followed by incubation with FITC-conjugated donkey anti-rat IgG or goat anti-mouse IgG at room temperature for 30 min. Cell nuclei were counterstained with propidium iodide. The coverslips were then mounted on slides before they were viewed with a confocal microscope.
Results
The Dimerization Site Resides in ECL3, TM5, and L0
Previously, we have shown that the site for homodimerization of ABCC1 is located in the amino-terminal 281 amino acids including MSD0L0 (11). To further map the dimerization site within these 281 amino acids of ABCC1, we first engineered a series of constructs with amino-terminal deletions of ABCC11–281N and generated ABCC133–281N, ABCC162–281N, ABCC195–281N, ABCC1120–281N, and ABCC1155–281N (Figure 1A). These constructs were then transiently transfected into HEK293 cells with stable expression of FLAG-tagged full-length ABCC1 (ABCC1F-FLAG). All constructs were well expressed as determined via Western blotting using antibody MRPr1 with a common epitope in all constructs (Figure 1B, lanes 1–6). The deletion from the amino terminus also does not appear to affect plasma membrane localization of the truncated proteins (Figure S1 of the Supporting Information). We next performed co-immunoprecipitations using the FLAG antibody followed by Western blot analysis probed using antibody MRPr1 to determine if the truncated proteins interact with full-length ABCC1F-FLAG. Figure 1B (lanes 7–12) shows that all the truncated constructs, including the shortest one (ABCC1155–281N), coprecipitate with ABCC1F-FLAG, suggesting that there is a common site in all deletion constructs responsible for binding to FLAG-tagged full-length ABCC1. Thus, it is possible that this putative common binding site resides within the region of amino acids 155–281, including ECL3, TM5, and L0.
Figure 1.
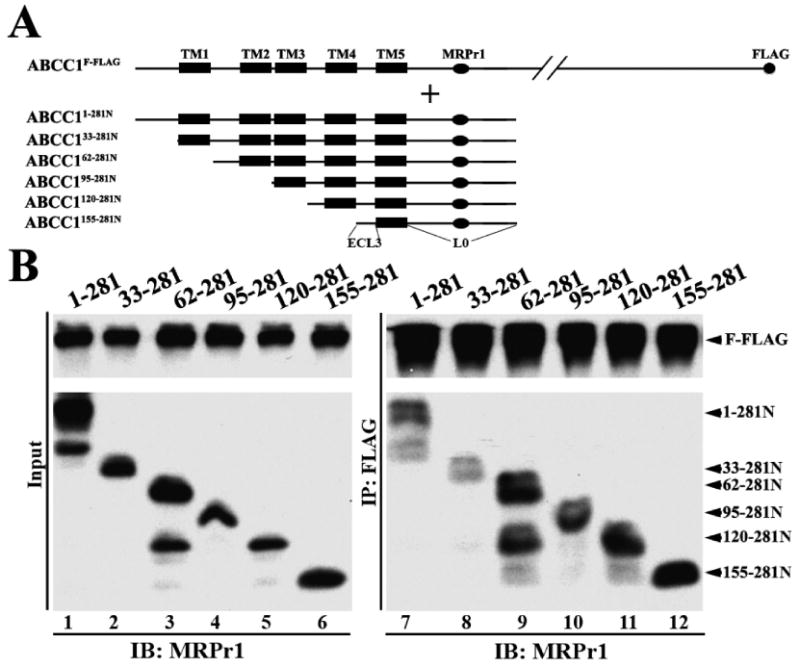
Effect of amino-terminal deletions on dimerization. (A) Schematic diagram of ABCC1F-FLAG and the NH2-terminally truncated ABCC1281N constructs. The TM segments are shown as boxes. The MRPr1 epitope and the FLAG tag are shown as an oval and circle, respectively. ECL3 and L0 are also indicated. (B) Expression (left) and co-immunoprecipitation (right) of the NH2-terminally truncated ABCC1281N constructs. The ABCC1281N constructs were transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG. Forty-eight hours following transfection, cells were harvested and lysates were prepared for immunoprecipitation using the anti-FLAG antibody followed by Western blot analysis probed using antibody MRPr1. The smaller band in lanes 3 and 9 is possibly due to degradation.
Both TM5 and ECL3 Are Essential but Insufficient for Dimerization
To further determine which of the ECL3, TM5, and L0 domains are essential components of the dimerization site, we first engineered a HA-tagged construct with deletion of the L0 domain in ABCC11–281N and generated ABCC11–191N-HA as well as a control HA-tagged ABCC11–281N (ABCC11–281N-HA) (see Figure 2A). These constructs were then transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG for co-immunoprecipitation analyses. As shown in Figure 2B, the level of ABCC11–191N-HA expression is greatly reduced in comparison with that of ABCC11–281N-HA as determined using the HA antibody. However, ABCC11–191N-HA coprecipitates with ABCC1F-FLAG although the amount of coprecipitated ABCC1F-FLAG is smaller than the amount of coprecipitated ABCC11–281N-HA. The decrease in the amount of coprecipitated ABCC1F-FLAG by ABCC11–191N-HA is proportionate with the decreased level of expression of ABCC11–191N-HA. Thus, we conclude that L0 is not required for the dimerization of ABCC1.
Figure 2.
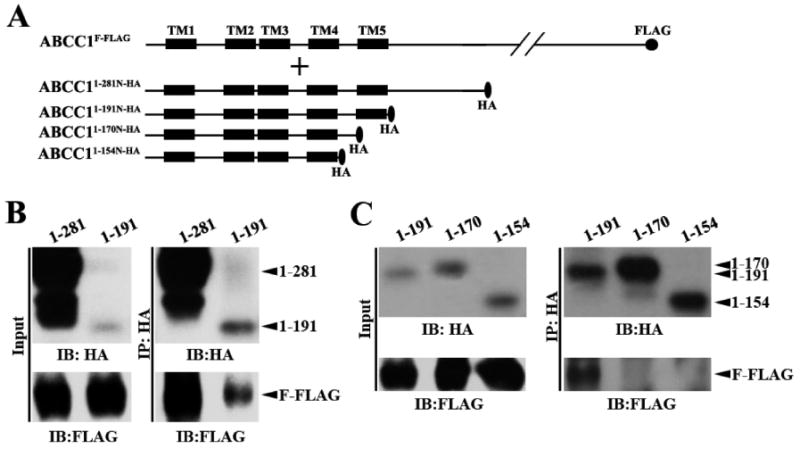
Effect of carboxyl-terminal deletions on dimerization. (A) Schematic diagram of ABCC1F-FLAG and the COOH-terminally truncated ABCC1281N-HA constructs. The TM segments are shown as boxes. The HA and the FLAG tags are shown as an oval and circle, respectively. (B and C) Expression (left) and co-immunoprecipitation (right) of the COOH-terminally truncated ABCC1281N-HA constructs. The COOH-terminally truncated ABCC1281N-HA constructs were transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG. Forty-eight hours following transfection, cells were harvested and lysed for co-immunoprecipitation using the anti-HA antibody followed by Western blot analysis probed with the anti-FLAG antibody. It is noteworthy that ABCC11–191N-HA appears to have a mobility similar to that of ABCC11–170N-HA, possibly due to the removal of a segment of 21 hydrophobic residues. Similar observations have been made with truncated constructs of ABCG2 in previous studies (27).
Next, we engineered two more HA-tagged constructs via deletion of TM5 (ABCC11–170N-HA) and ECL3 (ABCC11–154N-HA) (Figure 2A) followed by transient transfection into HEK293 cells with stable expression of ABCC1F-FLAG for co-immunoprecipitation analyses. As shown in Figure 2C (left panel), both constructs are expressed at levels comparable to that of ABCC11–191N-HA. The deletion from the carboxyl terminus does not appear to affect the membrane localization because the most distant deletion construct (ABCC11–154N-HA) appears to be located on plasma membranes (Figure S1 of the Supporting Information). Following co-immunoprecipitation with HA antibodies and Western blot analysis probed with the FLAG antibody for ABCC1F-FLAG or the HA antibody for the truncated constructs, we found that ABCC11–191N-HA coprecipitated with full-length ABCC1F-FLAG as expected and none of the two new constructs with deletion of TM5 and ECL3 could coprecipitate with ABCC1F-FLAG (Figure 2C, bottom right panel). Thus, it is likely that TM5 is an essential component of the dimerization site in MSD0 and that ECL3 without TM5 is insufficient to support the dimerization of ABCC1.
To determine if ECL3 is also essential for the binding to ABCC1F-FLAG, we took advantage of ABCC1155–281N, which interacts with ABCC1F-FLAG (see Figure 1B, lane 12), and engineered a series of constructs with deletions of ECL3, named ABCC1155–281N, ABCC1159–281N, ABCC1163–281N, ABCC1167–281N, ABCC1169–281N, and ABCC1171–281N (Figure 3A). These constructs were then transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG followed by immunoprecipitation with the FLAG antibody and detection of coprecipitated deletion mutants with antibody MRPr1. As shown in Figure 3B, the expression levels of these constructs vary with ABCC1155–281N at the lowest level (see X-281N input), although the deletions do not appear to affect membrane localization (Figure S1 of the Supporting Information). Nevertheless, ABCC1155–281N could be coprecipitated by ABCC1F-FLAG (Figure 3B, panel X-281N IP). Other deletion mutants could also be coprecipitated by ABCC1F-FLAG, and the coprecipitated level of the first four constructs has a trend of correlation with their expression level (Figure 3C). However, a detailed analysis of the amount of coprecipitated deletion mutants after normalization to their expression level shows that the relative level of coprecipitation decreases continuously with the sequential deletion of ECL3 (Figure 3D). The ABCC1171–281N construct with two amino acid residues of ECL3 had only ∼10% of the binding of the ABCC1155–281N construct with full ECL3. This observation suggests that ECL3 is also an essential component of the dimerization site in MSD0 and that TM5 without ECL3 is not sufficient to support dimerization of ABCC1.
Figure 3.
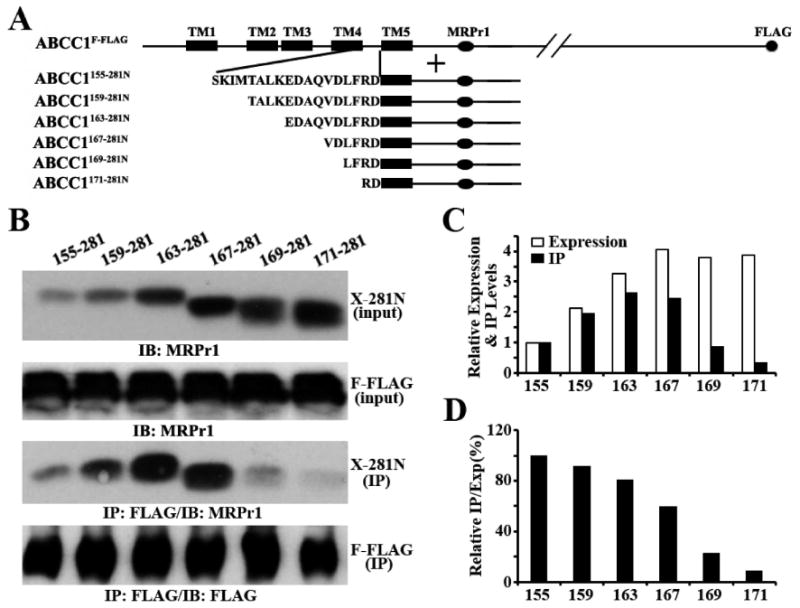
Role of ECL3 in dimerization. (A) Schematic diagram of ABCC1F-FLAG and the constructs containing ECL3 and TM5. TM5 and ECL3 are shown as a box and as a sequence of amino acids, respectively. The MRPr1 epitope and the FLAG tag are shown as an oval and circle, respectively. (B) Expression (input panels) and co-immunoprecipitation (IP panels) of the constructs containing ECL3 and TM5. The constructs containing ECL3 and TM5 were transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG followed by co-immunoprecipitation using the anti-FLAG antibody and Western blot analysis probed with the MRPr1 or FLAG antibody. (C and D) Quantification of expression and co-immunoprecipitation. The expression and co-immunoprecipitation levels of constructs containing ECL3 and TM5 in panel B were quantified using ScnImage and normalized to that of the shortest ABCC1155–281 construct (C) followed by calculation of the relative ratio of co-immunoprecipitation to expression level (D).
The Dimerization Ability of TM5 and ECL3 Is Not Sequence-Dependent
To further determine the features of TM5 and ECL3 that are essential for the dimerization ability, we first examined if replacing TM5 with other transmembrane segments affects the dimerization ability of ABCC1. For this purpose, TM5 in ABCC11–281N was replaced with TM3 or TM9, resulting in ABCC11–281N-TM5→3 or ABCC11–281N-TM5→9, respectively (Figure 4A). TM9 located in the core fragment does not appear to have dimerization ability because the core fragment of ABCC1 does not dimerize (11). Removal of TM3 as shown in Figure 1B (lane 11) did not affect the dimerization activity of ABCC11–281N. Thus, the replacement of TM5 with these TM segments may eliminate the dimerization ability of ABCC11–281N if the amino acid sequence of TM5 is important.
Figure 4.
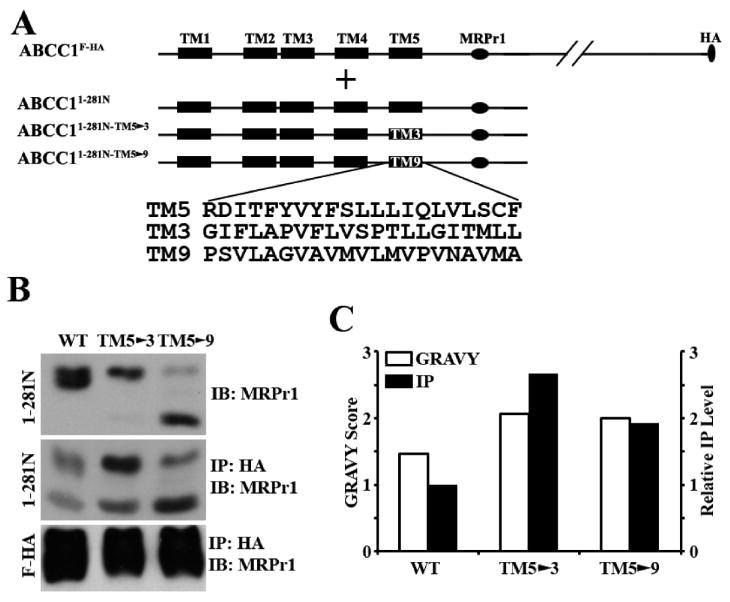
Effect of TM5 replacement on dimerization. (A) Schematic diagram of ABCC1F-HA and the ABCC1281N constructs with TM5 replaced with TM3 or TM9. The TMs are shown as boxes, and the amino acid sequences of TM3, TM5, and TM9 are shown in single-letter code. The MRPr1 epitope and the HA tag are shown as horizontal and vertical ovals, respectively. (B) Expression (top) and co-immunoprecipitation (middle and bottom) of the wild type and the mutant ABCC1281N constructs. The wild type and the mutant ABCC1281N constructs were transiently transfected into HEK293 cells with stable expression of ABCC1F-HA followed by co-immunoprecipitation with the HA antibody and Western blot analyses probed with antibody MRPr1. (C) Correlation of hydrophobicity with the level of co-immunoprecipitation. The hydrophobicity of TM3, TM5, and TM9 was calculated using an online program (http://www.expasy.ch/tools/protparam.html), and the GRAVY scores are shown. The co-immunoprecipitation levels of the wild type and the mutant ABCC1281N constructs in panel B were quantified using ScnImage and normalized to that of wild-type ABCC1281N.
These constructs were then transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG. As shown in Figure 4B (top panel), both constructs were well expressed like ABCC11–281N except that ABCC11–281N-TM5→9 was less glycosylated than the others, consistent with previous findings (11). These constructs were also properly localized on plasma membranes (Figure S1 of the Supporting Information). Unexpectedly, both mutant constructs could be coprecipitated by ABCC1F-FLAG, and it appears that more mutants were coprecipitated than wild-type ABCC11–281N (Figure 4B, middle panel, and Figure 4C). Next, we performed a hydropathy analysis of these TM segments using an online program (http://www.expasy.ch/tools/protparam.html). Figure 4C shows that both TM3 and TM9 are more hydrophobic than TM5, correlating with the enhanced ability of ABCC11–281N to bind to ABCC1F-FLAG when TM5 is replaced with TM3 or TM9. Thus, replacing TM5 with TM3 or TM9 does not decrease but increases the activity of binding of ABCC11–281N to ABCC1F-FLAG, and the hydrophobicity and geographic location, not the amino acid sequence, of TM5 may contribute to the dimerization ability.
To validate the finding described above, we engineered other constructs by replacing parts of TM5 with polyalanines and generated ABCC11–281N-TM5A1, ABCC11–281N-TM5A2, and ABCC11–281N-TM5A3 (Figure 5A). All mutants, except ABCC11–281N-TM5A2, were similarly expressed and glycosylated like wild-type ABCC11–281N, and all constructs again could be coprecipitated by ABCC1F-FLAG but at different levels (Figure 5, middle panel). ABCC11–281N-TM5A1 with the higher hydrophobicity of TM5 compared to the wild-type sequence also had more co-immunoprecipitation (Figure 5C), consistent with the findings shown in Figure 4. However, ABCC11–281N-TM5A2 and ABCC11–281N-TM5A3 with the similar hydrophobicity of TM5 like the wild-type sequence also had more co-immunoprecipitation, although the increase in the level of co-immunoprecipitation was much lower than that of the ABCC11–281N-TM5A1 mutant (Figure 5C). This smaller increase in the level of co-immunoprecipitation may possibly be due to the introduction of an AXXXA motif, which has also been shown to facilitate helix–helix interactions in membrane proteins (see Discussion).
Figure 5.
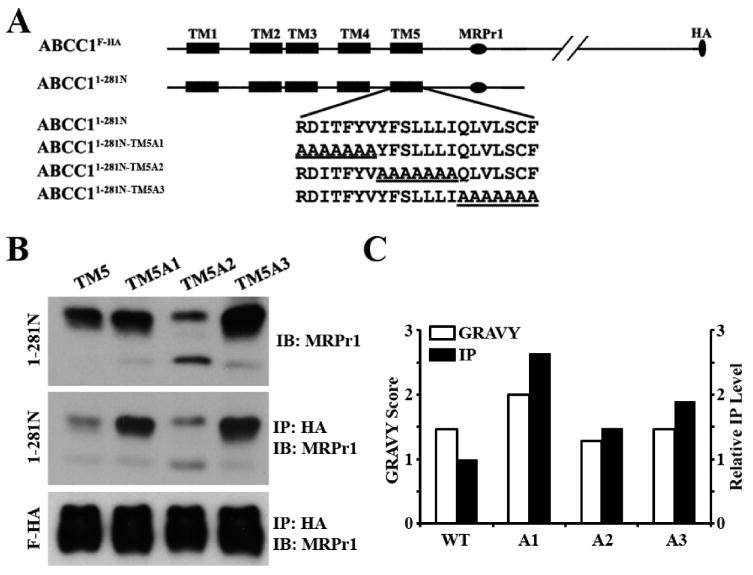
Polyalanine mutagenesis analyses of TM5. (A) Schematic diagram of ABCC1F-HA and the ABCC1281N constructs with TM5 containing polyalanine mutations. The TMs are shown as boxes, and the amino acid sequence of TM5 is shown in single-letter code. The MRPr1 epitope and the HA tag are shown as an oval and circle, respectively. (B) Expression (top) and co-immunoprecipitation (middle and bottom) of the wild type and the mutant ABCC1281N constructs. The wild type and the mutant ABCC1281N constructs were transiently transfected into HEK293 cells with stable expression of ABCC1F-HA followed by co-immunoprecipitation with the HA antibody and Western blot analyses probed with antibody MRPr1. (C) Correlation of hydrophobicity with the level of co-immunoprecipitation. The hydrophobicity of wild-type and mutant TM5 was calculated as described in the legend of Figure 4, and the GRAVY scores are shown. The co-immunoprecipitation levels of the wild type and the mutant ABCC1281N constructs in panel B were quantified using ScnImage and normalized to that of wild-type ABCC1281N.
We next determined if the sequence of ECL3 makes an important contribution to dimerization by replacing ECL3 with ECL7 (Figure 6A) of different lengths in ABCC11–281N, resulting in ABCC11–281N-ECL3→7A and ABCC11–281N-ECL3→7B. These constructs along with ABCC11–281N were transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG. Both mutant constructs were well expressed with a level slightly lower than that of ABCC11–281N (Figure 6B, bottom left panel), and membrane localization analysis of one of the constructs shows that it is correctly located on plasma membranes (Figure S1 of the Supporting Information). Both mutant constructs could also be co-immunoprecipitated by ABCC1F-FLAG (Figure 6B, bottom right panel). Analysis of the co-immunoprecipitated proteins relative to their expression level did not show major changes in the co-immunoprecipitation level by the sequence replacement (Figure 6C). Thus, we conclude that, like TM5, the amino acid sequence of ECL3 does not appear to make an important contribution to ABCC1 dimerization.
Figure 6.
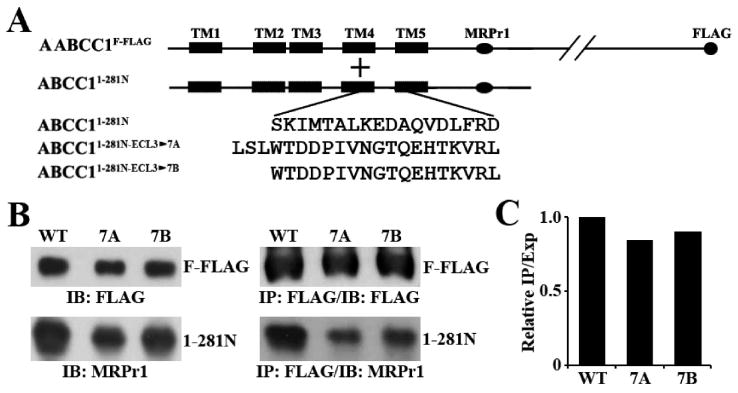
Effect of ECL3 replacement on dimerization. (A) Schematic diagram of ABCC1F-FLAG and the ABCC1281N constructs with ECL3 replaced by ECL7. The TMs are shown as boxes, and the amino acid sequence of ECL3 or the replacement sequence is shown in single-letter code. The MRPr1 epitope and the FLAG tag are shown as horizontal and vertical ovals, respectively. (B) Expression (left) and co-immunoprecipitation (right) of the wild type and the mutant ABCC1281N constructs. The wild type and the mutant ABCC1281N constructs were transiently transfected into HEK293 cells with stable expression of ABCC1F-FLAG followed by co-immunoprecipitation with the FLAG antibody and Western blot analyses probed with antibody MRPr1. (C) Quantification of the co-immunoprecipitated wild type and the mutant ABCC1281N constructs. The expression and co-immunoprecipitation levels of the wild type and the mutant ABCC1281N constructs in panel B were quantified using ScnImage, and the relative ratios of co-immunoprecipitation level to expression level were calculated.
TM5 and ECL3 in One Subunit of a Dimer Likely Interact with TM5 and ECL3 in the Opposing Subunit
To identify the domain in the opposing subunit where TM5 and ECL3 likely bind, we engineered a FLAG-tagged ABCC1155–281N [ABCC1155–281N-FLAG (Figure 7A)] and performed an interaction study by co-expressing ABCC1155–281N-FLAG with ABCC11–281N, ABCC162–281N, ABCC195–281N, or ABCC1120–281N in HEK293 cells (Figure 7B, left panel). Following co-immunoprecipitation with the FLAG antibody, ABCC1155–281N-FLAG could coprecipitate with all constructs that have amino-terminal deletions (Figure 7B, right panel), suggesting that TM5 and ECL3 likely interact with TM5 and ECL3 in the opposing subunit of the dimer.
Figure 7.
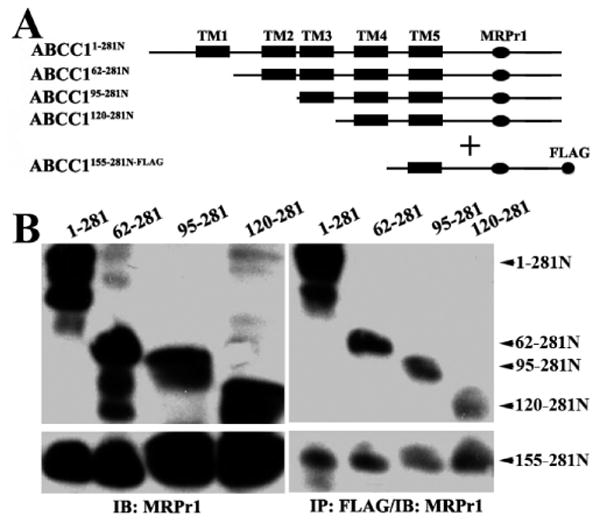
Mapping the interaction site in the opposing subunit. (A) Schematic diagram of ABCC1155–281N-FLAG and the NH2-terminally truncated ABCC1281N constructs. The MRPr1 epitope and the FLAG tag are shown as an oval and circle, respectively. (B) Co-expression (left) and co-immunoprecipitation (right) of ABCC1155–281N-FLAG with the NH2-terminally truncated ABCC1281N constructs. The NH2-terminally truncated ABCC1281N constructs were transiently cotransfected with ABCC1155–281N-FLAG into HEK293 cells followed by co-immunoprecipitation with the FLAG antibody and Western blot analyses probed with antibody MRPr1 to detect all constructs.
Discussion
In this study, we mapped the dimerization site of human ABCC1 to ECL3 and TM5 in MSD0. Both ECL3 and TM5 are essential for ABCC1 dimerization, but neither is sufficient for ABCC1 dimerization. The amino acid sequences of ECL3 and TM5 are not an important element contributing to ABCC1 dimerization. ECL3 and TM5 in one subunit of an ABCC1 dimer may interact with ECL3 and TM5 in the opposing subunit, respectively. Together with the findings in a previous study of the electron microscopy image of purified ABCC1 that ABCC1 dimerizes via a head–head interaction between the two subunits (15), we propose a dimeric ABCC1 model with possible interactions via TM5 and ECL3 (Figure 8).
Figure 8.
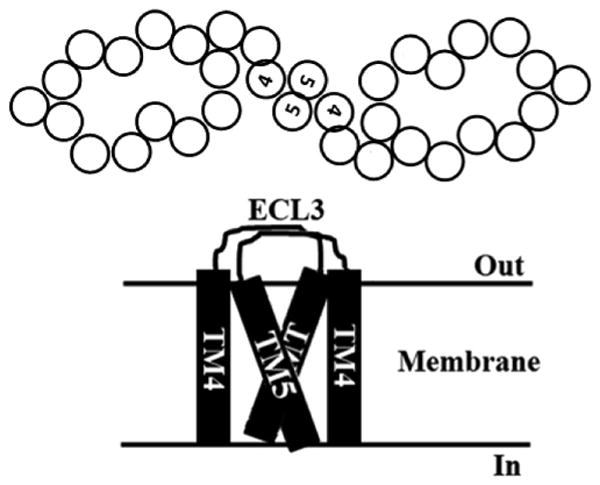
Model of ECL3–TM5 interaction and ABCC1 dimerization. The head-to-head dimerization was modeled on the basis of the findings from the study by Rosernberg et al. (15) and Yang et al. (11) as well as this study. The circles and bars represent TM segments with maximum TM5–TM5 and ECL3–ECL3 contacts, respectively.
Previously, the role of TM segments in membrane protein–protein interactions has been demonstrated. In one of the most well studied cases, glycophorin A, a membrane protein with a single TM segment, was shown to dimerize via the interactions of the TM segment in a sequence-dependent manner (16–19). The GXXXG element or motif was later demonstrated to be most crucial for dimerization (20). This motif was later shown to exist in many TM helices that possibly facilitate helix–helix interaction (21) and dimerization of other membrane proteins (22, 23). Later, other motifs of membrane protein–protein interactions have been identified, including QXXS (24) and AXXXA (25, 26) motifs.
Interestingly, TM5 of human ABCC1 does not have any of these motifs, and its contribution to protein–protein interaction appears to be sequence-independent. In fact, increasing the hydrophobicity of TM5 by replacing it with a different set of sequences increased the activity in dimer formation. Thus, the major factors of TM5 contributing to ABCC1 dimerization are possibly its physical location and hydrophobicity. However, two other constructs with partial replacement of TM5 with polyalanine (ABCC11–281N-TM5A2 and ABCC11–281N-TM5A3) also had enhanced dimerization ability without increases in hydrophobicity. This observation may be due to the introduction of an AXXXA motif that has also been shown previously to facilitate helix–helix interactions (25, 26). Nevertheless, the increased level of dimerization of these two constructs is much smaller than the increase in the dimerization ability of the ABCC11–281N-TM5A1 construct that has an increased hydrophobicity in addition to the artificially introduced AXXXA motif, and the magnitudes of their increases are proportionate with their hydrophobicities as well.
ECL3 also does not appear to have any known motif of protein interactions, and the contribution of ECL3 to dimerization ability is independent of its amino acid sequence. However, the length of the ECL3 may be an important factor as shortening ECL3 by sequential deletions from its amino terminus gradually weakened the dimerization ability (Figure 3). This observation is also consistent with the possibility that ECL3 in one subunit interacts with another ECL3 in the opposing subunit and shortening one ELC3 likely reduces its interactions with the opposing ECL3.
The finding that both ECL3 and TM5 are essential but neither is sufficient to support ABCC1 dimerization suggests that the dimerization requires both interacting sites. However, it is noteworthy that the interface between these two sites appears to represent a very small fraction of the total protein. Thus, the forces for ABCC1 dimerization may not be very strong. The weak interactions between the two ABCC1 subunits may cost less energy for ABCC1 to transit between dimer and monomers and, thus, provide another level of ABCC1 regulation.
Currently, it is still unknown if the dimeric or monomeric ABCC1 is the functional form. We have shown previously that the transport activity of ABCC1 is lost in the presence of a truncated MSD0, which suggests that ABCC1 dimerization is disrupted by truncated MSD0 and possibly dimeric ABCC1 is functional. However, these findings could not rule out the possibility that monomeric ABCC1 is functional and binding of a truncated MSD0 to monomeric ABCC1 eliminated its activity. Nevertheless, this finding suggests that either disrupting ABCC1 dimerization or binding of a ligand to the dimerization site will inhibit the function of ABCC1. Thus, the finding of the minimum dimerization site in this study may help future studies in targeting the dimerization site for drug discovery to sensitize ABCC1-mediated MDR in cancer chemotherapy.
Supplementary Material
Footnotes
This work was supported in part by grants from the National Institutes of Health (R01 CA113384 and R01 CA120221). Y.Y. was supported, in part, by Grant NRSA T32HL07910 from the National Institutes of Health. W.M. was supported, in part, by a predoctoral fellowship from Department of Defense Breast Cancer Research Program (W81XWH-08-1-0228).
Abbreviations: ABC, ATP-binding cassette; BCRP, breast cancer resistance protein; ECL, extracellular loop; MDR, multidrug resistance; MRP, multidrug resistance-associated protein; MSD, membrane-spanning domain; NBD, nucleotide-binding domain; Pgp, P-glycoprotein; TM, transmembrane.
Supporting Information Available: Supplemental data of indirect immunofluoresence staining of mutant ABCC1 (Figure S1) and supplemental Tables 1–3 of primers for PCR. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kim IW, Booth-Genthe C, Ambudkar SV. Relationship between drugs and functional activity of various mammalian P-glycoproteins (ABCB1) Mini-Rev Med Chem. 2008;8:193–200. doi: 10.2174/138955708783744100. [DOI] [PubMed] [Google Scholar]
- 2.Sharom FJ. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–127. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 3.Zhang JT. Use of arrays to investigate the contribution of ATP-binding cassette transporters to drug resistance in cancer chemotherapy and prediction of chemosensitivity. Cell Res. 2007;17:311–323. doi: 10.1038/cr.2007.15. [DOI] [PubMed] [Google Scholar]
- 4.Mo W, Liu JY, Zhang JT. Biochemistry and pharmacology of human ABCC1/MRP1 and its role in detoxification and in multidrug resistance of cancer chemotherapy. In: Pestka S, Shi Y, Liu XY, editors. Recent Advances in Cancer Research and Therapy. Springer; Berlin: 2010. [Google Scholar]
- 5.Deeley RG, Cole SP. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1) FEBS Lett. 2006;580:1103–1111. doi: 10.1016/j.febslet.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 6.Bakos E, Evers R, Szakacs G, Tusnady GE, Welker E, Szabo K, de Haas M, van Deemter L, Borst P, Varadi A, Sarkadi B. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem. 1998;273:32167–32175. doi: 10.1074/jbc.273.48.32167. [DOI] [PubMed] [Google Scholar]
- 7.Westlake CJ, Cole SP, Deeley RG. Role of the NH2-terminal membrane spanning domain of multidrug resistance protein 1/ABCC1 in protein processing and trafficking. Mol Biol Cell. 2005;16:2483–2492. doi: 10.1091/mbc.E04-12-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Chen Q, Zhang JT. Structural and functional consequences of mutating cysteine residues in the amino terminus of human multidrug resistance-associated protein 1. J Biol Chem. 2002;277:44268–44277. doi: 10.1074/jbc.M207003200. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Yang Y, Li L, Zhang JT. The amino terminus of the human multidrug resistance transporter ABCC1 has a U-shaped folding with a gating function. J Biol Chem. 2006;281:31152–31163. doi: 10.1074/jbc.M603529200. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Yang Y, Liu Y, Han B, Zhang JT. Cytoplasmic retraction of the amino terminus of human multidrug resistance protein 1. Biochemistry. 2002;41:9052–9062. doi: 10.1021/bi025634s. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, Liu Y, Dong Z, Xu J, Peng H, Liu Z, Zhang JT. Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. J Biol Chem. 2007;282:8821–8830. doi: 10.1074/jbc.M700152200. [DOI] [PubMed] [Google Scholar]
- 12.Gao M, Yamazaki M, Loe DW, Westlake CJ, Grant CE, Cole SPC, Deeley RG. Multidrug resistance protein. Identification of regions required for active transport of leukotriene c4. J Biol Chem. 1998;273:10733–10740. doi: 10.1074/jbc.273.17.10733. [DOI] [PubMed] [Google Scholar]
- 13.Dong Z, Zhang JT. EIF3 p170, a Mediator of Mimosine Effect on Protein Synthesis and Cell Cycle Progression. Mol Biol Cell. 2003;14:3942–3951. doi: 10.1091/mbc.E02-12-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. doi: 10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg MF, Mao Q, Holzenburg A, Ford RC, Deeley RG, Cole SPC. The Structure of the Multidrug Resistance Protein 1 (MRP1/ABCC1). Crystallization and Single-Particle Analysis. J Biol Chem. 2001;276:16076–16082. doi: 10.1074/jbc.M100176200. [DOI] [PubMed] [Google Scholar]
- 16.Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, Engelman DM. Glycophorin A dimerization is driven by specific interactions between transmembrane α-helices. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- 17.Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A dimerization motif for transmembrane α-helices. Nat Struct Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 18.MacKenzie KR, Engelman DM. Structure-based prediction of the stability of transmembrane helix-helix interactions: The sequence dependence of glycophorin A dimerization. Proc Natl Acad Sci USA. 1998;95:3583–3590. doi: 10.1073/pnas.95.7.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 20.Brosig B, Langosch D. The dimerization motif of the glycophorin A transmembrane segment in membranes: Importance of glycine residues. Protein Sci. 1998;7:1052–1056. doi: 10.1002/pro.5560070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russ WP, Engelman DM. The GxxxG motif: A framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 22.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, Ambudkar SV, Tarasova N, Bates SE. Mutational analysis of ABCG2: Role of the GXXXG motif. Biochemistry. 2004;43:9448–9456. doi: 10.1021/bi0497953. [DOI] [PubMed] [Google Scholar]
- 24.Sal-Man N, Gerber D, Shai Y. The identification of a minimal dimerization motif QXXS that enables homo- and hetero-association of transmembrane helices in vivo. J Biol Chem. 2005;280:27449–27457. doi: 10.1074/jbc.M503095200. [DOI] [PubMed] [Google Scholar]
- 25.Kleiger G, Grothe R, Mallick P, Eisenberg D. GXXXG and AXXXA: Common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry. 2002;41:5990–5997. doi: 10.1021/bi0200763. [DOI] [PubMed] [Google Scholar]
- 26.Schneider D, Engelman DM. Motifs of two small residues can assist but are not sufficient to mediate transmembrane helix interactions. J Mol Biol. 2004;343:799–804. doi: 10.1016/j.jmb.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT. Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res. 2007;67:4373–4381. doi: 10.1158/0008-5472.CAN-06-3169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.