

Abstract
This article describes the rational design of 1st generation systems for oxidatively-induced Aryl–CF3 bond-forming reductive elimination from PdII. Treatment of (dtbpy)PdII(Aryl)(CF3) (dtbpy = di-tert-butylbipyridine) with NFTPT (N-fluoro-1,3,5-trimethylpyridium triflate) afforded the isolable PdIV intermediate (dtbpy)PdIV(Aryl)(CF3)(F)(OTf). Thermolysis of this complex at 80 °C resulted in Aryl–CF3 bond-formation. Detailed experimental and computational mechanistic studies have been conducted to gain insights into the key reductive elimination step. Reductive elimination from this PdIV species proceeds via pre-equilibrium dissociation of TfO− followed by Aryl–CF3 coupling. DFT calculations reveal that the transition state for Aryl–CF3 bond formation involves the CF3 acting as an electrophile with the Aryl ligand acting as a nucleophilic coupling partner. These mechanistic considerations along with DFT calculations have facilitated the design of a 2nd generation system utilizing the tmeda (N,N,N’,N’-tetramethylethylenediamine) ligand in place of dtbpy. The tmeda complexes undergo oxidative trifluoromethylation at room temperature.
Introduction
The formation of C–CF3 bonds is an important transformation for the construction of pharmaceuticals and agrochemicals.1 Replacing a methyl group with a trifluoromethyl substituent can have a profound effect on the physical and biological properties of a molecule.2 As a result, there is high demand for versatile synthetic methods for generating carbon–CF3 bonds.3 While there has been significant progress in the construction of sp3-carbon–CF3 linkages,3 there are comparatively fewer methods for Aryl–CF3 bond-formation.4,5,6,7
Transition metal-catalyzed cross-coupling between Aryl–X and CF3–Y would serve as an attractive method for the synthesis of benzotrifluorides.8,9,10,11 The use of Pd-based catalysts for such transformations is of particular interest because they serve as versatile and widely used catalysts for a variety of other carbon–carbon bond-forming reactions.12 However, developments in this area have been limited by the challenges associated with a key step of the cross-coupling catalytic cycle, namely Aryl–CF3 bond-forming reductive elimination from PdII centers.4,13 The vast majority of known PdII(Aryl)(CF3) complexes are inert to Aryl–CF3 coupling at temperatures as high as 150 °C.24,21
Two strategies have been utilized in the literature to address this challenge. The first has focused on achieving Aryl–CF3 bond-forming reductive elimination from PdII via steric and electronic modification of the ancillary ligands (L) at (L)nPdII(Aryl)(CF3). For example, pioneering work by Grushin demonstrated that the Xantphos ligand (Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) facilitates high yielding formation of trifluorotoluene from (Xantphos)PdII(Ph)(CF3) at 80 °C (eq. 1).14 More recently, Buchwald has elegantly demonstrated that the sterically large monodentate phosphine ligand Brettphos (Brettphos = 2-(dicyclohexylphosphino)-3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl) promotes stoichiometric Aryl–CF3 coupling from (Brettphos)PdII(Aryl)(CF3) at 80 °C (eq. 2).15 Remarkably, this latter system was successfully applied to the catalytic trifluoromethylation of aryl chlorides with Et3SiCF3 at 130–140 °C.15
 |
 |
While this first approach has provided important breakthroughs, PdII-mediated Aryl–CF3 bond-forming reactions remain limited by the requirement for specialized and expensive phosphine ligands16,17 relatively high reaction temperatures (80-140 °C), and the need for expensive Et3SiCF318 in catalytic processes. As a result, several groups have focused on a second, complementary strategy for achieving Aryl–CF3 bond-forming reductive elimination from Pd.19,20,21 This approach hinges on changing the oxidation state, rather than the ancillary ligand environment, at the metal center. As shown in eq. 3, it was expected that palladium(IV) complexes of general structure (L)2(X)2PdIV(Aryl)(CF3) (A) would be highly kinetically and thermodynamically reactive towards Aryl–CF3 coupling. This hypothesis is predicated on literature reports showing that PdIV complexes participate in numerous carbon-heteroatom bond-forming reductive elimination reactions that remain challenging at PdII centers.22,23 A key advantage of this approach would be that PdIV intermediate A could potentially be accessed using nucleophilic (CF3−)24, electrophilic (CF3+)25, or free-radical (CF3·)26 based trifluoromethylating reagents.
 |
Two preliminary examples have shown the viability of this approach. First, Yu and coworkers have elegantly demonstrated the PdII/IV-catalyzed ligand-directed C–H trifluoromethylation with electrophilic trifluoromethylating reagents (CF3+ reagents).19 Subsequent stoichiometric studies implicated a mechanism involving: (a) C–H activation to form a cyclometalated PdII–Aryl intermediate, (b) oxidation with CF3+ to generate PdIV(Aryl)(CF3), and (c) Aryl–CF3 bond-forming reductive elimination from PdIV to release the product.20
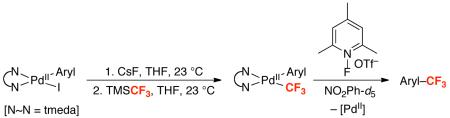
In a second example, our group has shown the viability of stoichiometric Aryl–CF3 bond-forming reductive elimination from PdIV centers bearing σ-aryl ligands that do not contain chelate directing groups.21 In this system, the key PdIV intermediate is generated via oxidation of a pre-assembled PdII(Aryl)(CF3) species with an N-fluoropyridinium reagent. We report herein a detailed mechanistic study of Aryl–CF3 bond-forming reductive elimination from PdIV in this latter system. This work provides valuable insights into the influence of ancillary ligands, the role of each coupling partner, and the nature of the transition state for this transformation. These mechanistic studies have also allowed us to rationally design the first examples of room temperature Aryl–CF3 bond-formation from palladium.
Results and Discussion
Development and Scope of Aryl–CF3 Coupling at PdIV.
Our studies began with the synthesis of a series of PdII–CF3 complexes of general structure (dtbpy)PdII(Aryl)(CF3) (1a-i, dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine). These compounds were prepared by the treatment of (dtbpy)PdII(Aryl)(I) with CsF followed by TMSCF3 in THF at 23 °C (Table 1). The products were isolated as yellow solids in 32-70% yield.21 X-ray quality crystals of 1a were obtained by vapor diffusion of pentanes into a dichloromethane solution of 1a, and the X-ray crystal structure of this complex is shown in Figure 1.
Table 1.
Synthesis of Complexes 1a-i
| Entry | Aryl | Compound | Isolated Yield |
|---|---|---|---|
| 1 | p-FC6H4 | 1a | 70% |
| 2 | p-CNC6H4 | 1b | 54% |
| 3 | p-CF3C6H4 | 1c | 63% |
| 4 | p- PhC(O)C6H4 | 1d | 51% |
| 5 | p-PhC6H4 | 1e | 58% |
| 6 | p-MeOC6H4 | 1f | 32% |
| 7 | p-MeC6H4 | 1g | 49% |
| 8 | C6H5 | 1h | 47% |
| 9 | m-MeC6H4 | 1i | 42% |
Figure 1.
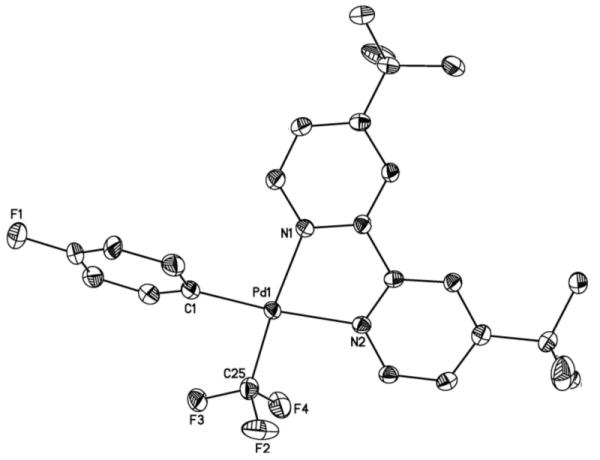
ORTEP drawing of complex 1a. Thermal ellipsoids are drawn at 50% probability, hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Pd–C(1) 2.005(3), Pd–C(25) 2.007(4), Pd–N(1) 2.107(2), Pd–N(2) 2.143(3). Selected bond angles (°): C(1)–Pd–C(25) 89.99(13), C(1)–Pd–N(1) 95.58(11), C(1)–Pd–N(2) 169.99(11), C(25)–Pd–N(1) 173.92(12), C(25)–Pd–N(2) 96.67(11).
The PdII complexes (1a-i) are inert towards direct Aryl–CF3 bond-forming reductive elimination. For example, heating 1a at 130 °C for 72 h produced <5% of 4-fluorobenzotrifluoride (2a), and the PdII starting material could be recovered in >80% yield (eq. 4). Importantly, similarly low reactivity has been reported in the literature for other (L~L)PdII(Aryl)(CF3) complexes (L~L = diphenylphosphinoethane, diphenylphosphinopropane, and diphenylphosphinobenzene).24a,b As discussed above, the only demonstrated examples of Aryl–CF3 bond-forming reductive elimination from PdII require relatively high temperatures (80 °C) and specialized phosphine ligands.14,15a
 |
We reasoned that the 2e− oxidation of (dtbpy)PdII(Aryl)(CF3) would yield a highly reactive PdIV adduct that might undergo more facile Aryl–CF3 bond-forming reductive elimination (eq. 5, i). Thus, we examined the reaction of 1a with N-bromosuccinimide (NBS), N-chlorosuccinimide (NCS), and PhI(OAc)2, which are all well-known to promote the oxidation of PdII to PdIV.22 As shown in Table 2, all three oxidants reacted rapidly with 1a in nitrobenzene-d5 at 80 °C. However, the desired trifluoromethylated product was not obtained; instead, the major organic product contained a nucleophile derived from the oxidant (Br, Cl, or OAc, respectively). These results are consistent with the formation of a PdIV intermediate, from which Aryl–X (X = OAc, Br, and Cl) bond-forming reductive elimination is significantly faster than Aryl–CF3 coupling (eq. 5, ii).21
Table 2.
Reaction of 1a with Diverse Oxidants (Oxidant–X)
| Entry | Oxidant–X | Yield 2a | Yield 3a (X) |
|---|---|---|---|
| 1 |
|
<5% | 75% (Br) |
| 2 |
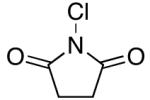
|
<5% | 70% (Cl) |
| 3 |
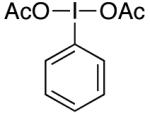
|
<5% | 20% (OAc) |
| 4 |
|
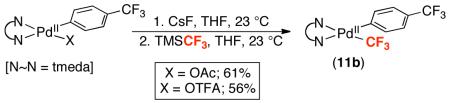
<5% | nd (F)a |
| 5 |
|
60% | <5% |
| 6 |
|
53% | <5% |
| 7 |
|
69% | <5% |
| 8 |
|
55% | <5% |
| 9 |
|
68% | <5% |
| 10 |
|
70% | <5% |
| 11 | XeF2 | 65% | 5% |
Yields were determined by 19F NMR spectroscopy and are an average of two runs. nd = not detected.
a1a accounted for the remaining mass balance.
 |
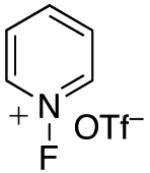
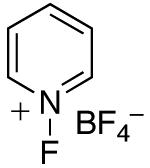
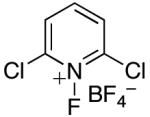
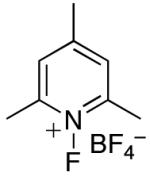
In an effort to avoid competing reductive elimination processes, we next examined the use of electrophilic fluorinating reagents (F+ sources). These reagents were selected based on the hypothesis that fluoride (the X-type ligand introduced to PdIV by F+ sources) might undergo slower reductive elimination than CF3 .22d,27,28 Gratifyingly, a variety of different F+ reagents reacted with 1a to afford modest to excellent yields of the trifluoromethylated product 2a after 3 h at 80 °C (Table 2, entries 4-10). The optimal electrophillic fluorinating reagent was N-fluoro-1,3,5-trimethylpyridium triflate (NFTPT), which provided 2a in 70% yield as determined by 19F NMR spectroscopy.29 Importantly, <5% of products derived from Aryl–F or Aryl–OTf coupling were observed under these conditions.
This transformation was next applied to complexes 1b-i, which contain sterically and electronically diverse aryl groups. As shown in Table 3, the yield of trifluoromethylated product was relatively insensitive to the electronic properties of the arene, and the reaction proceeded with good results in systems containing both electron withdrawing [e.g., CN, C(O)Ph] and electron donating (e.g., CH3, OCH3) para- and meta-substituents.30
Table 3.
NFTPT-Promoted Aryl–CF3 Coupling at Complexes 1a-i
| Entry | Compound | Aryl | Yield Aryl–CF3 |
|---|---|---|---|
| 1 | 1a | p-FC6H4 | 70% |
| 2 | 1b | p-CNC6H4 | 25% |
| 3 | 1c | p-CF3C6H4 | 55% |
| 4 | 1d | p-PhC(O)C6H4 | 56% |
| 5 | 1e | p-PhC6H4 | 70% |
| 6 | 1f | p-MeOC6H4 | 72% |
| 7 | 1g | p-MeC6H4 | 66% |
| 8 | 1h | C6H5 | 70% |
| 9 | 1i | m-MeC6H4 | 64% |
Yields were determined by 19F NMR spectroscopy and are an average of two runs.
At room temperature, the oxidation of 1a by NFTPT produced a single major intermediate. This species (4) was isolated from dichloroethane as a yellow solid in 53% yield (eq 6). Analysis of 4 by 19F NMR spectroscopy in MeCN-d3 showed four characteristic resonances: a doublet at −30.9 ppm (Pd–CF3), a singlet at −79.4 ppm (Pd–OTf), a multiplet at −117.1 ppm (Pd–ArF), and a quartet at −256.5 ppm (Pd–F) in a 3: 3: 1: 1 ratio. In nitrobenzene-d5, broad resonances were observed with similar chemical shifts in the same 3: 3: 1: 1 ratio.31 The 1H NMR spectrum of 4 showed two singlets at 8.47 and 8.40 ppm (5,5′ protons of dtbpy) as well as two singlets at 1.49 and 1.41 ppm (tBu groups of dtbpy).
 |
X-ray quality crystals were obtained via vapor diffusion of pentanes into a DCE solution of 4. As shown in Figure 2, the solid state structure of 4 shows the octahedral PdIV species (dtbpy)Pd(p-FC6H4)(CF3)(F)(OTf). Interestingly, the Pd–CF3 bond distance in 4 (2.009(4) Å) is nearly identical to that of the PdII–CF3 starting material 1a (2.005(3) Å). However, the Pd–N bond lengths of 4 (2.038(4) and 2.082(4) Å) are significantly shorter than those in 1a (2.107(2) and 2.143(3) Å).
Figure 2.
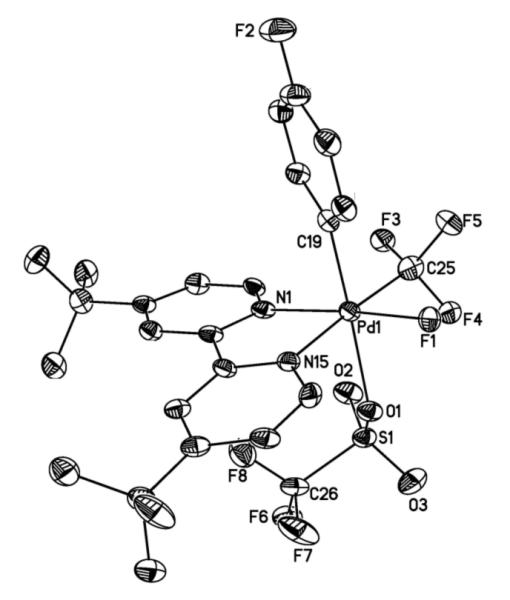
ORTEP drawing of complex 4. Thermal ellipsoids are drawn at 50% probability, hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Pd–C(1) 2.009(5), Pd–C(25) 2.018(5), Pd– N(1) 2.038(4), Pd–N(15) 2.082(4), Pd–O(1) 2.226(3). Selected bond angles (°): C(19)–Pd–C(25) 91.09(15), C(19)–Pd–N(15) 92.18(16), C(25)–Pd–N(15) 175.68(17), C(19)–Pd–F(1) 91.09(15), C(25)–Pd–F(1) 83.31(16), C(19)–Pd(1)–O(1) 175.74 (16), C(25)–Pd(1)–O(1) 95.15 (16).
Mechanistic Study of Aryl–CF3 Bond-Forming Reductive Elimination from PdIV.
There are at least three possible pathways for Aryl–CF3 bond-forming reductive elimination from 4, mechanisms A, B, and C (Figure 3). Mechanism A involves initial triflate dissociation to form a cationic five-coordinate PdIV intermediate I and subsequent Aryl–CF3 bond formation. Mechanism B involves fluoride dissociation, followed by Aryl–CF3 coupling from intermediate II. Mechanism C proceeds via concerted C–CF3 bond-forming reductive elimination. Notably, there is significant literature precedent for both ionic22e,Error! Bookmark not defined.,32,33 and concerted22e,34 reductive elimination mechanisms from octahedral group 10 metal complexes.
Figure 3.
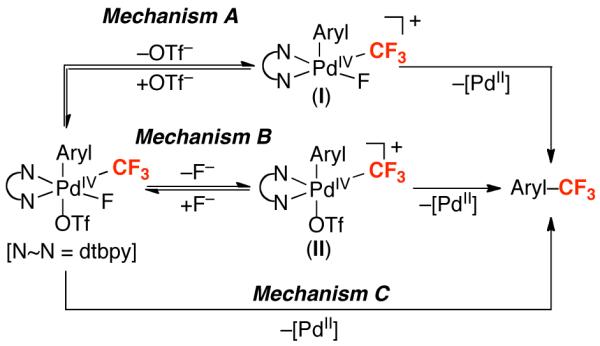
Three potential mechanisms for Aryl–CF3 bond formation from 4.
Order in triflate.
The addition of 1 equiv of NBu4OTf to the thermolysis of 4 in NO2Ph-d5 at 50 °C significantly slowed the initial rate of formation of 2a (from 2.21 × 10−5 M s−1 to 1.35 × 10−5 M s−1).35 Furthermore, an excellent linear fit was observed for a plot of initial rate versus 1/[NBu4OTf] (Figure 4).
Figure 4.
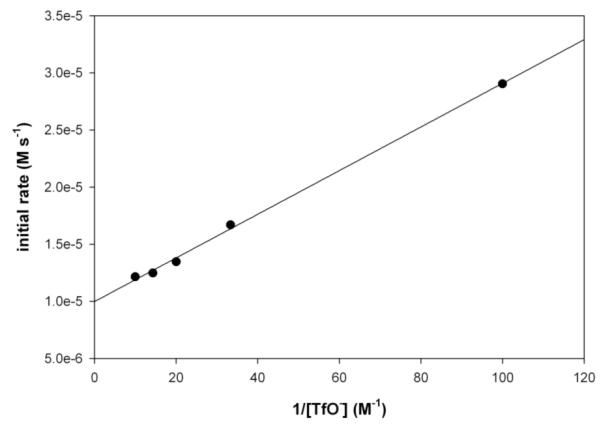
Plot of initial rate versus 1/[OTf] for reductive elimination from 4 to form 2a in PhNO2-d5 at 50 °C. y = 1.91 × 10−7 + 9.86 × 10−6; R2 = 0.998.
In order to confirm that the presence of TfO− was responsible for the observed effect, this reaction was next examined in the presence of NBu4PF6, which contains a non-coodinating anion. The use of 1 equiv of NBu4PF6 under otherwise identical conditions resulted in a >2-fold increase in the initial rate of reductive elimination to 5.57 × 10−5 M s−1. This result indicates that inibition by NBu4OTf is specifically due to the TfO− anion. Furthermore, it suggests that enhancing the polarity of the medium (by adding non-coordinating ions) accelerates Aryl–CF3 reductive elimination. Both pieces of data are consistent with ionic mechanism A operating in this system, as reflected by the rate expression in eq. 7.36
 |
Activation parameters.
The initial rate of C–CF3 bond-forming reductive elimination from 4 in NO2Ph-d5 was next examined as a function of temperature. An Eyring plot of the data showed that ΔH‡ is +29.1 ± 0.2 kcal/mol, while ΔS‡ = +9.48 ± 0.8 eu. The observed entropy of activation indicates a significant increase in disorder in the transition state for this reductive elimination reaction. Similar values have been observed for carbon-heteroatom bond-forming reductive elimination reactions from PdIV that proceed by ionic mechanisms.22e,23e,37 For example, C–OAc reductive elimination from PdIV complex (phpy)2PdIV(OAc)2 (phpy = 2-phenylpyridine), which is proposed to involve pre-equilibrium dissociation of an acetate ligand, showed ΔS‡ of +4.2 ± 1.2 eu.22e
Electronic effects.
Finally, electronic effects on reductive elimination were evaluated using PdIV complexes containing p-fluoro, p-methyl, and p-trifluoromethyl-substituted aryl groups (complexes 4-6). Thermolysis of 4, 5, and 6 at 80 °C in NO2Ph-d5 for 3 h afforded the corresponding benzotrifluorides in 77%, 93%, and 65% yield, respectively (Table 4). The initial rates of reductive elimination from these complexes at 50 °C in NO2Ph-d5 show that electron donating substituents accelerate the reaction. For example, with X = CH3 (5), the initial rate is more than 20 times greater than with X = F (4). Additionally, when X = CF3 (6), the initial rate is nearly two times slower than from 4 (Table 4).
Table 4.
Initial Rates of Aryl–CF3 Bond-Forming Reductive Elimination from Complexes 4-6
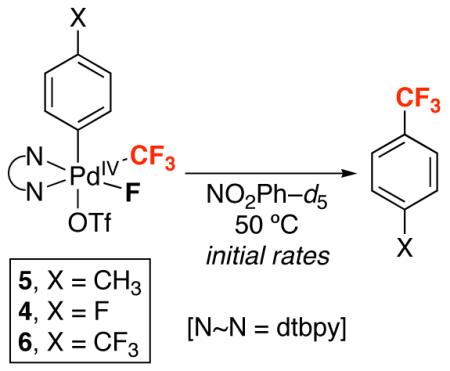
| Entry | X | Yield Aryl–CF3 | Initial rate (M s−1) × 105 |
|---|---|---|---|
| 1 | CH3 | 93% | 45.8 |
| 2 | F | 77% | 2.21 |
| 3 | CF3 | 65% a | 1.43 |
Reactions were run in duplicate and yields were determined by 19F NMR spectroscopy.
a1,4(bistrifluoromethyl)biphenyl formed in 21% yield.
While Table 4 clearly shows that electron-donating aryl substituents accelerate this reaction, it is challenging to definitively interpret this data in the context of the C–CF3 bond-forming event. Mechanism A is a two-step process involving triflate dissociation followed by Aryl–CF3 coupling. Thus, the faster rate with complex 5 may be due to the stronger trans effect of a more electron donating σ-aryl ligand and/or from a partial positive charge buildup on the aromatic ring in the transition state for Aryl–CF3 coupling.
DFT calculations.
We next turned to DFT calculations to more fully explore the mechanism of Aryl–CF3 bond formation. The complex [(bpy)PdIV(Ph)(CF3)(F)(OTf)] (7) (bpy = 2,2′-bipyridyl, eq. 8) was employed as a model for 5 (Table 4), and the CEP-31G(d)38,39 basis set and M0640,41 functional were used along with a single point solvent correction in nitrobenzene (SMD solvation model).42 As shown in eq. 8, the calculated ΔH298 for loss of TfO− from 7 in nitrobenzene to form cationic intermediate ([(bpy)PdIV(Ph)(CF3)(F)]+ (8)) is 15.0 kcal/mol. Furthermore, the activation enthalpy (ΔH‡298) for Ph–CF3 bond-forming reductive elimination from 8 is 13.7 kcal/mol. Thus, we calculate that mechanism A has an overall ΔH‡298 of 28.7 kcal/mol, which is in excellent agreement with the experimental value (+29.1 ± 0.2 kcal/mol, vide supra).
 |
As discussed above, we observe experimentally that trifluoromethylated products are formed selectively over the corresponding fluorinated compounds. To gain further insights into this selectivity, we used DFT to examine the transition state for Ph–F bond-forming reductive elimination from intermediate 8. As shown in eq. 8, ΔH‡298 (DFT) from Ph–F coupling is 14.7 kcal/mol. As such, this is a higher energy pathway than Ph–CF3 bond-formation (ΔΔH‡298 (DFT) = 1 kcal/mol), consistent with the experimental results.
The calculated charge distribution of intermediate 8 using Natural Bond Order (NBO) analysis43,44 indicates that the CF3 carbon carries a significant positive charge (+1.18), while the α-carbon of the phenyl ligand bears a charge of +0.07.45 This charge difference is amplified in the transition state for C–CF3 bond-forming reductive elimination, where the CF3 carbon carries an enhanced positive charge of +1.24, and the charge on the Ph carbon decreases to −0.11. These data imply that the Ph group is acting as the nucleophile during the bond-forming event. This is in sharp contrast to other reports of reductive elimination from PdIV in which the aryl or alkyl ligand typically serves as the electrophilic coupling partner.22,23
We next used DFT to determine the transition state enthalpies (ΔH‡298) for Aryl–CF3 coupling from complexes of general structure [(bpy)PdIV(p-XC6H4)(CF3)(F)]+. As expected based on the NBO analysis, ΔH‡298 was smallest with electron donating para substituents (X) on the aromatic ring, consistent with a transition state involving nucleophilic attack by σ-aryl ligand on the CF3 moiety (Table 5). Furthermore, the ΔH‡298 values showed better correlation with Hammett σ+ values for X than the corresponding σ or σ− parameters. This implicates significant resonance effects in the transition state for Aryl–CF3 coupling.46
Table 5.
Gas Phase Values of ΔH‡298 for C–CF3 Bond-Formation from [(bpy)PdIV(p-XC6H4)(CF3)(F)]+[a]
| X | ΔH‡298 (kcal/mol) | σ + |
|---|---|---|
| NMe2 | 6.89 | −1.70 |
| NH2 | 7.10 | −1.30 |
| OH | 7.71 | −0.92 |
| OMe | 7.86 | −0.78 |
| SMe | 7.79 | −0.60 |
| Me | 9.19 | −0.30 |
| F | 8.41 | −0.07 |
| H | 9.41 | 0 |
| CF3 | 9.43 | 0.53 |
| CN | 9.23 | 0.71 |
| NO2 | 9.23 | 0.78 |
Complexes are calculated using CEP-31G(d)/M06 level of theory.
Finally, we sought to identify supporting ligands that would lower the energy barrier for Ph–CF3 bond-forming reductive elimination in this system. Literature studies have shown that (tmeda)PdIV(CH3)2(Ph)(I) (tmeda = tetramethylethylenediamine) is significantly more reactive towards C–C bond-forming reductive elimination than its bipyridine analogue (bpy)PdIV(CH3)2(Ph)(I)47. Thus, we hypothesized that using tmeda in place of dtbpy in our system might impart a similar effect. Consistent with this hypothesis, DFT calculations show that the nitrobenzene solvent-corrected transition state enthalpy (ΔH‡298) for formation of trifluorotoluene from [(tmeda)PdIV(Ph)(CF3)(F)]+ is 0.8 kcal/mol lower than that for the analogous bpy complex (12.9 versus 13.7 kcal/mol) (eq. 9). Furthermore, ΔH298 for the loss of OTf− from [(tmeda)PdIV(Ph)(CF3)(F)(OTf)] is >10 kcal/mol lower than that from 7 (4.2 versus 15 kcal/mol). Thus, the calculated overall ΔH‡298 for reductive elimination from the tmeda complex is 17.1 kcal/mol, suggesting that Aryl-CF3 coupling should proceed at significantly lower temperatures than from 7.
 |
Room Temperature Arene Trifluoromethylation.
To assess the effect of diamine ligands experimentally, a series of (N~N)PdII(Ph)(CF3) complexes (9, 10, and 11e) were prepared by the reaction of (N~N)PdII(Ph)(I) with CsF followed by TMSCF3 (see Supporting Information for full details).48 Treatment of 9, 10, and 11e with NFTPT under our standard conditions (80 °C for 3 h in NO2Ph) resulted in clean formation of trifluorotoluene in modest to excellent yield (Table 6). The readily available and inexpensive tmeda ligand was particularly effective. Tmeda complex 11e afforded 90% yield of trifluorotoluene at 80 °C (entry 4), and, most remarkably, provided 83% yield at room temperature (entry 5). To our knowledge, this is first example of room temperature arene trifluoromethylation at a Pd center.49
Table 6.
NFTPT-Promoted Aryl–CF3 Coupling from Complexes 1f, 9, 10, and 11e
| Entry | Compound | N~N | Temp | Yield Ph–CF3 |
|---|---|---|---|---|
| 1 | 1f | dtbpy | 80 °C | 66% |
| 2 | 9 |
|
80 °C | 99% |
| 3 | 10 |
|
80 °C | 40% |
| 4 | 11e |
|
80 °C | 90% |
| 5 | 11e |
|
23 °C | 83% |
Reactions were run in duplicate and yields determined by 19F NMR spectroscopy.
The scope of room temperature oxidative trifluoromethylation from (tmeda)PdII(Aryl)(CF3) was found to be quite broad.50 These reactions proceeded efficiently and in high yield with electron donating and electron neutral substituents on the σ-aryl ligand (for example, Table 7, entries 4-7). The room temperature reactions were lower yielding with arenes bearing highly electron-withdrawing substituents like CF3 and CN (entries 2 and 3), which is consistent with the proposed transition state (vide supra). Finally, tmeda complexes containing ortho-substituted aryl groups underwent high yielding room temperature oxidative trifluoromethylation (entries 8 and 9). In contrast, these were poorly effective substrates in the dtbpy system, even at 80 °C.30
Table 7.
Synthesis and Reactivity of (tmeda)Pd(Aryl)(CF3) Complexes
| Entry | Compound | Aryl | Yield Aryl–CF3 (80 °C) |
Yield Aryl–CF3 (23 °C) |
|---|---|---|---|---|
| 1 | 11a | p-FC6H4 | 81% | 78% |
| 2 | 11b | p-CF3C6H4 | 76%a | 52%a |
| 3 | 11c | p-CNC6H4 | 60% | 22% |
| 4 | 11d | p-MeOC6H4 | 92% | 95% |
| 5 | 11e | C6H5 | 94% | 88% |
| 6 | 11f | p-MeC6H4 | 90% | 83% |
| 7 | 11g | m-MeC6H4 | 95% | 95% |
| 8 | 11h | o-MeC6H4 | 85% | 88% |
| 9 | 11i | o-MeOC6H4 | 90% | 99% |
These reactions were conducted at 80 °C for 3 h and at 23 °C for 1 h. Reactions were run in duplicate and all of the starting material was consumed. Yields were determined by 19F NMR spectroscopy.
At both temperatures, trifluorotoluene was formed in 13% yield.
Conclusion.
This article describes the rational design of 1st generation systems for oxidatively-induced Aryl–CF3 bond forming reductive elimination from Pd. Experimental mechanistic studies implicate Aryl–CF3 coupling from a cationic five-coordinate intermediate, and DFT suggests that the CF3 ligand serves as the electrophilic partner during bond formation. Our investigations into the scope and mechanism of this reaction have facilitated the development of a 2nd generation ligand system that enables Aryl–CF3 coupling at room temperature. This work provides the basis for the design of novel PdII/IV-catalyzed trifluoromethylation reactions of aryl metal species (metal = B, Sn, Si) or simple arene C–H bonds. Efforts in this area are currently underway in our group and will be reported in due course.
Supplementary Material
Acknowledgements.
We thank the NIH [GM073836 and F31GM089141 (fellowship to NDB)], the National Science Foundation Graduate Research Fellowship and Murrill Memorial Scholarship (fellowship to JBG) and the Research Corporation Cottrell Scholar Program for support of this research. Additional unrestricted support from Dupont is also gratefully acknowledged. We also thank Paul Lennon and Jim Windak for assistance with mass spectroscopy as well as Prof. Tom Cundari and Prof. Brian Yates for valuable discusions on DFT calculations.
Footnotes
Supporting Information Avaliable. Experimental and computational details and spectroscopic data for new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Kirk KL. Org. Process Res. Dev. 2008;12:305–321. [Google Scholar]
- 2.Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ma JA, Cahard D. Chem. Rev. 2004;104:6119–6146. doi: 10.1021/cr030143e. [DOI] [PubMed] [Google Scholar]; (b) Ma JA, Cahard D. Chem. Rev. 2008;108:PR1–PR43. doi: 10.1021/cr800221v. [DOI] [PubMed] [Google Scholar]; (c) Prakash GKS, Chacko S. Curr. Opin. Drug Disc. Dev. 2008;11:793–802. [PubMed] [Google Scholar]; (d) Shibata N, Mizuta S, Kawai H. Tetrahedron: Asymmetry. 2008;19:2633–2644. [Google Scholar]
- 4.Grushin VV. Acc. Chem. Res. 2010;43:160–171. doi: 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]
- 5.Swarts F. Bull. Acad. R. Belg. 1892;24:415–429. [Google Scholar]
- 6.(a) Henne AL, Whaley AM, Stevenson JK. J. Am. Chem. Soc. 1941;63:3478–3479. [Google Scholar]; (b) Furuta S, Kuroboshi M, Hiyama T. Bull. Chem. Soc. Jpn. 1999;72:805–819. [Google Scholar]
- 7.Yang JJ, Kirchmeier RL, Shreeve JM. J. Org. Chem. 1998;63:2656–2660. doi: 10.1021/jo972213l. [DOI] [PubMed] [Google Scholar]
- 8.For examples of Cu-mediated reaction of TMSCF3 with aryl halides, see: Kobayashi Y, Kumadaki I. Tetrahedron Lett. 1969;10:4095–4096. Konderatenko NV, Vechirko EP, Yagupolskii LM. Synthesis. 1980:932–933. Matsui K, Tobita E, Ando M, Kondo K. Chem. Lett. 1981;10:1719–1720. Suzuki H, Yoshida Y, Osuka A. Chem. Lett. 1982;11:135–136. Burton DJ, Wiemers DM. J. Am. Chem. Soc. 1985;107:5014–5015. Urata H, Fuchikami T. Tetrahedron Lett. 1991;32:91–94. Dubinina GG, Furutachi H, Vicic DA. J. Am. Chem. Soc. 2008;130:8600–8601. doi: 10.1021/ja802946s. Dubinina GG, Ogikubo J, Vicic DA. Organometallics. 2008;27:6233–6235.
- 9.For the Cu-mediated reaction of TMSCF3 with aryl boronic acids, see Chu L, Qing F-L. Org. Lett. 2010;12:5060–5063. doi: 10.1021/ol1023135. Senecal TD, Parsons AT, Buchwald SL. J. Org. Chem. 2011;76:1174–1176. doi: 10.1021/jo1023377.
- 10.Zhang C-P, Wang Z-L, Chen Q-Y, Zhang C-T, Gu Y-C, Xiao J-C. Angew. Chem. Int. Ed. 2011;50:1896–1900. doi: 10.1002/anie.201006823. [DOI] [PubMed] [Google Scholar]
- 11.(a) Oishi M, Kondo H, Amii H. Chem. Commun. 2009:1909–1911. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]; (b) Knauber T, Arikan F, Röschenthaler G, Gooßen LJ. Chem. Eur. J. 2011;17:2689–2697. doi: 10.1002/chem.201002749. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews see: Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r. Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318–5365. doi: 10.1021/cr068006f. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447–2464. doi: 10.1039/b805701j. Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074–1086. doi: 10.1021/ar9000058.
- 13.For reviews on [M]–CF3 and [M]–Rf (Rf = perfluoroalkyl) complexes, see: Hughes RP. Adv. Organomet. Chem. 1990;31:183–267. Morrison JA. Adv. Organomet. Chem. 1993;35:211–239.
- 14.Grushin VV, Marshall WJ. J. Am. Chem. Soc. 2006;128:12644–12645. doi: 10.1021/ja064935c. [DOI] [PubMed] [Google Scholar]
- 15.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524. Pd-catalyzed coupling between ArI and [Zn–CF3]: Kitazume T, Ishikawa N. Chem. Lett. 1982:137–140.
- 16.Current price for Xantphos = $18,122/mol. Determined based on the largest quantity of Xantphos available from Sigma-Aldrich on March 17, 2011 (25 g/$783.00).
- 17.Current price for Brettphos = $99,195/mol. Determined based on the largest quantity of Brettphos available from Strem Chemicals on March 17, 2011 (5 g/$924.00).
- 18.Price for the largest quantity of TESCF3 available from Sigma-Aldrich on March 17, 2011: 1 g/$72.90, $13,433/mol. Price for the largest quantity of TMSCF3 available from Sigma-Aldrich on March 17, 2011: 25 mL/$397.00, $2347/mol.
- 19.Wang X, Truesdale L, Yu JQ. J. Am. Chem. Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]
- 20.Ye Y, Ball ND, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2010;132:14682–14687. doi: 10.1021/ja107780w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ball ND, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2010;132:2878–2879. doi: 10.1021/ja100955x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Dick AR, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2005;127:12790–12791. doi: 10.1021/ja0541940. [DOI] [PubMed] [Google Scholar]; (b) Whitfield SR, Sanford MS. J. Am. Chem. Soc. 2007;129:15142–15143. doi: 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]; (c) Dick AR, Remy MS, Kampf JW, Sanford MS. Organometallics. 2007;26:1365–1370. [Google Scholar]; (d) Ball ND, Sanford MS. J. Am. Chem. Soc. 2009;131:3796–3797. doi: 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Racowski JM, Dick AR, Sanford MS. J. Am. Chem. Soc. 2009;131:10974–10983. doi: 10.1021/ja9014474. [DOI] [PubMed] [Google Scholar]; (f) Arnold PL, Sanford MS, Pearson SM. J. Am. Chem. Soc. 2009;131:13912–13913. doi: 10.1021/ja905713t. [DOI] [PubMed] [Google Scholar]
- 23.For examples, see: Alsters PL, Engel PF, Hogerheide MP, Copijn M, Spek AL, van Koten G. Organometallics. 1993;12:1831–1844. Lagunas MC, Gossage RA, Spek AL, van Koten G. Organometallics. 1998;17:731–741. van Belzen R, Elsevier CJ, Dedieu A, Veldman N, Spek AL. Organometallics. 2003;22:722–736. Canty AJ, Denny MC, Patel J, Sun H, Skelton BW, White AH. J Organomet Chem. 2004;689:672–677. Canty AJ, Denney MC, Skelton BW, White AH. Organometallics. 2004;23:1122–1131. Yamamoto Y, Kuwabara S, Matsuo S, Ohno T, Nishiyama H, Itoh K. Organometallics. 2004;23:3898–3906. Kaspi AW, Yahav-Levi A, Goldberg I, Vigalok A. Inorg. Chem. 2008;47:5–7. doi: 10.1021/ic701722f. Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, III, Ritter T. J. Am. Chem. Soc. 2010;132:3793–3807. doi: 10.1021/ja909371t. Vicente J, Arcas A, Julia-Hernandez F, Bautista D. Chem. Comm. 2010;46:7253–7255. doi: 10.1039/c0cc02660c. Williamson O, Zavalij PY, Zhang J, Khaskin E, Vedernikov AN. J. Am. Chem. Soc. 2010;132:14400–14402. doi: 10.1021/ja107185w. Zhao X, Dong VM. Angew. Chem. Int. Ed. 2011;50:932–934. doi: 10.1002/anie.201005489.
- 24.(a) Culkin DA, Hartwig JF. Organometallics. 2004;23:3398–3416. [Google Scholar]; (b) Grushin VV, Marshall WJ. J. Am. Chem. Soc. 2006;128:4632–4641. doi: 10.1021/ja0602389. [DOI] [PubMed] [Google Scholar]; (c) McReynolds KA, Lewis RS, Ackerman LKG, Dubinina GG, Brennessel WW, Vicic DA. J. Fluorine Chem. 2010;131:1108–1112. [Google Scholar]
- 25.(a) Umemoto T. Chem. Rev. 1996;96:1757–1778. doi: 10.1021/cr941149u. [DOI] [PubMed] [Google Scholar]; (b) Eisenberger P, Gischig S, Togni A. Chem. Eur. J. 2006;12:2579–2586. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]; (c) Kieltsch I, Eisenberger P, Stanek K, Togni A. Chimia. 2008;62:260–263. [Google Scholar]
- 26.Stanek K, Koller R, Togni A. J. Org. Chem. 2008;73:7678–7685. doi: 10.1021/jo8014825. [DOI] [PubMed] [Google Scholar]
- 27.For similar strategies to generate C–N or C–C bonds using F+ sources see: Mei T-S, Wang X, Yu J-Q. J. Am. Chem. Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J. Am. Chem. Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 28.Engle KM, Mei TS, Wang X, Yu J-Q. Angew. Chem. Int. Ed. 2011;50:1478–1491. doi: 10.1002/anie.201005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The reported yields in Tables 1 and 2 are ~10% lower than reported in our original communication (ref. 21), due to a minor error associated with the quantity of internal standard used for 19F NMR analysis. The correct yields for these transformations are reported in Table 3.
- 30.(dtbpy)PdII(Aryl)(CF3) complexes containing ortho-substituted Aryl ligands reacted with NFTPT to provide very low yields of the corresponding benzotrifluorides. For example (dtbpy)Pd(o-CH3Ph)(CF3) afforded 13% yield of 2-methylbenzotrifluoride. Interestingly, the major product of this reaction was the corresponding fluorinated product (2-fluorotoluene, formed in 62% yield).
- 31.In MeCN-d3, it is likely that the triflate ligand is replaced by a solvent molecule.
- 32.For examples of mechanisms like A and B for reductive elimination from PdIV, see: Byers PK, Canty AJ, Crespo M, Puddephatt RJ, Scott JD. Organometallics. 1988;7:1363–1367. Ducker-Benfer C, van Eldik R, Canty AJ. Organometallics. 1994;13:2412–2414. Canty AJ, Jin H, Skelton BW, White AH. Inorg. Chem. 1998;37:3975–3981. doi: 10.1021/ic9715005.
- 33.For examples of mechanisms similar to A and B for reductive elimination from PtIV, see: Goldberg KI, Yan J, Winter EL. J. Am. Chem. Soc. 1994;116:1573–1574. Goldberg KI, Yan J, Breitung EM. J. Am. Chem. Soc. 1995;117:6889–6896. Williams BS, Goldberg KI. J. Am. Chem. Soc. 2001;123:2576–2587. doi: 10.1021/ja003366k. Vedernikov AN, Binfield SA, Zavalij PY, Khusnutdinova JR. J. Am. Chem. Soc. 2006;128:82–83. doi: 10.1021/ja0575171. Khusnutdinova JR, Zavalij PY, Vedernikov AN. Organometallics. 2007;26:3466–3483. Pawlikowski AV, Getty AD, Goldberg KI. J. Am. Chem. Soc. 2007;129:10382–10393. doi: 10.1021/ja069191h. Khusnutdinova JR, Newman LL, Zavalij PY, Lam Y-F, Vedernikov AN. J. Am. Chem. Soc. 2008;130:2174–2175. doi: 10.1021/ja7107323. Smythe NA, Grice KA, Williams BS, Goldberg KI. Organometallics. 2009;28:277–288.
- 34.For examples of mechanisms similar to C for reductive elimination from PdIV and PtIV, see: Crumpton DM, Goldberg KI. J. Am. Chem. Soc. 2000;122:962–963. Crumpton-Bregel DM, Goldberg KI. J. Am. Chem. Soc. 2003;125:9442–9456. doi: 10.1021/ja029140u. Arthur KL, Wang QL, Bregel DM, Smythe NA, O’Neill BA, Goldberg KI, Moloy KG. Organometallics. 2005;24:4624–4628. (d) Reference 22e.
- 35.We initially examined Aryl–CF3 bond-forming reductive elimination by monitoring the disappearance of 4 and concomitant appearance of 2a over three half lives. These studies revealed that the rate of product formation decreases upon higher conversion to product (see Supporting Information for full details). We hypothesize that this is due to a build-up of TfO− as the reaction progresses, which then inhibits the 1st step of Aryl–CF3 bond-forming reductive elimination from 4. To avoid this apparent product inhibition, we used the initial rates method (measuring the first 10% conversion) for both the order and activation parameter studies.
- 36.The addition of 1 equiv of NMe4F to 4 at 23 °C in NO2Ph-d5 produced a complex mixture of Pd–F and Pd–CF3 containing products (as determined by 19F NMR spectroscopy). As a result, it was not possible to definitively assess the effect of F− on the rate of reductive eliminaton from 4.
- 37.The entropy of activation for reductive elimination reactions from MIV can be highly dependent on both the structure of the MIV complex and the solvent. This is particularly the case in ionic reductive elimination reactions, where solvent ordering around charged species can have a significant effect. See ref. 32a.
- 38.Stevens WJ, Basch H, Krauss MJ. J. Chem. Phys. 1984;81:6026–6033. [Google Scholar]
- 39.Stevens WJ, Krauss M, Basch H, Jasien PG. Can. J. Chem. 1992;70:612–630. [Google Scholar]
- 40.Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 41.The M06 functional was selected based its success in DFT calculations of related late transition metal organometallic complexes. For examples, see: Sieffert N, Bühl M. Inorg. Chem. 2009;48:4622–4624. doi: 10.1021/ic900347e. Benitez D, Tkatchouk E, Goddard WA., III Organometallics. 2009;28:2643–2645. Benitez D, Shapiro ND, Tkatchouk E, Wang YM, Goddard WA, III, Toste FD. Nat. Chem. 2009;1:482–486. doi: 10.1038/nchem.331. Ariafard A, Hyland CJT, Canty AJ, Sharma M, Brookes NJ, Yates BF. Inorg. Chem. 2010;49:11249–11253. doi: 10.1021/ic1020912.
- 42.Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 43.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO Version 3.1. [Google Scholar]
- 44.For original references of theory, see: Foster JP, Weinhold F. J. Am. Chem. Soc. 1980;102:7211–7218. Reed AE, Weinhold F. J. Chem. Phys. 1983;78:4066–4073. Reed AE, Weinstock RB, Weinhold F. J. Chem. Phys. 1985;83:735–746. Reed AE, Weinhold F. J. Chem. Phys. 1985;83:1736–1740. Carpenter JE. PhD thesis. University of Wisconsin; Madison, WI: 1987. Extension of Lewis structure concepts to open-shell and excited-state molecular species. Carpenter JE, Weinhold F. J. Mol. Struct. (Theochem) 1988;46:41–62. Reed AE, Curtiss LA, Weinhold F. Chem. Rev. 1988;88:899–926. Weinhold F, Carpenter JE. In: The Structure of Small Molecules and Ions. Naaman R, Vager Z, editors. Plenum; New York: 1988. pp. 227–236.
- 45.The ground state structure of the analogous CH3 complex ([(bpy)PdIV(Ph)(CH3)(F)]+ has calculated charges of −0.58 and +0.06 on the CH3 and Ph carbons, respectively.
- 46.Assuming a similar entropy component in each transition state, a Hammett plot of log(kX/kH) versus σ+ with rate constants calculated based on the electronic energies (ΔH‡298), provided a reasonably good linear correlation (ρ+ = −0.79; R2 = 0.84, Figure S8). Much poorer correlations were observed with σ and σ− (ρ value of −1.17 and R2 = 0.57; ρ− value of −0.86 and R2 = 0.52).
- 47.Markies BA, Canty AJ, Boersma J, van Koten G. Organometallics. 1994;13:2053–2058. [Google Scholar]
-
48.Complex 11b can also be prepared by reacting (tmeda)PdII(p-CF3C6H4)(X) (X = acetate or trifluoroacetate) with CsF/TMSCF3 in THF. This synthetic pathway is potentially useful and relevant for catalytic applications of these transformations. For example, C–H functionalization reactions often use Pd(OAc)2 or Pd(TFA)2 as catalysts. For examples, see:
Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.
- 49.Vicic has reported that the reaction of (NHC)CuI(CF3) (NHC = heterocyclic carbene) with aryl iodides provides Aryl–CF3 products at room temperature. The mechanism of the transformation is still under investigation. See refs 8g and h.
- 50.For the synthesis of these complexes, see Supporting Information.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.