

Abstract
Next-generation sequencing provides technologies which sequence whole prokaryotic and eukaryotic genomes in days, perform genome-wide association studies, chromatin immunoprecipitation followed by sequencing and RNA sequencing for transcriptome studies. An exponentially growing volume of sequence data can be anticipated, yet functional interpretation does not keep pace with the amount of data produced. In principle, these data contain all the secrets of living systems, the genotype–phenotype relationship. Firstly, it is possible to derive the structure and connectivity of the metabolic network from the genotype of an organism in the form of the stoichiometric matrix N. This is, however, static information. Strategies for genome-scale measurement, modelling and predicting of dynamic metabolic networks need to be applied. Consequently, metabolomics science—the quantitative measurement of metabolism in conjunction with metabolic modelling—is a key discipline for the functional interpretation of whole genomes and especially for testing the numerical predictions of metabolism based on genome-scale metabolic network models. In this context, a systematic equation is derived based on metabolomics covariance data and the genome-scale stoichiometric matrix which describes the genotype–phenotype relationship.
Keywords: Genotype–phenotype relationship, Systems biology, Metabolomics, Proteomics, Genome annotation, Gene models, Metabolic modelling, Flux balance analysis, Dynamic modelling, Stochastic differential equations, Covariance, Principal components analysis, Phenotypic plasticity
Introduction
We have witnessed an exponential growth of public genome sequence releases (http://www.genomesonline.org/). In principle, this amount of data will enable us to investigate any subtle aspect in living systems. However, the process of whole-genome assembly and functional gene annotation of de novo sequenced organisms is far behind the speed of data generation using next-generation sequencing (NGS) technologies [1–4]. Whole genome assembly and ab initio gene prediction is in the first instance dependent on algorithms. In recent studies, approaches have been presented for functional annotation of newly sequenced genomes combining complementary DNA [expressed sequence tag (EST), messenger RNA or RNA-sequencing data] with gene predictions [5]. More recently, proteogenomic studies have used proteomics data to reveal new gene models [6–8]. A truly systems biology approach is the integration of several layers of molecular information in conjunction with metabolic modelling [7]. In this study, genome annotation with metabolomics data and a structural modelling approach were combined for the first time [7].
Besides the qualitative or structural investigation of genome function and metabolic networks, the next aim is to explore the quantitative prediction. Here, dynamic modelling is the key approach. The final goal is genome-scale metabolic reconstruction and quantitative understanding and prediction of metabolism in a newly sequenced organism, the genotype–phenotype relationship. By reviewing the literature it becomes clear that this is the limiting step in the functional interpretation of whole genomes and organisms. Only an iterative cycle of improving genome annotation, structural and dynamic modelling and comparison of the predictions with experimental data will be successful, necessitating not only further development of computer-based annotation of gene functions and modelling algorithms but also the integration of whole metabolome profiling approaches. In the next sections I will explore the strategies and limitations of how to connect metabolomics data and genome-derived metabolic reconstruction and suggest a complete workflow. A systematic equation is derived for the genotype–phenotype relationship.
Next-generation sequencing, gene prediction and functional annotation
Recent developments in bioanalytical chemistry have led to an ongoing replacement of classical Sanger DNA sequencing technology. Sanger sequencing is one of the first-generation DNA-sequencing techniques which resulted in monumental achievements, such as the first human genome sequence. This technique provides long sequence reads and belongs to the high-quality methods (see later). Drawbacks are high costs and a relatively low throughput [9]. The demand for rapid and cost-effective sequencing technologies and a consequent funding policy for method development has led to the development of several alternative approaches which are different in the use of genomic template libraries, the number of reads, the read length, genome coverage, the scale of the application and many other parameters (for an overview, see [9]). More important, NGS platforms have dramatically increased the throughput and substantially lowered reagent costs. As a result of these developments, the limitations of DNA sequencing shifted from the hardware to the software. The strongest drawbacks of any of these technologies are short read lengths compared with Sanger sequencing (454/Roche approximately 400 bases; Illumina/ABI-SOLiD approximately 60–100 bases; Sanger sequencing approximately 1,000 bases [1–4]) as well as different error characteristics. As a result, the assembly of genome sequences from these short reads is difficult and demands high computer power, novel algorithms and partial complementation and verification with high-quality sequencing strategies such as third-generation long-read technology or Sanger sequencing [10–12].
After or during genome sequence assembly, gene prediction and functional annotation are the concomitant steps. Ab initio gene prediction allowing constraints is being increasingly favoured [5, 13]. Here, especially NGS transcriptomics data can be used for gene prediction and functional annotation. Longer contig and singleton sequences are assembled from short reads and analysed for homology with sequences in public databases using BLAST algorithms. Assembled contigs and singletons are subsequently translated into peptides and annotated with biological function using a homology search against various public databases [12].
Proteomics data can also be exploited for gene prediction and functional gene annotation in fully sequenced organisms [6–8, 14–16]. Here, very large proteomics datasets covering up to 60% or more of the predicted proteome are matched against genomics databases, especially six frame translations, to discover novel peptides which are not predicted by the assembled and functionally annotated genome sequence because of wrongly annotated intron–exon boarders or completely missing annotations.
Recently, we have also used metabolomics data for functional annotation of the newly sequenced organism Chlamydomonas reinhardtii [7]. The comparison of all metabolites with the reconstructed metabolic network of Chlamydomonas reinhardtii revealed missing reactions. This observation was combined with a structural modelling approach and demonstrated that several metabolites cannot be synthesized or generated with the existing metabolic draft network of Chlamydomonas reinhardtii, pointing towards missing reactions or alternative pathways.
The quality of full genome annotation depends on the functional characterization of orthologous genes in other organisms. With sequence homology, a function can only be postulated. Gene functions are usually derived by classic biochemical studies such as complementary assays, cloning, enzyme substrate and activity tests, protein interaction tests and gene knockouts or conditional knockouts in the organism of interest. Many of the data obtained are assembled in databases such as STRING (http://string-db.org/) and can be systematically searched for any gene sequence. Furthermore, the function of a gene can be estimated if functional domains such as ATP-binding sites or protein kinase domains can be characterized (see [17] and http://pfam.sanger.ac.uk/). However, one has to be aware that a gene function prediction by homology with other orthologues is only a first step and needs further confirmation.
After gene prediction and annotation of a newly sequenced genome, the next step in the functional understanding of the organism is the reconstruction of the metabolic network, the metabolism specific to this species. Nowadays this is a straightforward and routine procedure. The procedure is described in the following section.
Ab initio prediction of metabolic networks from full genome sequences—static genotype information needs to be translated into dynamic molecular phenotype information
The workflow to predict an initial metabolic network from an annotated genome sequence is shown in Fig. 1. First, the NGS short reads are assembled to form a complete genome sequence and these sequences are analysed for intron/exon structure, start and stop codons and homology with known sequences (see “Next-generation sequencing, gene prediction and functional annotation”). After a genome-scale functional annotation based on the homology with functionally characterized genes from other organisms, a gene list is assembled. On the basis of this gene list, enzymatic reactions are postulated. Educts and products participate in an enzymatic reaction. Pathways are structured so that the product of the former enzymatic reaction is the educt of the next enzymatic reaction, for instance in glycolysis. Thus, the list of reactions can be mapped to existing knowledge of pathways. Fragmentary pathways can be filled up with reactions if the corresponding gene is not annotated in the genome sequence. On the basis of this reaction list, a stoichiometric matrix can be built, also known from chemical reaction lists. Only one principle applies here, which is mass conservation, e.g. one molecule of glucose is converted into two molecules of triose phosphate. In Fig. 2 the principles of generating a stoichiometric matrix from a list of coupled enzymatic reactions are exemplified. These principles can be extended to whole-genome-scale metabolic reconstruction (see “Modelling approaches for metabolic networks” and [18, 19]). On the basis of this stoichiometric matrix, a metabolic network can be postulated for an organism (Fig. 1). Nowadays, the whole workflow can be automated [18].
Fig. 1.

The static genotype. The complete workflow for ab initio prediction of metabolic networks from genome sequences. Central information is the functional annotation of genes based on homology with genes from other organisms and the corresponding stoichiometric matrix N. The resulting reconstructed metabolic network provides not dynamic but static information (for details, see text)
Fig. 2.

Construction of a stoichiometric matrix. A list of genes is identified to encode enzymes for reactions v1–v3 and metabolites M1–M4 (i). The corresponding enzymatic reaction network is shown in ii. The reaction rates of metabolites M1–M4 are expressed as the product of the flux vector and the stoichiometric matrix N (iii). The stoichiometric matrix N is derived directly from the gene list and the predicted pathways
Although the strategy is very sound, there are several obstacles which have to be clarified. First, this metabolic network reconstruction produces static information. It cannot be assumed that all reactions are present or active at the same time, so we have to measure the active pathway network. The active pathway network is the short- and longterm molecular response of the organism to environmental perturbations, for instance a plant will change its metabolic activity in a day-night-rhythm or adapt in a longterm behaviour to environmental conditions [20] (see Fig. 3). Information on this active metabolic network can be achieved by integrative approaches combing metabolomics, proteomics and transcriptomics data as well as metabolic flux analysis [21–25], and this is further discussed later.
Fig. 3.

The dynamic phenotype. Principal components analysis of metabolite profiles of Arabidopsis thaliana showing an oscillating molecular phenotype in a day–night rhythm (i) (for further details, see Morgenthal et al. [20]). This analysis demonstrates that the same genotype will produce all sorts of different molecular phenotypes, indicating the plasticity of metabolism as a direct result of the genotype-phenotype relationship. The main task is to predict such behaviour from the genotype (for more details, see text). Another example of variable molecular phenotypes based on metabolite profiling in five different plant species is shown in ii. Metabolites were measured by gas chromatography coupled with time-of-flight mass spectrometry and analysed with independent components analysis (ICA). Each species is clearly distinguished from the others. All these species also show different metabolic responses to biodiversity. The most pronounced effects of this phenotypic plasticity are found in distinct metabolite markers for C/N partitioning of central metabolism (for more information, see [28] and text)
Second, there is a strong bias for metabolic network reconstruction based on our classical biochemical knowledge, the annotated gene functions in public databases and our incompetence to characterize a gene functionally without any homology match in the databases. In other words, one might observe that the reconstructed metabolic networks look very similar, although we would expect exactly the opposite. This was indeed observed in a comparative network topology study of metabolic networks from 43 organisms showing similar diameters [26]. At that time, in fact, the databases of reaction pathways, metabolic networks and genome sequences were only fragmentary; one should be aware that all our classical hypothesis-driven research is strongly biased by our present knowledge [27]. This, on the other hand, is a strong argument for the application of unbiased omics measurements, especially metabolomics with respect to metabolic networks. In Fig. 3, metabolite measurements of a recent environmental study are shown. Five different plant species were analysed by gas chromatography coupled with time-of-flight mass spectrometry (GC-TOF-MS) and independent components analysis [28]. All the plant species were classified differently using the same set of identified and quantified metabolites. These sets of altered metabolites are physiological markers pointing to various regulations in plant metabolism depending on the genotype.
In summary, next-generation genome sequencing and metabolic reconstruction will reveal static metabolism, yet the measurements demonstrated how dynamic and different these species are in their metabolic response to the environment [28]. Consequently, the combination of (1) systematic metabolite measurements and (2) modelling approaches which can predict dynamic metabolism on the basis of genome annotation and metabolic network reconstruction is urgently needed. In the following section, modelling approaches are briefly summarized.
Modelling approaches for metabolic networks
The initial phase of prediction for species-specific metabolism requires an understanding of the metabolic network or better the complete picture of metabolism in the targeted biological system. The genome-derived stoichiometric matrix N (see the previous section) provides the “structure” of the metabolic network and is the basis for almost all modelling approaches. Thus, structural analysis identifies entry points for the ab initio-modelling of pathway- and genome-derived metabolic networks. A plethora of methods exist to address structural modelling, kinetic modelling and control of metabolism, namely flux balance analysis (FBA) and elementary flux modes, kinetic modelling using complex differential equations and kinetic constants, and metabolic control analysis (for a review, see [29]).
The classic approach of modelling metabolic systems is the computer-based simulation of time-dependent metabolite concentrations using ordinary differential equations [30]. Kinetic modelling is severely hampered by the lack of knowledge of in vivo kinetic rate laws and enzymatic parameters, and is thus only applied in small-scale networks with well-characterized enzymatic reactions. Examples of these pathways are the glycolytic pathway and the red blood cell [31–36].
Predictions of metabolite concentrations from these modelling approaches have partly matched experimental data. For a genome-scale metabolic network prediction, however, there are still too many enzymatic parameters missing. A promising approach here is inverse parameter estimation (for a review, see [37]).
FBA provides a framework for metabolic reconstruction and constraint-based metabolic flux analysis of an organism without the need for detailed kinetic modelling [38–42].
On the basis of the reconstructed metabolic network for a particular organism derived from its genome sequence and bioinformatics (gene predictions, functional assignments based on homology) and experimental annotation (e.g. EST sequences, proteomics measurements) and arresting mass balance, it is relatively straightforward to write down all reactions and processes that alter the concentrations of a metabolite based on the stoichiometry of the metabolic reactions called the stoichiometric matrix N (see also the previous section and Fig. 2).
The steady-state solutions (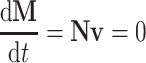 , where M is the matrix of metabolite concentrations, t is time, N is the stoichiometric matrix and v is a vector including all fluxes—metabolic, transport and usage fluxes) of this postulated metabolic network can be obtained by linear mathematics assuming mass conservation and constraints such as optimized biomass production or metabolite secretion, or both [43, 44].
, where M is the matrix of metabolite concentrations, t is time, N is the stoichiometric matrix and v is a vector including all fluxes—metabolic, transport and usage fluxes) of this postulated metabolic network can be obtained by linear mathematics assuming mass conservation and constraints such as optimized biomass production or metabolite secretion, or both [43, 44].
Kinetic modelling and FBA will reveal metabolite concentrations and metabolic fluxes, respectively, by numerical simulations solving the steady-state solutions of a metabolic network. Thus, in principle, it is possible to generate the complete stoichiometric matrix N from a newly sequenced and assembled genome of an organism by NGS and subsequently define metabolite dynamics in this postulated network. Several large-scale projects focus on this strategy [18, 19, 45].
However, the reality check reveals the complexity of such an approach. Most of the published metabolic network reconstructions of model organisms undergo permanent optimization and improvement for decades [46] using biochemist experts’ knowledge, proteogenomic methods [6, 7, 14] and supplementation of genome sequences with RNA sequencing and EST data [5, 47].
Most important, all these simulations of metabolite dynamics provide information of only a snapshot of the system. Any transitions of the organisms due to environmental perturbations will result in changes of the active pathway/regulation network (see Fig. 3); thus, they will also change the enzymatic and regulatory reaction rates. The most direct readout of this phenomenon is the measurement of metabolite concentrations and fluxes, in genome-scale metabolomics measurements. To test computer simulations in a useful manner, we need these metabolomics measurements also to reveal transient behaviour. Therefore, the combination of NGS, metabolic network reconstruction and metabolomics science—the quantitative measurement of metabolism and metabolic modelling—seems to be most suitable.
Proving the predictions—metabolomics science
Bioanalytical methods in metabolomics science provide the most direct tools for the quantitative measurement of metabolism in an organism (for reviews, see [48, 49]). The physicochemical diversity of small biological molecules in a biological organism still exceeds our analytical capacities, and the general estimation of the size and dynamic range of a species-specific metabolome is at a preliminary stage. In the plant kingdom the structural diversity is enormous, with new compounds being revealed on a daily basis. Estimates exceed five million putative structures. A combination of analytical techniques has to be used to cope with such diversity [48]. Mass spectrometry is one of the technologies which has developed rapidly and also revolutionized the field. In Table 1 different “hyphenated” technologies are presented. Each different technique provides different features. Therefore, it can be expected that by combining different technologies, we will substantially increase the coverage of a metabolome. Metabolic fingerprinting techniques using, for instance, NMR or IR spectroscopy achieve a high sample throughput and provide a global view on in vivo dynamics of metabolic networks [48, 50]. One of the gold standard techniques in terms of sample throughput, comprehensiveness and accuracy in metabolite identification is gas chromatography coupled with mass spectrometry [20, 51–56].
Table 1.
Mass analyzers, “hyphenated” techniques and their performances
| Mass analyzer | Ionization technique | Chromatography | Scan modes | Speediness/sensitivity/mass accuracy |
|---|---|---|---|---|
| Quadrupole | ESI, EI, CI, FI | GC, CE, LC | Full scan | Scan speed slow, faster with SIM mode, but mass range restricted |
| APCI, APPI | SIM | |||
| Triple quadrupole | ESI | GC, CE, LC | Full scan | Full scan slow |
| MRM very fast and sensitive | ||||
| APCI, APPI | SIM | |||
| MS2 | ||||
| SRM/MRM | Exact masses with internal calibration | |||
| MALDI | ||||
| Triple quadrupole linear trap | ESI | CE, LC | Full scan | Full scan medium |
| APCI, APPI | ||||
| MALDI | MS2, MS3 | |||
| SRM/MRM | ||||
| MS3 possible | ||||
| Ion trap | ESI, EI, CI | GC, CE, LC | Full scan | As for above |
| SIM | ||||
| APCI, APPI | ||||
| MS2, MSn | ||||
| MALDI | ||||
| Linear ion trap | ESI | CE, LC | Full scan | Very fast and sensitive full scan |
| APCI, APPI | SIM | |||
| MS2, MSn | Rest as above | |||
| MALDI | ||||
| TOF | ESI, EI, FI | GC, CE, LC | Full scan | Very sensitive full scan |
| APCI, APPI | ||||
| Exact masses with internal calibration | ||||
| Source fragmentation | ||||
| MALDI | ||||
| Quadrupole TOF | ESI | CE, LC | Full scan | Most sensitive full scan |
| Exact masses with internal calibration | ||||
| APCI, APPI | ||||
| MS2 | ||||
| MALDI | ||||
| Resolution 20,000 | ||||
| Orbitrap | ESI, | LC | Full scan | Exact masses (<2 ppm) without internal calibration |
| APCI, APPI | ||||
| MS2, MSn | ||||
| Resolution 100,000 | ||||
| MALDI | ||||
| FTICR | ESI, EI, FI | LC | Full scan | Exact masses (<1 ppm) without internal calibration |
| APCI, APPI | ||||
| Resolution 1,000,000 | ||||
| MS2, MSn | ||||
| MALDI |
TOF Time of flight, FTICR Fourier transform ion cyclotron resonance, ESI Electrospray ionization, EI Electron impact, CI Chemical ionization, FI Field ionization, APCI Atmospheric pressure chemical ionization, APPI Atmospheric pressure photoionization, MALDI Matrix-assisted laser desorption ionization, GC Gas chromatography, CE Capillary electrophoresis, LC Liquid chromatography, SIM Single ion monitoring, MS Mass spectrometry, SRM Single reaction monitoring, MRM Multiple reaction monitoring
A very recent development is the use of two-dimensional gas chromatography coupled with fast acquisition rate time-of-flight mass spectrometry (GC × GC-TOF-MS). The online coupling of two gas chromatography columns with different functionality, for instance a first, long hydrophobic and a second, short polar column, increases the separation efficiency of a complex metabolomic sample and improves spectral quality after deconvolution. However, the deconvolution process from such extended two-dimensional raw chromatograms is very complicated. Moreover, metabolite identification and data alignment is the bottleneck. Recently, we presented a complete strategy to perform a convenient data extraction and alignment using two-dimensional gas chromatography coupled with mass spectrometry (GCxGC-MS) technology [25]. Especially important is the introduction of a second retention index which can be used to increase the confidence in metabolite identification. One of the most promising platforms for metabolomics is the combination of gas chromatography coupled with mass spectrometry (GC-MS) and liquid chromatography coupled with mass spectrometry (LC-MS) (see Fig. 4) [28]. Because of the specific technology, both technologies provide a complementary view of the metabolome [28]—central metabolites such as amino acids, sugars, organic acids and free fatty acids by GC-MS, and higher molecular masses, e.g. secondary metabolites, cofactors and sugar phosphates by LC-MS. In Fig. 4 such a platform is shown combining GCxGC-MS and LC-MS for metabolome analysis. However, the reader should be aware that most of the metabolomics platforms still need further method validation and daily quality checks. This is an essential requirement to guarantee meaningful biological applications. Furthermore, improvement of databases, experimental standards and data exchangeability between laboratories is an urgent issue for further developments in metabolomics [57] (see “Metabolome coverage has to be improved by the combination of different analytical procedures, international cooperation and open source databases and software”).
Fig. 4.

Metabolomic platform combining the techniques of gas chromatography coupled with mass spectrometry (GC MS) and liquid chromatography coupled with mass spectrometry to cope with metabolomic complexity in biological systems [28]. GC MS is one of the current “gold standards” with respect to comprehensiveness, sample throughput and identification rates [52]. Two-dimensional gas chromatography (CC × GC) coupled with fast acquisition rate time-of-flight mass spectrometry increases further the resolution of ultracomplex metabolome samples [25] High-mass-accuracy/high-resolution mass spectrometry emerges in parallel with high-resolution ultraperformance liquid chromatography (UPLC) and increases the detection capacities by orders of magnitude. Data can be combined in one data matrix to reveal the covariance matrix for the detection of physiological biomarkers, metabolite correlation networks and network topologies (see the text for further details and [20, 21, 28, 58, 59]). MS mass spectrometry, RI retention index
In Fig. 5 the classic chemometric approach for the analysis of complex metabolomic data sets is shown. The analysis of hundreds of biological replicates of an organism under different controlled environmental treatments in the natural environment or with genotypic variation will result in a complex data matrix. This data matrix can be analysed by classic multivariate or univariate statistical tools, supervised or unsupervised methods (for an overview, see [60]. One of the central results of such an experimental design is the covariance matrix C of metabolite concentrations or fluxes [20, 22, 59, 60].
Fig. 5.

Chemometric workflow for the analysis of ultracomplex metabolome datasets. A multitude of samples are analysed and the complete set of samples versus detected, and quantified metabolites are assembled into a data matrix. Subsequently, the data are analysed by classic multivariate statistical tools (for more details, see [60]). The backbone of many of these supervised and unsupervised statistical tools is the covariance matrix C (or the normalized covariance matrix, resulting in a correlation matrix) of the metabolite concentrations (for in-depth analysis of the covariance/correlation matrix of metabolite concentrations, see [20–22, 58, 59]). For the systematic connection between C and the stoichiometric matrix of a genome-scale network, see “A systematic genotype–phenotype equation: connecting metabolomics covariance data (C) and genome-scale metabolic network reconstruction (N)”
The major question is now how this covariance matrix C of metabolite concentrations and fluxes is related to the underlying metabolic network. I will address this question in the next sections and will also demonstrate that there is a direct relationship between the covariance matrix C of metabolite concentrations and fluxes and the genome-scale reconstruction of the underlying metabolic network.
A systematic genotype–phenotype equation: connecting metabolomics covariance data (C) and genome-scale metabolic network reconstruction (N)
Recently, we proposed a systematic approach to connect the observed covariance matrix C of metabolite concentrations with the underlying biochemical system and the corresponding genotype, respectively [21, 58]. This relationship is characterized by the following equation [61]:
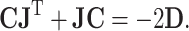 |
1 |
Here, J is the Jacobian matrix (for the relationship between metabolic networks and the Jacobian, see [62]), D is the fluctuation or diffusion matrix (for more information, see [64]), the diagonal entries D ii characterize the magnitude of fluctuations of each metabolite, whereas off-diagonal entries D ij (i ≠ j) represent the fluctuation of metabolites caused by the interaction between enzymes i and j, and C is the covariance matrix obtained from the metabolite concentrations (see “Proving the predictions—metabolomics science” and Figs. 4 and 5). The entries of the Jacobian represent the elasticities of reaction rates (via enzymes or other regulatory principles) to any change of the metabolite concentrations. On the basis of the Jacobian solution, one can estimate differences in biochemical regulation in the system. The Jacobian itself is characterized by the following equation [62]:
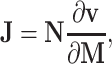 |
2 |
where N is the stoichiometric matrix of the metabolic network derived from a genome sequence (see “Ab initio prediction of metabolic networks from full genome sequences” and Figs. 1 and 2), v are the rates for each reaction, and M are the concentrations of each metabolite in vector notation (for details of Eq. 2, see [62]). Equations 1 and 2 together provide a conceptual basis for treating the observed covariance matrix C of metabolite concentrations (see Fig. 5) as the dynamic molecular phenotype related to the genotype characterized by the genomic stoichiometric matrix N (see Figs. 1 and 2). Consequently, this equation is the systematic description for the genotype–phenotype relationship.
If the covariance matrix of metabolite concentrations is measured (see “Proving the predictions–metabolomics science” and Figs. 4, 5), Eq. 1 is furthermore the conceptual basis for the estimation of the Jacobian in the dynamic genome-scale metabolic network using reverse or inverse modelling and optimization approaches (for a review of inverse modelling approaches, see [37]). The Jacobian entries reflect regulatory properties, the elasticities of the reaction rates to any change in the metabolite concentrations as discussed above. Any solution of the Jacobian is therefore a signature of metabolic—and phenotypic—plasticity of the corresponding genotype. In Fig. 3 an example of metabolic plasticity is given. In principal components analysis, the day–night plasticity trajectory is visualized.
Many limitations, however, have to be overcome to apply this principle in a routine manner. Metabolomics data cover, by definition, many different pathways and—in the optimal case—represent a genome-scale metabolism. However, owing to the restrictions of analytical methods, only a fraction of all the metabolites present are typically identified and quantified. In the following section I will describe the limitations of classic and advanced analytical methods for metabolomics and future strategies of how to increase metabolome coverage.
Metabolome coverage has to be improved by the combination of different analytical procedures, international cooperation and open source databases and software
A major limitation of metabolomics science is the vast amount of detected but structurally not characterized or putatively classified “features”, a chromatographic peak in LC-MS analysis, an m/z ratio or complex mass spectrum or a chemical shift in NMR analysis. This is accompanied by the habit in literature, especially in the abstract of the full study, to count detected “features” as metabolites, somewhat hiding the fact that only 30–50% or fewer are indeed identified chemical structures. Although it is fair to assume that detected features in a complex mixture of a metabolomics sample are indeed real metabolites, we have to be aware that analytical procedures also produce many artefacts. In recent work by Giavalisco et al. [63], a reality check was performed by using plants fully labelled with 13C and high-accuracy mass spectrometry analysis. On the basis of this experimental setup, metabolite “features” with 13C incorporation were distinguished from unlabelled 12C “features” in the analysis, indicating thousands of analytical artefacts, chemical or electronic noise. From 20,000 to 40,000 detected m/z signals in positive electrospray ionization mass spectrometry and negative electrospray ionization mass spectrometry analysis, about 1,000–3,000 peaks gave database hits using the exact mass for the generation of a chemical formula. However, only 1,024 13C/12C m/z pairs were identified from all the spectra, leading to unambiguous database hits. The results are more than challenging with respect to separate chemical artefacts from real metabolites.
Another typical example of how misleading numbers can be is typical practice in GC-MS analysis. Although not so sensitive to contamination as electrospray ionization mass spectrometry, in a classic untargeted approach 1,000 or even 3,000 (GC-TOF-MS and GC × GC-TOF-MS analysis, respectively) “features” or deconvoluted spectra can be detected. From these only a small fraction can be structurally identified using reference compound libraries and spectral matching procedures. The remaining “metabolites” are characterized by spectra which can be found reproducibly in the GC-MS analyses, however without unambiguous identification. In summary, the number of reproducibly identified and quantified metabolites in a batch of samples is in the range of 100–120, 200 perhaps in a single sample. Indeed, these numbers are the average identification rates using the GC-MS analysis demonstrated in many studies. This low identification rate is mainly due to the inherent strategy used for metabolite identification from GC-MS data. After a complex deconvolution process of the gas chromatography–electron impact-coupled with mass spectrometry data, mass spectra are reconstructed and sent to a library of reference spectra of chemically known compounds [65]. Thus, the identification rate depends on the size and quality of the library. There is a strong need to extend and to combine existing libraries such as the NIST [66], GMD [67] and the FiehnLib [68] libraries to enable higher identification rates. One approach would be to complement chemical structures with chemical synthesis. The whole approach is also dependent on the quality of the software. Therefore, further development of algorithms, software tools and databases for the interpretation of mass spectra are necessary for both GC-MS and LC-MS analysis [69–72].
Further accuracy is introduced by “targeted” approaches. Although metabolomics as a classic omics science is by definition an untargeted analytical discovery procedure to screen for unexpected effects [21], targeted approaches are helpful to complement the set of metabolites which cannot be detected by untargeted analysis, as well as to improve the accuracy of quantification.
In Fig. 6 the combination of discovery and targeted metabolomic analysis is shown. Classic discovery methods include “full scan” analysis of LC-MS instruments such as liquid chromatography coupled with quadrupole time-of-flight mass spectrometry [73, 74] and liquid chromatography coupled with Fourier transform Orbitrap/Fourier transform ion cyclotron resonance mass spectrometry [28] instruments as well as GC-MS instruments, especially GC-TOF-MS and GC × GC-TOF-MS instruments [20, 25, 51–55]. These measurements give unmatched resolution and fingerprints of metabolome samples; however, the simultaneous analysis of thousands of compounds demands compromises with respect to accuracy of quantification. Thus, a more targeted approach using classic multiple reaction monitoring-based triple-quadrupole mass spectrometry instruments ensures a very accurate quantification procedure for both GC-MS and LC-MS [75, 76]. The combination of both analytical procedures comprises the iterative strategy depicted in Fig. 6. Discovery phases enable rapid diagnostic analysis and identification of putative biomarkers and physiological markers. Moreover, large libraries of metabolites and putative structures are generated (Fig. 6). A subsequent or parallel targeted approach covers interesting compounds and targeted pathways, thus dissecting the complex system into smaller parts which can be investigated in more detail. Interestingly, this strategy also coincides with metabolic modelling strategies that subdivide metabolic networks which are too complex into smaller subunits (see “Conclusions and perspectives”).
Fig. 6.

Overall strategy combining full-scan mass spectrometry analyses of metabolites and targeted analysis. Full-scan mass spectrometry analysis provides a completely unbiased identification of metabolites and metabolite dynamics. This information is used for sample classification and biological interpretation and the setting up of metabolite libraries as well as knowledge bases. Physiological markers are selectively identified and quantified from complex metabolomics samples in a high-sample-throughput manner with multiple reaction monitoring mass spectrometry technology
The combination of different analytical techniques is of the utmost importance in the metabolomics field as already discussed in “Proving the predictions—metabolomics science” and illustrated in Fig. 4. Many different combinations can be imagined, for instance the combination of NMR spectroscopy and LC-MS and online coupling of liquid chromatography with NMR spectroscopy and LC-MS, especially used for structural elucidation of unknown peaks in LC-MS [77]. All the developments in analytical procedures for metabolomics should be accompanied by chemical synthesis of reference compounds to extend existing libraries. These reference compounds can be analysed with the respective methods to generate libraries compatible with the respective analytical method. Most important, these libraries need to be open source, as do the corresponding databases.
Only the active collaboration of many groups can cope with these current limitations of metabolomic analysis. This is already recognized by the research community and is reflected by the initiatives of the Metabolomics Society (http://www.metabolomicssociety.org/). An active international collaboration in metabolomics science might be as important as the development of novel analytical strategies and will exploit the full potential of this relatively young technology [57].
Conclusions and perspectives
NGS will enable the systematic and comparative investigation of the genotype–phenotype relationship. However, before this relationship reveals its secrets, a comprehensive strategy of metabolic modelling and metabolic measurements has to be established. Here, I have presented a systematic and conceptual equation connecting the genotype and the molecular dynamic phenotype. This equation can be exploited in future for the inverse modelling of the dynamic molecular phenotype and will be instrumental in the interpretation of the corresponding genotype. To achieve these accurate predictions of a dynamic metabolism in newly sequenced organisms, the following improvements are essential.
For validating models of metabolism metabolomics will play a key role; however, metabolome coverage needs to be enhanced by combining different analytical procedures and novel technologies. Furthermore, we need to aim for improved cellular resolution of metabolite profiles.
Because of the complexity of metabolism it can be helpful to dissect the system into smaller parts and analyse these discretely. This procedure coincides with targeted pathway analysis in metabolomics (see also Fig. 6 and earlier). The complete structure can be reconstructed by defining biochemical modules and assembling these modules into large-scale networks. Two recent studies demonstrated how genotyping and metabolite profiling can be combined on a robust statistical basis [78, 79]. In the study by Gieger et al. [79] genome-wide association studies with the human metabolic phenotype were performed using a commercial metabolite profiling platform. The observation from this study was that common genetic polymorphisms induce major differentiations in the metabolism of the individuals. These results strongly support the general strategy of personalized health care and nutrition in combination with metabolite profiling and genotyping [79]. In the study of Chan et al. [78] a large panel of Arabidopsis plants were genotyped and investigated by GC-TOF-MS metabolite profiling. One of the conclusions from this study was that genotype–metabolite associations are sensitive to environmental fluctuations. This opens up a completely new avenue for environmental studies combining rapid NGS genotyping and molecular profiling using omics technologies such as metabolomics and proteomics.
Finally, the integrative approach combining multilevel measurements and modelling approaches in the targeted organism (see Fig. 7) [21] is the conclusive goal. The combination of transcript, protein and metabolite data is especially relevant since there is no initial, as yet readable information from the genome sequence on which enzyme is active or inactive. However, active or inactive enzymes will give different biochemical states and will result in a different stoichiometric matrix N and a different Jacobian J (see earlier). Thus, we need knowledge of the activity or presence and absence of messenger RNAs and proteins. Furthermore, metabolic flux analysis is crucial to reveal active pathways and flux distributions. Techniques such as metabolic labelling with stable isotopes can be exploited in combination with genome-scale metabolite profiling to reveal the in vivo activity of whole pathways and enzymes [23–25]. In combination with the genome-scale investigation of the molecular network of an organism to understand its networking properties, it is as important to continue classic biochemical studies to elucidate protein functions on a much smaller scale and case by case. Integrating this knowledge into the information about the network dynamics of the molecular components might finally result in a functional understanding of the system in relation to the genotype.
Fig. 7.

Integrative approach combining genome sequencing, dynamic modelling and omics analysis to reveal a mechanistic and predictive understanding. EFM elementary flux modes, FBA flux balance analysis, MCA metabolic control analysis
In conclusion, NGS in combination with metabolomics science will be a powerful tool for the investigation of the genotype-phenotype relationship and the ab initio prediction of metabolism in newly sequenced organisms.
Acknowledgments
I thank Anke Bellaire and Xiaoliang Sun for all our fruitful discussions. I apologize to all colleagues who have been cited incompletely due to space problems.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any non-commercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- EST
Expressed sequence tag
- FBA
Flux balance analysis
- GC × GC-TOF-MS
Two-dimensional gas chromatography coupled with fast acquisition rate time-of-flight mass spectrometry
- GC-MS
Gas chromatography coupled with mass spectrometry
- GC-TOF-MS
Gas chromatography coupled with time-of-flight mass spectrometry
- LC-MS
Liquid chromatography coupled with mass spectrometry
- NGS
Next-generation sequencing
References
- 1.454. http://www.454.com/
- 2.SOLiD. http://www3.appliedbiosystems.com/AB_Home/applicationstechnologies/SOLiDSystemSequencing/index.htm
- 3.Illumina. http://www.illumina.com/
- 4.Helicos. http://www.helicosbio.com/
- 5.Stanke M, Morgenstern B. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellana NE, Payne SH, Shen ZX, Stanke M, Bafna V, Briggs SP. Proc Natl Acad Sci USA. 2008;105:21034–21038. doi: 10.1073/pnas.0811066106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.May P, Wienkoop S, Kempa S, Usadel B, Christian N, Rupprecht J, Weiss J, Recuenco-Munoz L, Ebenhoh O, Weckwerth W, Walther D. Genetics. 2008;179:157–166. doi: 10.1534/genetics.108.088336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch-Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, Baginsky S. Science. 2008;320:938–941. doi: 10.1126/science.1157956. [DOI] [PubMed] [Google Scholar]
- 9.Metzker ML. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 10.Nagarajan N, Pop M. Methods Mol Biol. 2010;673:1–17. doi: 10.1007/978-1-60761-842-3_1. [DOI] [PubMed] [Google Scholar]
- 11.Alkan C, Sajjadian S, Eichler EE. Nat Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantacessi C, Jex AR, Hall RS, Young ND, Campbell BE, Joachim A, Nolan MJ, Abubucker S, Sternberg PW, Ranganathan S, Mitreva M, Gasser RB. Nucleic Acids Res. 2010;38:e171. doi: 10.1093/nar/gkq667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawkins RD, Hon GC, Ren B. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wienkoop S, Weiss J, May P, Kempa S, Irgang S, Recuenco-Munoz L, Pietzke M, Schwemmer T, Rupprecht J, Egelhofer V, Weckwerth W. Mol Biosyst. 2010;6:1018–1031. doi: 10.1039/b920913a. [DOI] [PubMed] [Google Scholar]
- 15.Brunner E, Ahrens CH, Mohanty S, Baetschmann H, Loevenich S, Potthast F, Deutsch EW, Panse C, de Lichtenberg U, Rinner O, Lee H, Pedrioli PG, Malmstrom J, Koehler K, Schrimpf S, Krijgsveld J, Kregenow F, Heck AJ, Hafen E, Schlapbach R, Aebersold R. Nat Biotechnol. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- 16.Jungblut PR, Muller EC, Mattow J, Kaufmann SHE. Infect Immun. 2001;69:5905–5907. doi: 10.1128/IAI.69.9.5905-5907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. Nat Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 19.Schellenberger J, Park JO, Conrad TM, Palsson BO. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgenthal K, Wienkoop S, Scholz M, Selbig J, Weckwerth W. Metabolomics. 2005;1:109–121. doi: 10.1007/s11306-005-4430-9. [DOI] [Google Scholar]
- 21.Weckwerth W. Annu Rev Plant Biol. 2003;54:669–689. doi: 10.1146/annurev.arplant.54.031902.135014. [DOI] [PubMed] [Google Scholar]
- 22.Weckwerth W. Physiol Plant. 2008;132:176–189. doi: 10.1111/j.1399-3054.2007.01011.x. [DOI] [PubMed] [Google Scholar]
- 23.Wiechert W. Metab Eng. 2001;3:195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 24.Zamboni N, Sauer U. Curr Opin Microbiol. 2009;12:553–558. doi: 10.1016/j.mib.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Kempa S, Hummel J, Schwemmer T, Pietzke M, Strehmel N, Wienkoop S, Kopka J, Weckwerth W. J Basic Microbiol. 2009;49:82–91. doi: 10.1002/jobm.200800337. [DOI] [PubMed] [Google Scholar]
- 26.Jeong H, Tombor B, Albert R, Oltval ZN, Barabasi AL. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 27.Pfeiffer T, Hoffmann R. PLoS One. 2009;4:e5996. doi: 10.1371/journal.pone.0005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scherling C, Roscher C, Giavalisco P, Schulze ED, Weckwerth W (2010) PLoS One 5:e12569 [DOI] [PMC free article] [PubMed]
- 29.Kempa S, Walther D, Ebenhoeh O, Weckwerth W. In: Molecular biology and biotechnology. 5. Walker JM, Rapley R, editors. Royal Society of Chemistry: Cambridge; 2008. [Google Scholar]
- 30.Heinrich R, Schuster S. Biosystems. 1998;47:61–77. doi: 10.1016/S0303-2647(98)00013-6. [DOI] [PubMed] [Google Scholar]
- 31.Garfinkel D, Hess B. J Biol Chem. 1964;239:971. [PubMed] [Google Scholar]
- 32.Rapoport TA, Heinrich R, Rapoport SM. Biochem J. 1976;154:449–469. doi: 10.1042/bj1540449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werner A, Heinrich R. Biomed Biochim Acta. 1985;44:185–212. [PubMed] [Google Scholar]
- 34.Joshi A, Palsson BO. J Theor Biol. 1989;141:515–528. doi: 10.1016/S0022-5193(89)80233-4. [DOI] [PubMed] [Google Scholar]
- 35.Rizzi M, Baltes M, Theobald U, Reuss M. Biotechnol Bioeng. 1997;55:592–608. doi: 10.1002/(SICI)1097-0290(19970820)55:4<592::AID-BIT2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 36.Mulquiney PJ, Bubb WA, Kuchel PW. Biochem J. 1999;342:567–580. doi: 10.1042/0264-6021:3420567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engl HW, Flamm C, Kugler P, Lu J, Muller S, Schuster P (2009) Inverse Probl 25. doi:10.1088/0266-5611/1025/1012/123014.
- 38.Kauffman KJ, Prakash P, Edwards JS. Curr Opin Biotechnol. 2003;14:491–496. doi: 10.1016/j.copbio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Lee JM, Gianchandani EP, Papin JA. Brief Bioinform. 2006;7:140–150. doi: 10.1093/bib/bbl007. [DOI] [PubMed] [Google Scholar]
- 40.Selkov E, Maltsev N, Olsen GJ, Overbeek R, Whitman WB. Gene. 1997;197:GC11–GC26. doi: 10.1016/S0378-1119(97)00307-7. [DOI] [PubMed] [Google Scholar]
- 41.Feist AM, Palsson BO. Nat Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuster S, Klamt S, Weckwerth W, Moldenhauer F, Pfeiffer T. Bioprocess Biosyst Eng. 2002;24:363–372. doi: 10.1007/s004490100253. [DOI] [Google Scholar]
- 43.Varma A, Palsson BO. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varma A, Palsson BO. Biotechnology. 1994;12:994–998. doi: 10.1038/nbt1094-994. [DOI] [Google Scholar]
- 45.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, Herrgard MJ. Nat Protoc. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 46.Herrgard MJ, Swainston N, Dobson P, Dunn WB, Arga KY, Arvas M, Bluthgen N, Borger S, Costenoble R, Heinemann M, Hucka M, Le Novere N, Li P, Liebermeister W, Mo ML, Oliveira AP, Petranovic D, Pettifer S, Simeonidis E, Smallbone K, Spasic I, Weichart D, Brent R, Broomhead DS, Westerhoff HV, Kirdar B, Penttila M, Klipp E, Palsson BO, Sauer U, Oliver SG, Mendes P, Nielsen J, Kell DB. Nat Biotechnol. 2008;26:1155–1160. doi: 10.1038/nbt1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tisserant E, Da Silva C, Kohler A, Morin E, Wincker P, Martin F. New Phytol. 2011;189:883–891. doi: 10.1111/j.1469-8137.2010.03597.x. [DOI] [PubMed] [Google Scholar]
- 48.Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Trends Biotechnol. 2004;22:245–252. doi: 10.1016/j.tibtech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Hall RD. New Phytol. 2006;169:453–468. doi: 10.1111/j.1469-8137.2005.01632.x. [DOI] [PubMed] [Google Scholar]
- 50.Nicholson JK, Lindon JC, Holmes E. Xenobiotica. 1999;29:1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- 51.Weckwerth W, Tolstikov V, Fiehn O (2001) Metabolomic characterization of transgenic potato plants using GC/TOF and LC/MS analysis reveals silent metabolic phenotypes. Proceedings of the 49th ASMS Conference on Mass spectrometry and Allied Topics 1–2
- 52.Weckwerth W, Wenzel K, Fiehn O. Proteomics. 2004;4:78–83. doi: 10.1002/pmic.200200500. [DOI] [PubMed] [Google Scholar]
- 53.Shellie RA, Welthagen W, Zrostlikova J, Spranger J, Ristow M, Fiehn O, Zimmermann R. J Chromatogr A. 2005;1086:83–90. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 54.Kusano M, Fukushima A, Arita M, Jonsson P, Moritz T, Kobayashi M, Hayashi N, Tohge T, Saito K. BMC Syst Biol. 2007;1:17. doi: 10.1186/1752-0509-1-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jonsson P, Johansson AI, Gullberg J, Trygg J, A J, Grung B, Marklund S, Sjostrom M, Antti H, Moritz T. Anal Chem. 2005;77:5635–5642. doi: 10.1021/ac050601e. [DOI] [PubMed] [Google Scholar]
- 56.Fiehn O. Trends Anal Chem. 2008;27:261–269. doi: 10.1016/j.trac.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sansone SA, Fan T, Goodacre R, Griffin JL, Hardy NW, Kaddurah-Daouk R, Kristal BS, Lindon J, Mendes P, Morrison N, Nikolau B, Robertson D, Sumner LW, Taylor C, van der Werf M, van Ommen B, Fiehn O. Nat Biotechnol. 2007;25:846–848. doi: 10.1038/nbt0807-846b. [DOI] [PubMed] [Google Scholar]
- 58.Weckwerth W, Loureiro ME, Wenzel K, Fiehn O. Proc Natl Acad Sci USA. 2004;101:7809–7814. doi: 10.1073/pnas.0303415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wienkoop S, Morgenthal K, Wolschin F, Scholz M, Selbig J, Weckwerth W. Mol Cell Proteomics. 2008;7:1725–1736. doi: 10.1074/mcp.M700273-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weckwerth W, Morgenthal K. Drug Discov Today. 2005;10:1551–1558. doi: 10.1016/S1359-6446(05)03609-3. [DOI] [PubMed] [Google Scholar]
- 61.Steuer R, Kurths J, Fiehn O, Weckwerth W. Bioinformatics. 2003;19:1019–1026. doi: 10.1093/bioinformatics/btg120. [DOI] [PubMed] [Google Scholar]
- 62.Heinrich R, Schuster S (1996) The Regulation of Cellular Systems. Chapman & Hall, New York
- 63.Giavalisco P, Hummel J, Lisec J, Inostroza AC, Catchpole G, Willmitzer L (2008) Analytical Chemistry 80:9417–9425 [DOI] [PubMed]
- 64.Paulsson J. Phys Life Rev. 2005;2:157–175. doi: 10.1016/j.plrev.2005.03.003. [DOI] [Google Scholar]
- 65.Stein SE. J Am Soc Mass Spectrom. 1999;10:770–781. doi: 10.1016/S1044-0305(99)00047-1. [DOI] [PubMed] [Google Scholar]
- 66.Stein SE, Ausloos P, Clifton CL, Klassen JK, Lias SG, Mikaya AI, Sparkman OD, Tchekhovskoi DV, Zaikin V, Zhu D. Abstr Pap Am Chem Soc. 1999;218:U368–U368. doi: 10.1016/S1044-0305(98)00159-7. [DOI] [PubMed] [Google Scholar]
- 67.Kopka J, Schauer N, Krueger S, Birkemeyer C, Usadel B, Bergmuller E, Dormann P, Weckwerth W, Gibon Y, Stitt M, Willmitzer L, Fernie AR, Steinhauser D. Bioinformatics. 2005;21:1635–1638. doi: 10.1093/bioinformatics/bti236. [DOI] [PubMed] [Google Scholar]
- 68.Kind T, Wohlgemuth G, Lee DY, Lu Y, Palazoglu M, Shahbaz S, Fiehn O. Anal Chem. 2009;81:10038–10048. doi: 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neumann S, Bocker S. Anal Bioanal Chem. 2010;398:2779–2788. doi: 10.1007/s00216-010-4142-5. [DOI] [PubMed] [Google Scholar]
- 70.Hummel J, Strehmel N, Selbig J, Walther D, Kopka J. Metabolomics. 2010;6:322–333. doi: 10.1007/s11306-010-0198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kovacik V, Patoprsty V, Oksman P, Mistrik R, Kovac P. J Mass Spectrom. 2003;38:924–930. doi: 10.1002/jms.506. [DOI] [PubMed] [Google Scholar]
- 72.Akiyama K, Chikayama E, Yuasa H, Shimada Y, Tohge T, Shinozaki K, Hirai MY, Sakurai T, Kikuchi J, Saito K. In Silico Biol. 2008;8:339–345. [PubMed] [Google Scholar]
- 73.De Vos RC, Moco S, Lommen A, Keurentjes JJ, Bino RJ, Hall RD. Nat Protoc. 2007;2:778–791. doi: 10.1038/nprot.2007.95. [DOI] [PubMed] [Google Scholar]
- 74.Zhao X, Fritsche J, Wang J, Chen J, Rittig K, Schmitt-Kopplin P, Fritsche A, Haring HU, Schleicher ED, Xu G, Lehmann R. Metabolomics. 2010;6:362–374. doi: 10.1007/s11306-010-0203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fragner L, Weckwerth W, Huebschmann H-J (2010) Thermo Application Note 51999
- 76.Sawada Y, Akiyama K, Sakata A, Kuwahara A, Otsuki H, Sakurai T, Saito K, Hirai MY. Plant Cell Physiol. 2009;50:37–47. doi: 10.1093/pcp/pcn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Willman, J, Thiele H, Leibfritz D (2011) J Biomed Biotechnol. doi:10.1155/2011/385786 [DOI] [PMC free article] [PubMed]
- 78.Chan EK, Rowe HC, Hansen BG, Kliebenstein DJ. PLoS Genet. 2010;6:e1001198. doi: 10.1371/journal.pgen.1001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gieger C, Geistlinger L, Altmaier E, de Angelis MH, Kronenberg F, Meitinger T, Mewes HW, Wichmann HE, Weinberger KM, Adamski J, Illig T, Suhre K (2008) PLoS Genet 4. doi:10.1371/journal.pgen.1000282 [DOI] [PMC free article] [PubMed]