

Abstract
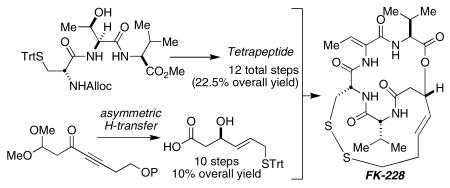
A scaleable synthesis of the potent histone deacetylase (HDAC) inhibitor FK228 is described. A reliable strategy for preparing the key β-hydroxy mercapto heptenoic acid partner was accomplished in nine steps and 13% overall yield. A Noyori asymmetric hydrogen-transfer reaction established the hydroxyl stereochemistry in >99:1 er via the reduction of a propargylic ketone.
The depsipeptide, FK228 (1) (previously named FR-901228), was isolated from the fermentation broth of Chromobacterium violaceum (No. 968) by Fujisawa Pharmaceutical Co., Ltd. in 1991 along with a closely related depsipeptide, FR-901375 (2) (Figure 1).1 The potent activity of FK228 (IC50=1.0 nM for FK228 with DTT for HDAC1) against a range of murine and human solid tumor cells, and its ability to target epigenetic silencing mechanisms crucial to carcinogenesis have culminated in its advancement into clinical trials as a potential anticancer therapy.2 During a phase II study, serious adverse cardiac events were observed, which precipitated termination of the trial and indicates that a better understanding of the biochemistry and cell biology of this unique natural product is critical.3 These investigations have been hindered by a lack of available material within the scientific community. The only reported total synthesis of FK228 by Simon, et al., has proven to be capricious and difficult to reproduce, and we were thus compelled to develop a new synthesis amenable to a larger scale preparation.4,5,6
Figure 1. Structures of FK228 (1) and FR-901375 (2).

FK228, acting as a prodrug, undergoes a disulfide reduction within the cell to release a zinc-binding thiol. The butenyl thiol is known to interact with zinc in the binding pocket of Zn-dependent histone deacetylase (HDAC) reversibly.7 Inhibitors of HDAC are under intense investigation as potential ‘transcriptional therapies’ for the treatment of cancer.8 They are able to restore normal expression of genes, which may result in cell cycle arrest, differentiation, and apoptosis. HDAC inhibitors are known to affect expression of the following genes: VEGF, ErbB2, Cyclin D1, Cyclin E, and Gelsolin.9 The mechanism of action for FK228 has been linked to the reversal of prodifferentiation of the ras oncogene pathway via blockade of p21 protein-mediated signal transduction. Its cytotoxicity is associated with the upregulation of Rap1, a small GTP-binding protein of the Ras family.10 Rap1 is an intrinsic regulator of the Ras-Raf-MAP kinase signaling pathway in melanoma cells.
The structure of FK228 is intriguing when compared to other HDAC inhibitors. It contains a 16-membered cyclic depsipeptide bridged by a 15-membered macrocyclic disulfide. The d-Cys residue of the tetrapeptide portion enables FK228 to act as a pro-drug, which is reduced in-vivo by glutathione reductase to reveal the Zn-binding butenyl thiol.2 This thiol is connected to the depsipeptide by a four-carbon tether, which is shorter than the typical tether length of 5-6 atoms observed to be optimal.11 The potent biological activity of FK228 with respect to its unique structure as an HDAC inhibitor has yet to be thoroughly elucidated.
The key challenges in preparing FK228 are: (1) formation of the delicate macrocyclic depsipeptide and, (2) preparation of the β-hydroxy mercapto heptenoic acid (5) intermediate. Simon and co-workers reported in their total synthesis of FK228 that a Mitsunobu-based macrolactonization provides a good yield of the 16-membered cyclic depsipeptide.4 The other key challenge, the synthesis of β-hydroxy acid 5, has been addressed by two other research groups.5,6,12 Acid 5 is a key intermediate for the synthesis of FR-901375 and spiruchostatin A. Simon applied Carreira's catalytic asymmetric aldol reaction to establish the C3-hydroxyl stereochemistry (99% yield, >98% ee).4 In the course of synthesizing FR-901375 (2) Wentworth and Janda noted that they encountered difficulties reproducing the enantioselectivity in the key Carreira-type asymmetric aldol reaction.5 Likewise, Ganesan noted the same difficulty in their recent synthesis of spiruchostatin A.6 Both groups developed alternative asymmetric aldol approaches to 5. Wentworth and Janda mimicked an acetate aldol by using 2-chloro-acetyl-oxazolidinone in an asymmetric aldol reaction. This provided 5 in six steps and 26% yield from commercially available materials. Although efficient, it requires chromatographic separation of diastereomers (>90% de), which may be tedious on a large-scale. Ganesan's synthesis of 5 using a SmI2-promoted Reformatsky reaction of an N-(α-bromoacetyl)-oxazolidin-2-one (Seebach's chiral auxiliary) and also mandates chromatographic separation of diastereomers (86% de).12 These workers obtained 5 in nine steps and approximately 9.5% yield from commercially available materials.13 It is apparent from the literature, that compound 5 is a deceptively challenging substance to access on a preparative scale. We report herein, a catalytic, enantioselective, and scaleable synthesis of β-hydroxy acid 5, which is comparable in the number of steps to that reported by others and in our hands, constitutes a robust and reproducible method.
Our synthesis of FK228 follows the pioneering route established by Simon for completion of the synthesis, in which the disulfide of FK228 is disconnected to the trityl-protected bis-mercaptan 3, followed by opening of the macrocyclic depsipeptide at the lactone. Intermediate 3 is further disconnected to reveal tetrapeptide 4 and β-hydroxy mercapto acid 5 (Scheme 1). Our synthesis of 5 involves late-stage mercaptan installation and conversion of a dimethyl acetal to the desired acid. The asymmetric allylic alcohol is derived from a propargylic ketone (7) by a Noyori catalytic asymmetric ketone reduction and an (E)-selective alkyne reduction.
Scheme 1. Retrosynthetic Analysis.

Our synthesis of β-hydroxy mercapto acid 5 commences with the preparation of amide 9 from commercially available methyl 3,3-dimethoxy propionate (Scheme 2). Standard conditions14 provide Weinreb amide 9. Addition of the lithium acetylide of 10 to amide 9 gives propargylic ketone 11 in a straightforward manner.
Scheme 2. Preparation of propargylic ketone 11.

Noyori's asymmetric hydrogen transfer conditions reduced ketone 11 to the (R)-propargylic alcohol in 71% yield and 99:1 er as determined by HPLC.15 Treatment with sodium bis(2-methoxyethoxy) aluminum hydride (Vitride®) selectively affords (E)-alkene 13.16 It was determined that conversion of the silyl ether to tosylate 14 must be done prior to dimethyl acetal hydrolysis. Similarly, oxidation of the mercaptan is avoided by revealing the carboxylic acid before introduction of the trityl mercaptan. Therefore, the dimethyl acetal of tosylate 14 was hydrolyzed to the aldehyde using LiBF4, which was immediately oxidized to the carboxylic acid using Pinnick oxidation conditions.17 Finally, the trityl mercaptan was introduced by tosylate displacement to provide 5 in nine steps (13.3% overall yield) from commercially available materials and has been prepared on a multi-gram scale.
We initially envisioned preparing tetrapeptide 4 using Simon's strategy. Dipeptide 15, prepared as previously described, was to be coupled with N-Fmoc-d-Cysteine(STrt). However, N-Fmoc- d-Cysteine(STrt) could only be obtained in 16% yield from d-Cysteine. Furthermore, in our hands the overall yield using the existing synthetic route starting from 15, N-Fmoc-d-Cysteine(STrt), Fmoc-l-Thr, and Fmoc-d-Val resulted in a mere 23% yield of 4 rather than the reported 78% overall yield. The most significant decrease in yield was observed for the simultaneous threonine dehydration-Fmoc deprotection step, in which we obtained a 39% yield of 4 and could not realize the reported 96% yield. To address these low yielding and irreproducible steps, we then examined an alternative protecting group strategy. Fortunately, N-Alloc-d-Cysteine(STrt) can be obtained in high yield from d-Cysteine as described by Kruse,18 which prompted us to investigate its use in the synthesis of 4 (Scheme 4). Coupling of N-Alloc-d-Cysteine(STrt) with l-Thr-l-Val affords a 67% yield of 16. We observed an improved yield (95% over two steps) by dehydrating the threonine residue of tripeptide 16 earlier in the synthesis, prior to coupling with N-Fmoc-d-Valine. The N-Alloc deprotection and coupling with N-Fmoc-d-Val proceeded smoothly to provide 10 grams of tetrapeptide 18. Deprotection of the terminal nitrogen quantitatively affords tetrapeptide 4 in seven steps and 53% overall yield from 15 and readily available amino acid derivatives.
Scheme 4. Synthesis of tetrapeptide 4.

With tetrapeptide 4 and β-hydroxy mercapto acid 5 in hand we were poised to complete the synthesis of FK228. Following Simon's precedent, the two portions were coupled using BOP to provide 19 in quantitative yield. After methyl ester hydrolysis, the linear FK228 precursor was subjected to known Mitsunobu macrolactonization conditions. Despite extensive effort, we were unable to realize the 62% yield reported4 for this cyclization. Depsipeptide 3 was isolated in 24% yield after multiple purifications to remove triphenyl phosphine oxide. It was treated with I2/MeOH, which promoted disulfide formation and afforded FK228 (1).
In conclusion, an improved synthesis of FK228 (1) has been completed from simple, commercially available materials. An efficient, multi-gram scale synthesis of the tetrapeptide portion (4), using N-Alloc-d-cysteine, was developed. The key β-hydroxy mercapto acid (5), was prepared in nine steps and >99:1 er via an asymmetric Noyori hydrogen transfer reaction and late-stage mercaptan introduction. The weak step in this synthesis, which follows the pioneering route first developed by Simon,4 is the macrolactone-forming cyclization reaction. We are currently investigating alternative conditions and synthetic strategies for closing the bicyclic ring system with the objective of further improving the throughput to the natural product.
Supplementary Material
Scheme 3. Preparation of β-hydroxy mercapto acid 5.

Scheme 5. Completion of the synthesis of FK228 (1).

Acknowledgments
This paper is dedicated to the memory of Professor Albert I. Meyers. We thank the National Institutes of Health (GM068011) for financial support. DMJ thanks the American Cancer Society, New England Division Broadway on Beachside for a postdoctoral fellowship. We also acknowledge Dr. Julian Simon of the Fred Hutchinson Cancer Research Center for many helpful discussions and for graciously providing additional experimental details.
Footnotes
Supporting Information Available Experimental procedures and characterization of all key compounds. This material is available free of charge via the Internet at http://www.pubs.acs.org.
References
- 1.Fujisawa Pharmaceutical Co., Ltd. Jpn. Kokai Tokkyo Koho JP. 1991 03141296. [Google Scholar]
- 2.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 3.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Clin Cancer Res. 2006;12:3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]
- 4.Li KW, Wu J, Xing W, Simon JA. J Am Chem Soc. 1996;118:7237–7238. [Google Scholar]
- 5.Chen Y, Gambs C, Abe Y, Wentworth P, Jr, Janda KD. J Org Chem. 2003;68:8902–8905. doi: 10.1021/jo034765b. [DOI] [PubMed] [Google Scholar]
- 6.Yurek-George A, Habens F, Brimmell M, Packham G, Ganesan A. J Am Chem Soc. 2004;126:1030–1031. doi: 10.1021/ja039258q. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. J Antibiot. 1994;47:301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]; (b) Shigematsu N, Ueda H, Takase S, Tanaka H, Yamamoto K, Tada T. J Antibiot. 1994;47:311–314. doi: 10.7164/antibiotics.47.311. [DOI] [PubMed] [Google Scholar]; (c) Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K. J Antibiot. 1994;47:315–323. doi: 10.7164/antibiotics.47.315. [DOI] [PubMed] [Google Scholar]
- 8.Somech R, Izraeli S, Simon AJ. Cancer Treat Rev. 2004;30:461–472. doi: 10.1016/j.ctrv.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Marks PA, Richon VM, Miller T, Kelly WK. Adv Cancer Res. 2004;5:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi Y, Ohtsuki M, Murakami T, Kobayashi T, Sutheesophon K, Kitayama H, Kano Y, Kusano E, Nakagawa H, Furukawa Y. Oncogene. 2006;25:512–524. doi: 10.1038/sj.onc.1209072. [DOI] [PubMed] [Google Scholar]
- 11.TSA A analogs. Woo SH, Frechette S, Khalil EA, Bouchain G, Vaisburg A, Bernstein N, Moradei O, Leit S, Allan M, Fournel M, Trachy-Bourget MC, Li Z, Besterman JM, Delorme D. J Med Chem. 2002;45:2877–2885. doi: 10.1021/jm020154k.SAHA analogs. Remiszewski SW, Sambucetti LC, Atadja P, Bair KW, Cornell WD, Green MA, Howell KL, Jung M, Kwon P, Trogani N, Walker H. J Med Chem. 2002;45:753–757. doi: 10.1021/jm015568c. FK228 analogs: Yurek-George A, Cecil ARL, Mo AHK, Wen S, Rogers H, Habens F, Maeda S, Yoshida M, Packham G, Ganesan A. J Med Chem. 2007;50:5720–5726. doi: 10.1021/jm0703800.
- 12.Doi T, Iijima Y, Shin-ya K, Ganesan A, Takahashi T. Tetrahedron Lett. 2006;47:1177–1780. [Google Scholar]
- 13.The yield for N-acylation of Seebach's chiral auxiliary was not reported. A yield of 80% was reported for a similar N-acylation in the following article: Fukuzawa Si, Matsuzawa H, Yoshimitsu Si. J Org Chem. 2000;65:1702–1706. doi: 10.1021/jo9914317.
- 14.Denmark SE, Fujimori S. J Am Chem Soc. 2005;127:8971–8973. doi: 10.1021/ja052226d. [DOI] [PubMed] [Google Scholar]
- 15.(a) Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J Am Chem Soc. 1997;119:8738–8739. [Google Scholar]; (b) Haack KJ, Hashiguchi S, Fujii A, Ikariya T, Noyori R. Angew Chem Int Ed. 1997;36:285–287. [Google Scholar]
- 16.Jones TK, Denmark SE. Org Syntheses, Coll. Vol. 7. John Wiley & Sons; New York: 1990. p. 524. [Google Scholar]; 1986;64:182–186. [Google Scholar]
- 17.(a) Wong LSM, Sherburn MS. Org Lett. 2003;5:3603–6. doi: 10.1021/ol0353058. [DOI] [PubMed] [Google Scholar]; (b) Raach A, Reiser O. J Prakt Chem. 2000;342:605–608. [Google Scholar]
- 18.Kruse CH, Holden KG. J Org Chem. 1985;65:1192–1194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.