

Abstract
While initiation of transcription has attracted the most attention in the field of gene regulation, it has become clear that additional stages in the gene expression cascade including post-transcriptional events are under equally exquisite control. The seminal discovery that short RNAs (microRNA, small interfering RNA, Piwi-interacting RNA), play important roles in repressing gene expression has spurred a rush of new interest in post-transcriptional gene silencing mechanisms. The development of affinity tags and high-resolution tandem mass spectrometry (MS/MS) has greatly simplified the analysis of proteins that regulate gene expression. Further, the use of DNA microarrays and ‘second generation’ nucleic acid sequencing (‘deep sequencing’) technologies has facilitated the identification of their regulatory targets. These technological advancements mark a significant step towards a comprehensive understanding of gene regulatory networks. The purpose of this review is to highlight several recent reports that illustrate the value of affinity-purification (immunoprecipitation) followed by mass spectrometric protein analysis and nucleic acid analysis by deep sequencing (AP-MS/Seq) to examine mRNA after it has been transcribed. The ability to identify the direct nucleic acid targets of post-transcriptional gene regulatory machines is a critical first step towards understanding the contribution of post-transcriptional pathways on gene expression.
Keywords: AP-MS, mass spectrometry, deep sequencing, microRNA, post-transcriptional gene silencing, Argonaute
INTRODUCTION
Continued technological advancements in the fields of functional genomics and proteomics have secured these approaches as central methodologies in determining gene regulatory networks. An important first step in achieving this goal is to identify the protein complexes that regulate all stages of the process, namely transcription, mRNA processing, directed mRNA localization, mRNA stability and translation [1]. Over the past 30 years, many regulatory machines have been identified with the majority being involved at the level of mRNA transcription (e.g. initiation and elongation). Comparable studies of machines that regulate post-transcriptional processes have generally lagged behind.
Mass spectrometry (MS) has been successfully applied to many important biological questions, arguably the most fruitful application has been to identify co-purifying proteins from affinity purified (AP) samples, referred to as ‘AP-MS’ [2]. AP-MS experiments often identify novel interactions and unexpected relationships among proteins, and they have become a common approach to gain insight into protein function. The reduced complexity of immunopurified samples and improved MS technologies, which include faster scan speeds and improved mass accuracy, allow sequencing of virtually all of the polypeptides present in a complex including components at extremely low concentrations (attomolar). Such analyses can yield a wealth of information regarding not only the constituents of a single protein complex, but can also reveal potential interactions between protein complexes. From this, an investigator may draw information about linked functions between the co-purifying complexes. A large-scale AP-MS survey in budding yeast has revealed cross-talk between many protein complexes from different functional categories. For example, several connections were found between protein complexes involved in transcription and metabolism, or metabolism and defense [3].
After years of focusing on the identification and characterization of protein factors that regulate gene expression, many groups are now extending these investigations by performing high-throughput (or whole genome) analysis of their target nucleic acids. Specifically, the utilization of chromatin immunoprecipitation in combination with DNA microarrays (ChIP-on-chip) and more recently ‘second-generation’ DNA sequencing technologies (ChIP-Seq) has proven effective in identifying target genes of various transcription factors (TFs) [4, 5]. The new generation DNA sequencing is a large-scale parallel sequencing system known for unbiased, long, accurate sequence reads at high speed and comparatively low cost. In combination with bioinformatic analysis, these complementary approaches have proven valuable for identification of the cis-acting sequence(s) recognized by TFs and the specific genes that they target for activation or repression. Several excellent reviews describe the importance of ChIP-on-chip analysis for the determination of transcriptional regulatory networks [6].
This review will discuss recent reports that highlight the merit of analyzing the nucleic acid and protein components of immunopurified post-transcriptional regulatory complexes, and how these approaches aid the construction of gene regulatory networks. MicroRNA (miRNA) and small interfering RNA (siRNA) down-regulate gene expression by various mechanisms involving translational repression and accelerated mRNA turnover [7]. Increased mRNA turnover is mediated by at least two distinct mechanisms. If the siRNA or miRNA is fully complementary to the target mRNA, then mRNA is targeted for endonucleolytic cleavage by the RNA-induced silencing complex (RISC, see below). By contrast, an imperfectly base-paired miRNA/mRNA is subject to accelerated turnover by the removal of the 3′ poly(A) tail [8, 9]. The details of how miRNAs target mRNAs for translational repression are controversial and at least four mechanisms have been proposed [7]. The identification of the components of RISC and additional RISC-interacting proteins has provided much insight into the mechanisms by which miRNAs regulate gene expression.
Post-transcriptional gene silencing (PTGS) contributes to gene regulatory networks
The utility of the AP-MS/Seq approach to uncover gene regulatory networks is well illustrated by recent studies of the RISC. The core RISC proteins are the Argonaute (Ago) family members, which bind mi/siRNA. Ago2 additionally possesses the endonucleolytic ‘slicer’ activity (for details on RISC, see ref. [10]). The identification of additional proteins that help Ago to repress gene expression has been an area of intense research. RISC most often targets mRNA through specific base pairing between its bound small RNA and the 3′ untranslated region (UTR) of the mRNA, which leads to translational repression and/or RNA degradation [7]. Exactly how RISC selects specific miRNAs and their targets, and what determines their functional outcome is an active area of research. Affinity purification has proven valuable in providing answers to these important biological questions. RISC proteins are often isolated by immunoprecipitation of endogenous or epitope-tagged Ago proteins (Figure 1A). The Ago cofactors are identified by MS/MS and the miRNA and mRNA components analyzed by ‘second-generation’ DNA sequencing. By examining the mRNA sequences for matched miRNA seed sequences, miRNA/mRNA pairs associated with RISC have been identified.
Figure 1:
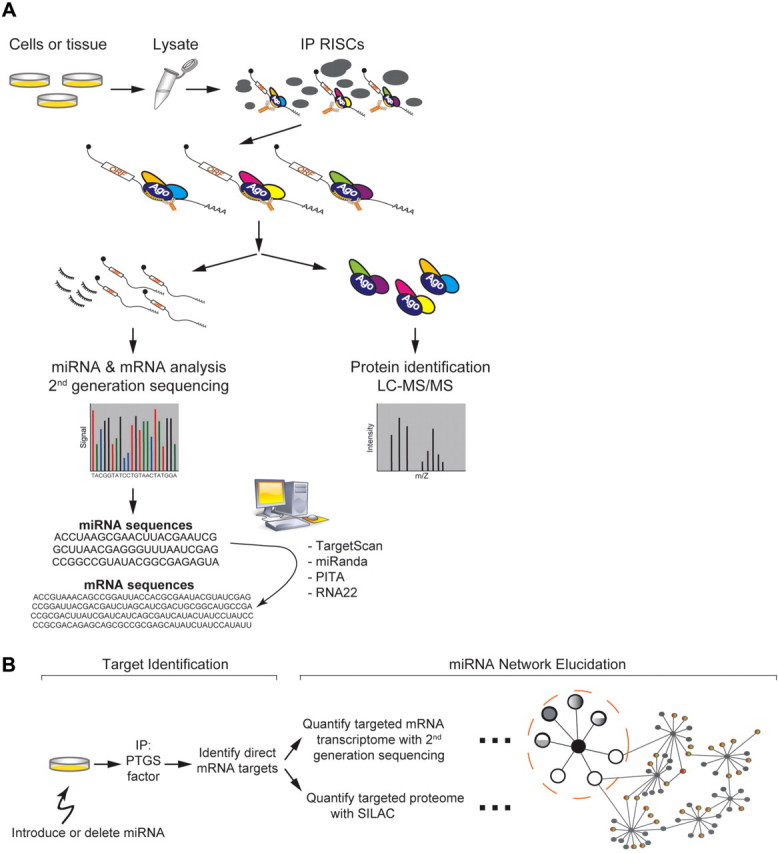
Biochemical elucidation of post-transcriptional gene silencing networks (A) A combined affinity purification, MS and nucleic acid sequencing approach to determine RISC protein factors, miRNAs and target mRNAs. Immunoprecipitations of endogenous or tagged proteins specific for various RISC components facilitate the isolation of RISCs. Protein components are identified by tandem MS. In parallel, RNA is extracted and analyzed by second-generation sequencing or by cDNA microarrays. The identified mRNA sequences are searched for corresponding miRNA seed matches by computational methods such as TargetScan, miRanda, PITA and RNA22 all available online. (B) A method to elucidate potential miRNA gene regulatory modules, one miRNA and one PTGS factor at a time. Cellular gene expression networks are perturbed by the introduction of a single miRNA and the mRNA targets are identified after immunoprecipitation as described earlier. Once the target mRNAs are identified, the miRNA’s effect on the mRNA’s abundance can be determined by quantification using cDNA microarrays or second-generation DNA sequencing. The miRNA’s effect on the corresponding protein levels can be interrogated by SILAC quantitative MS. The incorporation of many such experiments with various miRNAs allow for the determination of miRNA gene regulatory networks. Black circles in the center represent individual miRNAs and white/grey circles, target genes. White circle represents target genes whose translation is repressed without changes in mRNA levels, grey circle represents targets with decreased mRNA levels, and mixed white/grey circle represents targets with decreased mRNA levels and repressed translation. The large dashed circle represents a network module (a single miRNA and its target genes). The modules are linked by target genes, which are repressed by more than one miRNA.
Argonaute family members and their protein cofactors
After the discovery of Agos as the primary effectors of the miRNA and siRNA pathways, multiple groups immunopurified Ago and used MS to identify associated factors such as Dicer, TRBP, MOV10, IMP8 and GW182/TNRC6 as critical components of the pathway [11–14] (Table 1). The Ago family proteins are at the core of all RISCs; Dicer and TRBP play critical roles in the biogenesis/maturation of miRNAs, while TNRC6 proteins likely function within RISCs during the effector stage of the PTGS pathway. Interestingly, in many higher eukaryotes the core PTGS factors Ago and TNRC6 are present in multiple isoforms that can be distinguished through mass spectrometric identification of distinct peptides. It is possible that select mRNAs are targeted by RISCs composed of specific Ago or TNRC6 isoforms, or so-called ‘RISC isoforms’. It is possible that the expression of mRNAs targeted by specific RISC complex isoforms may be repressed by distinct mechanisms. Ago proteins have also been unexpectedly identified in AP-MS experiments with proteins of controversial function, such as the Huntington’s disease protein (Htt), for which there were previously no hints of an involvement in PTGS. Furthermore, Htt was found to localize at cytoplasmic processing (P)-bodies, sites of miRNA-mediated silencing, and shown to contribute to PTGS [15]. This discovery highlights the likely existence of additional yet undiscovered Ago cofactors.
Table 1:
Components of the RISC complexes and associated proteins identified by immunopurification and MS
| AP protein | Tagged/endogenous | Source | Proteins identified by MS | Reference |
|---|---|---|---|---|
| Gemin 3 | Endogenous | HeLa | Ago2, Gemin 4 | [36] |
| Ago1, 2, 3, 4 | Flag/HA | HeLa, HEK293 | Dicer, MOV10, TNRC6B, Gemin 3, Gemin 4, TRBP, Htt, IMP8 | [12, 37, 17, 11, 15, 38] |
| Huntingtin | Flag | HeLa | Ago1, 2 | [15] |
| AIN-1, -2 (TNRC) | GFP | Worm extract | DCR-1, ALG-1, -2 (Ago) | [39, 13] |
| TNRC 6A, 6B, 6C | Flag/HA | HEK293 | Ago1-4 | [17, 38] |
| Dicer | Flag/HA | HEK293 | TRBP, Ago1, 2 | [17, 40] |
| TRBP | Flag | HEK293 | Dicer, eIF6, MOV10 | [40, 14] |
All the data reported in the indicated references are combined for each affinity-purified protein.
To ask if interaction with Ago-associated proteins (e.g. Htt) is mediated through an RNA intermediate, co-immunoprecipitation (co-IP) experiments are performed in the absence and presence of RNase treatment. Such analysis has revealed that Ago’s interactions with Dicer, p54/RCK and GW182/TNRC6B are not mediated by RNA since their recovery in the co-IP experiments was unaffected in the presence of RNase, whereas the interactions with DCP2 and MOV10 are likely mediated by RNA [16, 17]. It should be noted that while the identification of RNase sensitive Ago-associated proteins is likely informative, an insensitive interaction needs to be interpreted with caution, because the interaction may still be mediated by an RNA intermediate that is protected from RNAse digestion. Additionally, the AP-MS approach has proven more successful at identifying novel proteins associated with the PTGS pathway than yeast-2-hybrid (Y2H) screens, consistent with previous reports suggesting that AP-MS outperforms Y2H in the identification of proteins present in multi-subunit complexes [18]. Moreover, because RISCs are composed of multiple protein and ribonucleic acid components, they are more likely to be identified by direct purification than by binary protein interaction screens such as the Y2H. The successful identification of many PTGS protein factors by AP-MS offers a shining example of the power of this approach and further discoveries are anticipated.
microRNAs target RISCs to specific mRNAs
High throughput analysis of co-purifying nucleic acids using cDNA/oligonucleotide microarrays and more recently by deep sequencing has facilitated an in-depth look at the small RNA components of several RISC complexes from various model organisms. Careful examination of these small RNA species has provided insight into the specificity of various PTGS factors with respect to distinct small RNA classes. For example, deep sequencing of immunoprecipitates containing AIN1 and AIN2 (TNRC6 family proteins) from Caenorhabditis elegans revealed a large repertoire of mature miRNAs, but significantly fewer siRNAs and 21-URNAs, suggesting that these proteins work specifically with miRNAs and are unlikely to play a role in their biogenesis [13]. The Arabidopsis thaliana (At) genome encodes 10 Argonaute proteins (for Ago phylogenic analysis, see ref. [19]) and a large number of small RNAs. To ask whether individual Agos show any specificity for small RNAs of a particular class, At Ago1, At Ago2, At Ago4 and At Ago5 immunoprecipitates were analyzed by deep sequencing [20]. Interestingly, miRNAs that co-purify with At Ago1 often favored a uridine nucleotide at the 5′ end. In contrast, the At Ago2 and At Ago4 precipitates were enriched for small RNAs with a 5′ terminal adenosine. At Ago5 also favored small RNAs but tended to select those with a cytosine at the 5′ end. These results suggest that the 5′ terminal nucleotide confers specificity for incorporation of small RNAs into distinct RISCs. These two examples demonstrate that biochemical purification followed by small RNA analysis provides a highly effective approach to determine which small RNAs are targeted by a given protein factor. Additionally, this approach offers a glimpse of the extraordinary complexity of the small RNA components involved in PTGS pathways.
mRNAs targeted by RISCs
After sorting out the protein factors and small RNA components of RISCs, biochemistry has been used to effectively identify their mRNA substrates, a critical step toward the elucidation of any gene regulatory network. Identification of mRNA targets remains a formidable challenge, and has been most often addressed through computational searches for miRNA-binding elements or ‘seeds’ [21–23]. Most often these bioinformatic approaches use evolutionary conservation of the seed as the criteria for the identification of miRNA-targeted mRNAs. Although this approach has been productive, different prediction algorithms have produced divergent results as well as many false positives. Recently there have been several successful biochemical ventures for the identification of mRNAs that physically co-purify with RISCs. An approach taken in Drosophila melanogaster (Dm) cells was to analyze the mRNAs and miRNAs from Dm Ago1 immunoprecipitates by reverse transcription and DNA microarray analysis [24]. Comparison of the identified miRNA seeds with the 3′UTR sequence of mRNAs revealed a significant enrichment for those mRNAs with miRNA seed matches. This analysis revealed a strong correlation between several miRNAs and their likely mRNA targets.
High-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP) has provided new and exciting data regarding mammalian Ago2 miRNA and mRNA targets from mouse brain [25]. This approach identified 454 miRNAs and 829 mRNA transcripts as direct Ago2 targets. By treating the crosslinked immunopurified sample with RNase, the investigators were able to obtain fragments of bound nucleic acids for high-throughput sequencing. The mRNA sequences obtained were subsequently mapped to the genome, and ‘peak clusters’ (stretches with the highest density) of sequence reads were obtained. This methodology provided sufficient data to determine where and how often Ago2 binds (‘footprints’) a given mRNA. The authors found that Ago2 binds most often (60%) within the 3′UTR and especially near stop codons (peaks at ∼50 nucleotides downstream). Bioinformatic analysis also revealed that on average there are ∼2 potential Ago2-binding sites per regulated transcript. Interestingly, these Ago2 footprints correlated well with the miRNA seed sequences and seed matches identified. To date, this approach has provided the most complete Ago2 miRNA–mRNA ternary interaction maps.
Another fruitful approach has been to use biochemically identified miRNA/mRNA pairs as benchmarks to determine the rules responsible for targeting. Computational RNA analysis of 3404 mRNAs identified from C. elegans AIN-1 and AIN-2 immunoprecipitates has revealed that these miRNA targets are enriched for several defining characteristics [26]. Enriched features include enhanced structural accessibility of target sequences (likelihood of being single stranded), total free energy of the miRNA/mRNA hybridization and base-pairing configuration to the 5′ seed region of the miRNA. This analysis suggests that the presence of miRNA binding elements within the 3′UTR is not the only feature of an mRNA that determines the likelihood of targeting. Indeed, incorporation of these characteristics into a revised computational method provided a more robust search engine that identified miRNA seeds with a reduced false positive rate compared with current computational methods alone.
An insightful variation on this theme has been to analyze the mRNAs of Ago2 immunoprecipitates from human cells transfected with a well-characterized miRNA, miR124a [27]. This analysis revealed a shift of the co-purifying mRNA pool towards a population that contained miR-124a seed sequences. In a separate study, immunopurified Ago2 from HEK293 cells transfected with miR-124a also revealed an enrichment of mRNAs that contained the corresponding seeds [28]. Interestingly, quantitative microarray analysis of these putative target mRNAs revealed sets that were reduced in abundance as well as sets that were unaffected by transfection of miR-124a. This observation suggests that some mRNAs targeted by miR-124a are likely repressed at the level of translation without significant changes in mRNA levels. Taken together, these studies demonstrated that high throughput identification of miRNA targets by biochemical means compliments and augments bioinformatic approaches and helps to instruct the development of better algorithms, which will improve the certitude of seed sequence prediction.
microRNAs contribute to gene networks
More than 174 miRNA genes have been identified in C. elegans, 157 in D. melanogaster and 721 in humans based on the miRNA database miRBase (September 2009). Each miRNA is predicted to target hundreds of mRNAs, and based on the presence of seed sequences, an ‘average’ mRNA has been predicted to be regulated by 15 miRNAs [29–31]. Because a single miRNA can target multiple mRNAs, and many mRNAs are targeted by several different miRNAs, miRNAs possess a significant regulatory potential and are thus likely to play central roles in gene expression networks. It is easy to imagine a scenario in which miRNAs target a class of mRNAs coding for TFs and would indirectly reduce the activation of their target genes and therefore also reduce their protein production. In fact, a large-scale computational analysis of miRNA targets has revealed that miRNAs target TFs more commonly than genes of other functional classes [32]. Consistent with this hypothesis, the combination of high-resolution ChIP-Seq data, the identification of miRNA promoters, and quantitative sequencing of short RNA transcripts have connected miRNAs to mouse embryonic stem cell transcriptional regulatory circuits [33].
Furthermore, a recent investigation in C. elegans has uncovered a composite miRNA/TF network characterized by high flow of information and feedback loops that provide coordinated control of gene expression [34]. First, to identify TFs that regulate miRNA promoters, a yeast one-hybrid approach was employed. This approach was successful in identifying 347 high confidence interactions between 63 miRNA promoters and 116 proteins (putative TFs). Next, a post-transcriptional network was created by computationally analyzing the candidate TF mRNAs for the presence of matched miRNA elements. Integration of these data revealed that a number of TFs that bind a miRNA promoter are themselves regulated post-transcriptionally by the transcribed miRNA. These studies provide an indication of the contribution of miRNAs on the integration of transcriptional regulatory circuits.
Recently, quantitative-mass-spectrometry by stable isotope labeling with amino-acids in cell culture (SILAC) and quantitative mRNA analysis by DNA microarrays have been combined to measure the simultaneous response of thousands of proteins and mRNAs after introducing or deleting a single miRNA [35]. This systems level analysis revealed that most mRNAs that were repressed contained seed matched sites for the overexpressed miRNA within their 3′UTRs. Hundreds of genes were found moderately repressed by a single miRNA. Comparison of mRNA and protein abundance revealed that mRNA destabilization was the most deterministic factor for gene repression. While this approach has yet to uncover regulatory networks, it highlights the potential rewards of performing large-scale analysis of thousands of miRNA targets. In-depth time-course experiments could further reveal the specific effects of a single miRNA not only on primary targets (those which contain seed matches) but their downstream effects on protein or mRNA levels.
CONCLUSION
Whilst it remains challenging to determine the relative contribution of PTGS to the overall function of a gene expression network, its inclusion bestows a more robust network than one governed by TFs alone. First, PTGS facilitates enhanced temporal regulation of gene expression by promoting turnover of key mRNAs. Second, the destruction of mRNAs provides the means for a ‘revised’ mRNA population, which may be subjected to differential regulation. Lastly, PTGS provides a key mechanism for spatially coordinated gene regulation at the level of local translation, a process known to be critical for cellular homeostasis in many biological contexts.
The essential first step towards a comprehensive understanding of PTGS and its influence on gene networks is the identification of the protein factors, miRNAs and target mRNAs. The addition or deletion of a single miRNA followed by the identification of miRNA and mRNA targets and downstream bioinformatic analysis should elucidate miRNA gene regulatory networks (Figure 1B). If the past findings are any indication of future success, then AP-MS/Seq should continue to provide us with valuable insights into the components and targets of gene regulatory machines and will likely strengthen the existing miRNA seed identification algorithms. It is worth noting that in order to successfully utilize AP-MS/Seq, sufficient quantities of copurifying protein and RNA must be recovered. Once we have determined a complete catalog of the factors and RNA components involved, bioinformatics will allow us to integrate the components into meaningful functional networks.
Key Points.
AP-MS/Seq has been used to identify both the protein and RNA components of RISCs.
Characterization of the RNA (miRNA and mRNA) components from affinity purified samples offers a powerful approach to determine the guides and targets of PTGS pathways and will aid in the development of accurate computational miRNA element identification algorithms.
Large-scale quantitative analysis of miRNA targets has revealed that gene expression is reduced by a combination of mRNA destabilization and translational inhibition.
Inclusion of PTGS into transcriptional networks will reveal a more complete view of gene expression circuitry.
FUNDING
The work in the authors’ laboratory was supported in part by the Hereditary Disease Foundation and CHDI Foundation, Inc. J.N.S. was supported in part by the Vilcek Endowment Fellowship Award.
Acknowledgments
The authors thank Christopher Parkhurst, Angus Wilson, Michael Garabedian, Bin Ma and Brady Culver for critical reading of the review and Emily Larrimer for editorial assistance.
Biographies
Jeffrey Savas recently received a PhD from NYU School of Medicine. He is currently a post-doctoral fellow in the laboratory of John Yates, III at the Scripps Research Institute in La Jolla. He is interested in applying advanced technologies in proteomics to explore new research areas in neuroscience.
Naoko Tanese is an Associate Professor at NYU School of Medicine. Her interests include regulation of gene transcription in mammalian cells and more recently, the role of normal and mutant Huntington’s disease protein in post-transcriptional pathways.
References
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–6. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, et al. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–34. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD. Constructing transcriptional regulatory networks. Genes Dev. 2005;19:1499–511. doi: 10.1101/gad.1325605. [DOI] [PubMed] [Google Scholar]
- Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell. 2008;29:1–7. doi: 10.1016/j.molcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, et al. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–9. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA. 2006;103:4034–9. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007;26:611–23. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Weinmann L, Hock J, Ivacevic T, et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136:496–507. doi: 10.1016/j.cell.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Peters L, et al. Identification of novel argonaute-associated proteins. Curr Biol. 2005;15:2149–55. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ding L, Cheung TH, et al. Systematic identification of C. elegans miRISC proteins, miRNAs, and mRNA targets by their interactions with GW182 proteins AIN-1 and AIN-2. Mol Cell. 2007;28:598–613. doi: 10.1016/j.molcel.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada TP, Finn KJ, Ji X, et al. MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007;447:823–8. doi: 10.1038/nature05841. [DOI] [PubMed] [Google Scholar]
- Savas JN, Makusky A, Ottosen S, et al. Huntington's; disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci USA. 2008;105:10820–5. doi: 10.1073/pnas.0800658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landthaler M, Gaidatzis D, Rothballer A, et al. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. Rna. 2008;14:2580–96. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner A, Polge C, Lentze N, et al. Yeast two-hybrid, a powerful tool for systems biology. Int J Mol Sci. 2009;10:2763–88. doi: 10.3390/ijms10062763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock J, Meister G. The Argonaute protein family. Genome Biol. 2008;9:210. doi: 10.1186/gb-2008-9-2-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Cai T, Hu Y, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133:116–27. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38:S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Easow G, Teleman AA, Cohen SM. Isolation of microRNA targets by miRNP immunopurification. Rna. 2007;13:1198–204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, et al. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell M, Long D, Zhang L, et al. mirWIP: microRNA target prediction based on microRNA-containing ribonucleoprotein-enriched transcripts. Nat Methods. 2008;5:813–9. doi: 10.1038/nmeth.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, Herschlag D, et al. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS ONE. 2008;3:e2126. doi: 10.1371/journal.pone.0002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Conaco C, Xuan Z, et al. A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci USA. 2007;104:19291–6. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Ritchie W, Flamant S, Rasko JE. Predicting microRNA targets and functions: traps for the unwary. Nat Methods. 2009;6:397–8. doi: 10.1038/nmeth0609-397. [DOI] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Lieber D, Oren M, et al. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–33. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Ow MC, Barrasa MI, et al. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008;22:2535–49. doi: 10.1101/gad.1678608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, et al. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–8. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock J, Weinmann L, Ender C, et al. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007;8:1052–60. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Shiekhattar R. Functional dissection of the human TNRC6 (GW182-related) family of proteins. Mol Cell Biol. 2009;29:4144–55. doi: 10.1128/MCB.00380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Spencer A, Morita K, et al. The developmental timing regulator AIN-1 interacts with miRISCs and may target the argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol Cell. 2005;19:437–47. doi: 10.1016/j.molcel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–4. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]