

Abstract
In this work, the utilization of matrix-assisted laser desorption/ionization mass spectrometric imaging (MALDI-MSI) for capillary electrophoresis (CE) analysis of peptides based on a simple and robust off-line interface has been investigated. The interface involves sliding the CE capillary distal end within a machined groove on a MALDI sample plate, which is precoated with a thin layer of matrix for continuous sample deposition. MALDI-MSI by TOF/TOF along the CE track enables high-resolution and high-sensitivity detection of peptides, allowing the reconstruction of a CE electropherogram while providing accurate mass measurements and structural identification of molecules. Neuropeptide standards and their H/D isotopic formaldehyde-labeled derivatives were analyzed using this new platform. Normalized intensity ratios of individual ions extracted from the CE trace were compared to MALDI-MS direct analysis and the theoretical ratios. The CE-MALDI-MSI results show potential for sensitive and quantitative analysis of peptide mixtures spanning a wide dynamic range.
INTRODUCTION
Featured by high separation efficiency and low sample consumption, capillary electrophoresis (CE), coupled to electrospray ionization mass spectrometry (ESI-MS) represents a significant breakthrough for its applications in biological sciences.1, 2 The on-line CE-MS has emerged as a powerful tool for the structural identification of proteins and peptides in complex mixtures.3, 4 The off-line coupling of CE with matrix-assisted laser desorption/ionization mass spectrometry (CE-MALDI-MS) offers an attractive alternative with increased flexibility for the independent optimization of CE and MS experiments, and makes the CE fractions available for reanalysis or further biochemical characterization.5, 6 Recently, the continuous development in off-line CE-MALDI-MS has established it as a versatile technology for proteomics,7, 8 metabolomics,9 and neuropeptidomics studies. 5, 10–12
To maximize the performance of off-line CE-MALDI-MS, the interface’s effectiveness proves to be a critical factor. In review of the interface development, a number of reports have utilized a T-junction with sheath-flow 6, 13, 14 for the coupling since this approach appears to be robust; however, a significant loss of sensitivity occurs when the flow rates of the sheath liquid are high. In the meantime, several robotic dispensing devices, such as electrospray 15–17 or inkjet spotting 18, have also been employed to deposit sample, matrix or CE effluent 19 onto MALDI targets. While high sensitivity has been reported, these schemes may induce a negative pressure or suction effect to the capillary exit, thus has decreased the column efficiency by introducing parabolic flow to electroosmotic flow (EOF). An electrocoupling 20 and recently an iontophoretic fraction collection 21 approach utilizing coupling droplets to deposit sample has eliminated the sheath-flow and suction effect. However, a current breakdown may occur due to the evaporation of the coupling droplet either being placed on the plate or hung at the capillary end that serves as a “mini” buffer reservoir, which leads to electrical disconnection. In addition, its application to high-throughput analysis has been limited because the spots need to be re-wetted or re-constituted individually after sample deposition onto the target plate.21
To minimize the loss of chromatographic resolution caused by the discrete spotting method with off-line CE-MALDI-MS, more studies have attempted to decrease the time intervals between fraction collection, e.g., 10 s/spot 21 or even 750 ms/spot.19 These studies began to approach a continuous deposition method.9, 22–25 Zhang et al. first described an off-line coupling by continuous depositing an effluent “track” on a matrix (α-cyano-4-hydroxycinnamic acid)-precoated cellulose membrane, which was then used as the MALDI-MS target.22 MALDI mass spectra were taken every 250 μm along the track by manually moving the target under mass spectrometer video view, which made the data acquisition rate relatively low. It is notable that, extracted ion electropherogram was for the first time successfully generated in this study by plotting the signal from spots verse track length (convertible to migration time). However, this interface was basically an earlier variant of electrocoupling design 20, in which the CE ground was connected to the target, poor electrical connections may lead to current breakdown during the CE separation and deposition processes. Later on, Karger and co-workers 25 designed an off-line deposition interface to continuously form 100-μm-wide traces on standard MALDI plate in a chamber with the aid of a rough vacuum. In this report, however, a vacuumed pressure was applied to the capillary exit, a wide liquid junction (20 μm) interface was used to transfer CE samples to deposition capillary, and the deposition was achieved by direct contact of a capillary tip with a bare target. These additional steps might cause some potential problems. First, the clean stainless-steel target surface strongly repels the aqueous solution while spreads organic solvent, making it difficult to form uniform trace on the target. The vacuum pressure also significantly affected the trace’s topography.25 Second, the negative pressure and large junction could significantly deteriorate CE separation efficiency.
Since its introduction by Caprioli and co-workers,26 the MALDI-mass spectrometric imaging (MSI) has been widely applied to investigate molecular distributions in biological tissues (see review papers 27–30). With MALDI laser beam rastered at microns or lower scale, the molecular profiles and associated ion intensities of various species generated from individual spots (“pixels”) provide high-resolution molecular mapping in a defined region. However, limited work has focused on developing MALDI-MSI for CE separation. In 1999, this concept was first attempted by Zhang et al. 23 The same deposition interface using membrane strip with precoated matrix 22 was applied. A significant technical breakthrough was that MALDI imaging software had been developed to automate the MALDI scanning of the sample plate along the deposited CE track. During the next decade, however, the general adoption of this technique has not occurred, probably due to the relatively poor performance of instrumentation at that time (e.g., limited MS/MS capability), which made it less attractive to researchers. The emergence of the TOF/TOF analyzer in MALDI has greatly promoted the MS imaging technique because of its wide mass range, powerful tandem MS capability, high sensitivity and fast acquisition rate. Here, we seek to promote the MALDI-MSI for CE by taking advantage of the power of MALDI-TOF/TOF and our continuous creation in the off-line coupling interface. We also attempted to demonstrate the application of CE-MALDI-MSI for quantitative neuropeptide analysis in this work.
Tailored for imaging applications, here we modified our previous off-line CE-MALDI-MS interface12 and combined the design of a grooved MALDI sample plate by Amantonico et al, 9 producing a robust interface with high sensitivity and high efficiency for the coupling. Proof-of-concept assessment of this design was performed by injecting a peptide mixture into a CE system equipped with the new interface. An electropherogram was successfully constructed based on the extracted ion intensities from MALDI imaging. The ability for relative quantitation was evaluated by using a pair of peptide mixtures at 2:1 concentration ratio pairwise labeled with isotopic formaldehyde (hydrogen vs. deuterium). The ratios of each pair from imaging intensity and electropherogram were found correlated well with the high-resolution MALDI-FTMS analysis results and with the theoretical ratios. Therefore, MALDI-MSI has been initially demonstrated for generating CE electrophoregrams for quantitative analysis of peptides. This technique may open a new avenue for the application of CE in comparative peptidomics and proteomics studies.
EXPERIMENTAL SECTION
Chemicals and Materials
Methanol, acetonitrile (ACN), ammonium hydroxide, trifluoroacetic acid (TFA) and acetic acid were purchased from Fisher Scientific (Pittsburgh, PA). Hydrogen fluoride (48%), Formaldehyde-H2 (FH2, 37% in H2O), and formaldehyde-D2 (FD2, Isotec, ~20% in D2O) were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO). 2,5-dihydroxybenzoic acid (DHB) was obtained from ICN Biomedicals Inc (Costa Mesa, CA). C18 Ziptip was manufactured by Millipore and all water used in this study was doubly distilled on a Millipore filtration system (Bedford, MA). The physiological saline consisted of (in mM): NaCl, 440; KCl, 11; MgCl2, 26; CaCl2, 13; Trizma base, 11; maleic acid, 5; pH 7.45.
Peptide Standards
Some of the neuropeptide standards used in this work were synthesized at the Biotechnology Center of the University of Wisconsin at Madison, including SGGFAFSPRLamide, GAHKNYLRF, FVNSRYamide, and KHKNYLRFamide, the remaining were purchased from the American Peptide Company (Sunnyvale, CA).
Animal dissection and pericardial organs tissue extraction
Blue crabs, Callinectes sapidus for a testing study to evaluate the new interface’s performance were ordered through Midway Asian market (Madison, Wisconsin, USA) and maintained without food in an artificial seawater tank at 10–12 °C. Details of the animal treatment and dissection were described previously. Briefly, animals were cold-anesthetized by packing in ice for 15–30 min prior to dissection, and the pericardial organs (PO) were dissected in chilled physiological saline. The organs were combined and homogenized and peptides were extracted using ice-cold acidified methanol (methanol: glacial acetic acid: water/90:9:1). The extract was dried down and resuspended with 5–10 μL of water containing 0.1% formic acid.
In solution formaldehyde labeling of peptide standard
In solution formaldehyde labeling was performed as described previously.31 Briefly, the two aliquots of 10-peptide mixtures (concentration) at 2:1 ratio were pairwise labeled with formaldehyde-H2 and formaldehyde-D2, respectively. 10 μl of peptide mixture at 2× concentration was added with formaldehyde-H2 (FH2, 4% in H2O, 2 μl) and then vortexed prior to the addition of sodium cyanoborohydride (NaCNBH3, 26 mM, 2 μl, freshly prepared). The mixture was vortexed again and then reaction was allowed to proceed at room temperature to complete. Deuterium labeling was performed with 1× concentration mixture (10 μl), which was treated identically except formaldehyde-D2 (FD2) was added in place of FH2 in the procedure. Two equal aliquots (5 μl each) of the above labeled products were mixed together prior to analysis.
Agilent (HP) CE-UV System, home-built CE system and interface reservoir
CE experiments with UV detection were carried out on an HP G1600AX 3D-CE system, consisting of a 0 to ± 30 kV power supply and a UV/Vis diode-array detector (190–600 nm) using a deuterium lamp as light source. The home-built CE apparatus consists of a high voltage power supply (HV30KVD, 0 to ±30 kV, Unimicro Technologies Inc., Pleasanton, CA), the capillary assembly described below and some small accessories. The running buffer (background electrolyte) for both CE experiments was ammonium acetate (0.5%, pH=4.9), prepared by adjusting 0.5% acetic acid with ammonium hydroxide to pH 4.9. Buffer solution was filtered with 0.45 μm filter (Millipore, Billerica, MA) to remove particulates.
The procedure for construction of an on-column open fracture was modified from our previous report by omitting the step of coating with the cellulose acetate membrane.12 Briefly, the fused-silica capillary of ~60-cm-long (50 μm i.d./360 μm o.d. Polymicro Technologies, Phoenix, AZ) was placed over a 1-cm × 0.3-cm glass slide, on which two small drops of QuickGrip® glue (Beacon Adhesives Co., Mt Vemon, NY) were preloaded on each end to form a V-shaped notch in the center. The capillary was affixed near one end (about 4 cm) by the glue onto the glass slide by leaving a distance of ca. 1–2 mm from glass surface. After glue dried, a small scratch was carefully made on top of the exposed column using a capillary cutter (Chromatography Research Supplies, Inc. Louisville, KY). The capillary was then pushed up gently from the bottom, directly under the scratch, thereby forming the fracture. Our previous report applied a cellulose acetate coating to seal the fracture, which formed an ion-permeable channel on the capillary. Here, the coating was skipped and the fracture was made open for next step of assembly.
A buffer reservoir cell was made with plexiglass (Figure 1C) in the machine shop of Chemistry Department at the University of Wisconsin-Madison. The buffer reservoir has an inner diameter of 1.2 cm, height of 3.0 cm, which holds about 6 mL of liquid. Three ports were made on the side wall to fit the nuts from Upchurch Scientific (Oak Harbor, WA, USA) of type 10-32, 1/4-28 threaded Fingertight fittings, for use on 1/16″ OD and 1/8″ OD tubing in 1/4-28 coned ports. One port located in the middle was used for inserting the platinum wire cathode. One located close to the bottom was for “buffer-in” line, which was connected to a buffer syringe, and the other one close to the top was for “buffer-out” line. The two “-in” and “-out” lines (2 mm ID tubing) formed a “U-shape tube” when they were elevated above the buffer cell. Two additional ports were centrally located on the cap and bottom for installing the separation capillary through two fingertight fittings, the cap was able to seal the cell body with O-ring and screws. The open fracture on the capillary was placed in the chamber, with two centimeters of deposition section of the capillary protruded out of the bottom (Figure 1B). The reservoir was filled with buffer solution and then the nut fittings on the top and bottom for holding the capillary were hand tightened after the air in the reservoir was completely expelled by the liquid. The relative heights of the buffer solution in syringe to the fracture were typically 10 to 30 cm, yielding a hydrostatic pressure of about 10 to 30 mbar towards the fracture.
Figure 1.

(A) Photograph of the MALDI-MS target plate with grooves (250 μm wide, 100 μm deep) and sprayed DHB matrix. The distal end of the capillary slides in the grooves, bearing an angle (60°) with the target plate. (Inset A1) Photograph of the distal end of the etched capillary, the outside diameter was narrowed to ~150 μm for obtaining increased flexibility to fit in the groove. (Inset A2) Close-up photograph showing grooves and the matrix distribution. (B) Schematic drawing of the off-line CE-MALDI-MSI interface with an open fracture on the end section of the capillary (~3 μm gap) for ions to exchange and for cathodic background electrolyte (BGE) to enter the deposition section. 1. Open fracture; 2. Buffer line to hydrostatic height; 3. Separation capillary; 4. Buffer reservoir; 5. Pt cathode. (C) Photograph of the BGE buffer reservoir assembly.
Capillary Treatment and Sampling
Prior to use, new capillary was rinsed/flushed with 1) 75:25/NaOH (1.0 M):MeOH, 2) water, 3) 0.1 M NaOH, 4) air, 5) water again, and 6) running buffer under ~0.5 psi in sequence for 5 min in each step, followed by electrophoretic equilibration with the separation buffer for 10 min prior to injection of the sample. Except for the first two steps, the remaining steps were repeated between CE runs to remove any residual peptides adsorbed on the capillary wall.
Grooved MALDI sample plate and sample continuous deposition
Grooved MALDI plate was also fabricated in the machine shop of Chemistry Department at the University of Wisconsin-Madison. Six lanes of grooves (250 μm wide × 100 μm deep × 10 cm long) were machined on the MALDI target plate (Figure 1A). Figure 1, inset A1 shows the capillary end of the interface assembly etched with hydrogen fluoride from outside with coating removed by flame and outlet sealed by wax. The outer diameter was inspected every 20 mins under microscope, until it reached ~ 150 μm (total about 45 min). The MALDI plate was sprayed with 100 mg/ml DHB by airbrush, filling the grooves with matrix (Figure 1, inset A2) followed by etched capillary end sliding within the grooves for sample deposition.
MALDI-FTMS
Mass spectra of the labeled peptides and CE fractions of blue crab PO extract were recorded on a Varian/IonSpec Fourier transform mass spectrometer (Lake Forest, CA) equipped with a 7.0 T actively shielded superconducting magnet. The FTMS instrument consisted of an external high-pressure MALDI source. A 355 nm Nd: YAG laser (Laser Science, Inc., Franklin, MA) was used to produce ions that can be accumulated in the external hexapole storage trap before being transferred through a quadrupole ion guide to the ICR cell. All mass spectra were acquired in the positive ion mode. The ions were excited prior to detection with an rf sweep beginning at 7050 ms with a width of 4 ms and amplitude of 150 V base to peak. The filament and quadrupole trapping plates were initialized to 15 V, and both were ramped to 1 V from 6500 to 7000 ms to reduce baseline distortion of peaks. Detection was performed in broadband mode from m/z 108.00 to 2500.00.
MALDI-MSI data acquisition and data processing
A model 4800 MALDI-TOF/TOF analyzer (Applied Biosystems, Framingham, MA) equipped with a 200 Hz, 355 nm Nd:YAG laser (spot diameter of 75μm) was operated in the positive ion reflectron mode for all mass spectral analyses. Instrument parameters were set using the 4000 Series Explorer Software (Applied Biosystems). The CE track to be imaged and the raster step size were controlled using the 4800 Imaging application software (Novartis, Basel, Switzerland) available through the MALDI-MSI web site (www.maldi-msi.org). To generate images, mass spectra were collected at 100 μm intervals in both the x and y dimensions within the CE effluent groove. Each mass spectrum was generated by averaging 200 laser shots over the mass range m/z 500–2000. Individual spectra were acquired using 1.0 ns binning. Mass spectra were externally calibrated using peptide standards applied onto the MALDI sample plate.
Image files were processed and extracted ion images were created using the TissueView software package v. 1.0 (Applied Biosystems). TissueView enables construction of MS images by reconstituting the x and y coordinates of the spectra in the acquired image file with their original locations within the groove. Images can be extracted over an m/z window and assigned an intensity-based color scale for optimal visualization. The same intensity scales were used for the MS images of different peptides from the same image file. To reconstruct CE electropherogram, the actual peak intensity of the peptide of interest was retrieved pixel by pixel from each MS spectrum and plotted manually against the migration time. The x coordinate of each spectrum can be translated into the migration time of the analyte of interest, while two spectra acquired at y dimension are viewed as two replicates.
RESULTS AND DISCUSSION
Robust off-line CE-MALDI-MSI interface
It is highly desirable to develop a sensitive, easy to operate, and robust CE-MALDI-MS platform for high throughput neuropeptidomics studies. In this work, we implemented a similar groove design reported by Amantonico et al 9 for the MALDI sample plate with additional modifications. We found that the 400-μm width grooves 9 were too wide to minimize the radial diffusion of the CE effluent within the channel, which could lead to the peak shape distortion in the reconstructed electrophoregram. At the same time, the matrix loading by pipette is not sufficiently homogenous for imaging experiments. Here we narrowed the width of the groove down to 250 μm, and etched the outside of the capillary distal end to ~150 μm by hydrofluoric acid. This modification resulted in a more flexible capillary that fit well within the grooves, preventing it from straying away from the channel while sliding (Figure 1A). The bare MALDI sample plate was sprayed with 100 mg/mL 2, 5-dihydroxybenzoic acid (DHB) by airbrush, filling the grooves with matrix (Figure 1, inset A2) to absorb the liquid to the crystal. A platform was constructed to hold the grooved MALDI target plate, and this platform was attached to a Model NE-300 syringe pump (New Era Pump Systems, Inc, Wantagh, NY). The syringe pump provided a uniform drive mechanism to move at a rate of ~0.11 mm/s, allowing the grooved MALDI plate travel at a uniform speed against the capillary tip placed within the groove. Additionally, the capillary interface was tilted to form an angle (60°) with the MALDI target plate to ensure the capillary slid smoothly in the groove. This configuration enabled the CE effluent to be uniformly and continuously deposited along the total length of the groove (10 cm). Afterwards, the groove was re-sprayed with MALDI matrix.
We previously introduced a sensitive CE-MALDI-MS coupling interface with a cellulose acetate membrane-coated fracture on the terminal section of the capillary.12 The fracture decouples the capillary into a separation section (50–60 cm) and a deposition (post-separation) section (~3 cm). The whole interface was constructed with a single capillary, and it avoided the sheath-flow or additional devices. This design was robust and sensitive, but compromised the CE separation efficiency somewhat because it introduced a pressure at the inlet. Here, an improved design removes the membrane and leaves the break open (~3 μm wide)32 for ion exchange and for cathodic background electrolyte (BGE) to enter the deposition section (Figure 1B). The BGE flow rate was controlled by the level of EOF (approximately 50 nL/min), which is just enough to keep the analyte and running buffer mobilized to pass through the break. The pressure was generated by elevating a U-shape tube 10–30 cm higher than the position of the fracture. The gravity-induced pressure can be finely controlled. Every 1 cm of height change produces 0.014 psi (< 1 mbar) of pressure change, which exceeds the precision of most pumping devices. The liquid flow movement against the EOF is negligible and its influence on separation is minimal since the deposition section is much shorter than the separation section (3 cm vs. 60 cm). Also, the flow towards the deposition section end is almost free from blockage, whereas that towards the CE inlet is overwhelmed by the EOF. Taken together, the interface is capable of eliminating many negative factors of other approaches to off-line CE-MALDI coupling, such as severe sample dilution as a result of large sheath-flow, the suction effect by any robotic spray device, the pressure-induced parabolic flow, and current breakdown due to a poor electrical connection. The improved interface was evaluated for a neuropeptidomics study with neuronal organ extracts. A MALDI-Fourier transform mass spectrometer was utilized for the detection of CE fraction spots using the method we previously reported.12 The interface demonstrated reproducible separations for extremely complex mixtures of biological replicates (Figure S1, Supporting Information). In addition to its reliable performance, capillaries having the open fracture interface can last weeks to months and are limited primarily by the natural degradation of the capillary itself. This new interface provides a robust means to couple CE to MALDI-MS and MALDI-MSI experiments.
Reconstruction of CE electropherogram from MALDI-MSI
A mixture of seven neuropeptide standards was used for the methodology evaluation. Five to fifteen femtomoles of the peptides (~20 nL) were injected for separation. An ABI 4800 MALDI-TOF/TOF mass spectrometer was used to image the CE effluent trace. The total imaging trace is shown in Figure 2A. Based on a pre-run and quick discrete spot profiling by MALDI-FTMS, a time span from 6 to 21 min was set as the effluent and imaging data collection window, which covered an effective distance of 10 cm on the MALDI sample plate. The relative coordinates for each spectrum taken along the sample track were recorded during acquisition and were converted to electrophoretic migration times.
Figure 2.

MALDI-MSI total ion intensity trace (A), and extracted-ion trace (EIT) of peptides after CE separation. The migration zones of peptides on the trace are shown by red arrows from left to right. The EITs were plotted by zooming the original imaging trace by 10-fold. All lanes have two pixel lines (100 μm × 2). These imaging bins were centrally cut and displayed from (B) to (H). CE conditions: running buffer at both ends, ammonium acetate, 0.5%, pH 4.9; capillary, L (effective) 58 cm, i.d. 50 μm; Injection 8 kV × 10 s; Separation, 18 kV, i = 40 μA. The migration zones of peptides on the track are shown by red arrows from left to right.
As we know the resolution of MALDI imaging is influenced by a number of factors, such as laser beam size, the matrix crystal size and the sample preparation quality. In this study, the DHB matrix was applied by airbrush manual deposition, the crystal size of this preparation is about 15–20 μm according to the literature33, and the MALDI laser beam size is 75 μm. Thus, the achievable resolution is limited by the laser beam diameter, 75 μm. But this limit was further altered by the radial diffusion of CE effluent. Our initial experimental with double-line scan (100 μm raster increment) in the groove showed that near 50% of the pixels have signal overlapping on y axis, indicating that the CE radial diffusion was roughly 50 to 100 μm. To shorten the acquisition time while to keep high coverage of these pixels, we set the raster increment as 100 μm for both x and y coordinates, generating a 1,000 pixels × 2 pixels imaging area.
The plate mounted on the XY stage was moved at a speed of ~110 μm/s, slightly greater than one shot (pixel) per second with spatial resolution being set at 100 μm. Thus, a 10-second CE peak, a common peak width for CE, can generate a bin having enough time for collecting up to 10 data points in a one pixel line. The 250 μm width allows at least two pixel lines of scan in a single groove, making an even narrower time window (e.g., 5 s) sufficient for plotting the CE trace. The extracted-ion traces (EITs) of the peptides are illustrated in Figures 2B to 2H, and the bin widths are seen to vary from approximately 6 s to 20 s.
One purpose of our efforts is to minimize the loss of chromatographic resolution caused by the discrete spotting method. CE is often used for relative or absolute quantitation based on peak heights or areas obtained in the electropherogram. The faster the acquisition rates of the detector, the more accurate the CE quantitation. Here, the high-definition and high-speed sampling capability of MALDI-TOF/TOF imaging makes it possible to extract high density ion signals and to reconstruct the peak trace. A reconstructed CE electropherogram is shown in Figure 3A. The estimated separation efficiency was roughly 100,000 plates per meter. The same sample mixture was analyzed previously by discrete spotting and detection with MALDI-FTMS.34 In that report, multiple peptides in one fraction or the splitting of single peptide into multiple fractions were common by using 1-min interval collection (data not shown here). It was noted that visible peak splitting was also observed for two peptides in the current study, as reflected in the electropherogram (peaks 3 and 5 in Figure 3A) or the imaging traces (Figure 2D and 2F). This splitting was not associated with the deposition approach; rather, this was more likely caused by the somewhat inhomogenous distribution of matrix within the groove. We are in the process of exploring a new approach to apply the matrix that combines the use of airbrush spray and priming the matrix into the channel with a capillary. Alternatively, nanostructure-initiator mass spectrometry (NIMS),35 a matrix-free, sensitive MS imaging technique, might be a good solution to this problem.
Figure 3.

(A) Reconstructed CE electropherogram from MALDI-TOF/TOF-MSI of seven peptide standards. The peak IDs are (1) GAHKNYLRF, m/z 1105.59 (0.70 μM), (2) IARRHPYFL, m/z 1172.67 (0.75 μM), (3) SGGFAFSPRLamide, m/z 1037.55 (0.65 μM), (4) CYFQNCPRGamide, m/z 1084.45 (0.37 μM), (5) APSGAQRLYGFGLamide, m/z 1335.72 (0.25 μM), (6) AGCKNFFWKTFTSC, m/z 1637.72 (0.55 μM), (7) PFCNAFTGCamide, m/z 956.37 (0.5 μM). CE conditions are the same as Figure 1. (B) CE-UV electropherogram. Agilent G1600AX 3D CE system, capillary: 50cm (43cm effective); UV/Vis: 214 nm, injection: 9 kV × 10s, Separation, 22 kV, i = 65 μA. (C) MS/MS spectrum identification of peptide GAHKNYLRF (peak 1).
Figure 3B illustrates a similar electropherogram recorded using a UV detector, without the assignment of peak identities. A typical way to identify these peaks is to run each peptide standard individually and then to confirm by the migration order matching. This comparison demonstrates a great advantage of the MS detection for CE, from which the peptides are easily identifiable based on accurate mass measurement. In the meantime, the sequences of the peptides can be determined by TOF/TOF tandem mass spectra with characteristic fragmentation patterns. Figure 3C shows the identification of peptide GAHKNYLRF (m/z 1105.59) (peak 1) with MS/MS fragmentation. Representative spectra showing the isolation of peptides and more MS/MS data are presented in Figure S2. To obtain optimal separation using different CE systems, slightly different experimental conditions were employed for the HP3D CE and home-built CE respectively, including the separation voltage and capillary length. It appeared that the separation efficiency on CE-UV was ~10% higher or at least similar to that using continuous deposition method for CE-MALDI-MSI. This comparison indicates that the off-line deposition has caused only a slight loss of electrophoretic resolution.
Evaluating CE-MALDI-MSI quantitation of peptides using isotopic formaldehyde labeling
It has become increasingly important to determine the relative abundances of endogenous protein or peptide levels in different biological events using mass spectrometry. Formaldehyde labeling is a widely-used mass-difference quantitation method thanks to its low cost, fast reaction and high labeling efficiency.36, 37 This method has been extensively employed in our lab for neuropeptide relative quantitation.38, 39 This binary isotopic labeling technique labels sample with light formaldehyde (FH2, +28 Da for each incorporated label) and heavy formaldehyde (FD2, +32 Da for each incorporated label), respectively, resulting in 4.02 Da mass difference for each peptide pair, and gives their relative abundance.
Here, we employed isotopic formaldehyde labeling for the evaluation of quantitative CE-MALDI imaging. Peptide standards at a 2:1 ratio of concentration were labeled with FH2 and FD2, respectively. Afterwards, the labeled samples were combined for two different experiments: CE-MALDI-MSI and direct MALDI-FTMS analysis. Then we compare several ratios generated in different ways: peak pair ratios of the extracted ion intensity from MALDI imaging (a), peak area ratios from the reconstructed electropherogram (b), and the peak pair ratios from MALDI-FTMS (c). How well these observed ratios (a–c) correlate with the theoretical ratio (d) will give us an idea if CE-MALDI-MSI could become a useful technique for comparative quantitation. Figure 4, the reconstructed CE electropherogram, shows a 10-peptide mixture in a 2:1 ratio with labels. The wide overall dynamic range of peak intensities (~25 fold), roughly correlates with the concentration ladder of ~20 fold. The individual ratio for each pair can be calculated based on the electropherogram, as light and heavy derivatives being labeled in green and red, respectively.
Figure 4.

Reconstructed CE electropherogram from MALDI-TOF/TOF-MSI of labeled peptides. A mixture containing 10 peptides originally at 2:1 ratio were pairwise labeled with FH2 (green trace) and FD2 (red trace) for analysis. B) is the 5 fold y-axis expansion of A). The peak IDs are (1) FMRFamide, m/z 599.31 (0.35 μM), (2) FVNSRYamide, m/z 784.41 (0.25 μM), (3) GAHKNYLRF, m/z 1105.59 (1.0 μM), (4) SGFYANRYamide, m/z 976.46 (2.0 μM), (5) APSGAQRLYGFGLamide, m/z 1335.72 (0.1 μM), (6) DRVYVHPFHL, m/z 1282.67 (0.30 μM), (7) DRVYIHPF, m/z 1046.54 (0.52 μM), (8) KHKNYLRFamide, m/z 1104.64 (0.09 μM), (9) GDGRLYAFGLamide, m/z 1067.56 (0.47 μM), and (10) DLWQK, m/z 689.36 (1.5 μM). CE conditions: capillary length, 66 cm; separation 20 kV. Other parameters are the same as in Figure 2.
Figure 5 shows the extracted ion electropherogram of peak pair m/z 1161/1169 that originated from peptide GAHKNYLRF (m/z 1105.59, peak 3 in Figure 4), giving an example how to correlate the intensity ratio from the imaging data (a) with that from the CE electropherogram (b). As shown in Figure 5 inset, the extracted ion traces (EITs) of this pair are displayed in lanes 1 and 3. The region of interest (ROI) covering the signal area was drawn with the software TissueView 1.0, as shown in lanes 2 and 4, respectively. The ROIs were carefully aligned with each other (± 1 pixel). The “population” multiplied by the “means” in these two areas are used to represent the total imaging intensities of the extracted ions from the peak pair. The ratio of m/z 1161 to m/z 1169 was calculated to be about 1.67 (a) in this experiment according to the above described calculation method (Figure S3A), whereas the area ratio for the two peaks from the electropherogram was observed to be as high as 2.3 (b). The similar variations can be observed from peptides to peptides, the ratios are biased by 10–20% compared to the theoretical ratio of 2:1. At the same time, the MALDI-FTMS spectrum (Figure S3B) was analyzed and the experimental ratio was calculated to be 1.75 (c). The two ratios (a) and (c) correlate well with each other, and both are about 15% below the theoretical ratio (d) of 2:1. Relative abundance ratios from imaging intensity for other peptides are shown in Table S2, varying from 1.67 to 2.27. These results collectively demonstrate that, in combination with isotopic formaldehyde labeling, we could develop either the reconstructed CE electropherogram or the imaging total intensity into a quantitative analysis method for our future comparative peptidomic studies.
Figure 5.
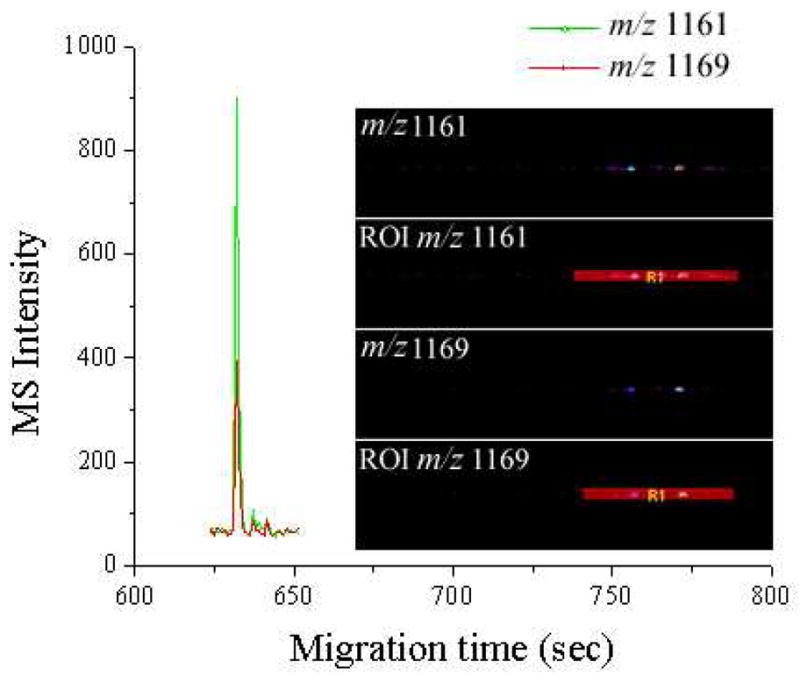
Extracted ion electropherogram of peak pair at m/z 1161 and 1169, corresponding to peak 3 in Figure 4. Inset, the extracted ion imaging trace and region of interest (ROI) plot of two mass spectral peaks.
CONCLUSIONS
A robust off-line interface combines spatial mapping and accurate identification of peptides by MALDI-MSI with high efficiency and sensitivity of CE separation, allowing the reconstruction of a CE electropherogram based on extracted MALDI ion image abundances. The utility of CE-MALDI-MSI for quantitative analysis of peptide mixtures with a wide dynamic range is demonstrated. One of our goals in the development of the CE-MALDI-MS imaging is to evaluate whether we can employ this hyphenated technique as a label-free method for relative quantitation. The pilot experiments described in this report have verified that it is feasible by comparing the ratios of normalized ROI signal intensities of individual ion peaks from labeled peptide pairs. This quick comparison could even eliminate the step of extracting ion intensities to plot the CE electropherogram in order to get their ratios. On the other hand, additional algorithms need to be developed to expedite data processing for quantitative analysis. Further improvements in CE effluent deposition and matrix application methods are currently underway to enhance the performance and expand the utility of CE-MALDI-MSI methodology.
Supplementary Material
Acknowledgments
The authors wish to thank the University of Wisconsin-Biotechnology Mass Spectrometry Facility for access to the MALDI TOF/TOF instrument. We also want to thank the University of Wisconsin School of Pharmacy Analytical Instrumentation Center for access to the MALDI FTICR MS instrument. This work was supported in part by a National Science Foundation grant (CHE-0957784) and National Institutes of Health through grant 1R01DK071801. L.L. acknowledges an Alfred P. Sloan Research Fellowship, a Vilas Associate Fellowship, and an H. I. Romnes Faculty Fellowship.
Footnotes
SUPPORTING INFORMATION AVAILABLE Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smith RD, Olivares JA, Nguyen NT, Udseth HR. Analytical Chemistry. 1988;60:436–441. [Google Scholar]
- 2.Valaskovic GA, Kelleher NL, McLafferty FW. Science. 1996;273:1199–1202. doi: 10.1126/science.273.5279.1199. [DOI] [PubMed] [Google Scholar]
- 3.Moini M. Analytical and Bioanalytical Chemistry. 2002;373:466–480. doi: 10.1007/s00216-002-1283-1. [DOI] [PubMed] [Google Scholar]
- 4.Fonslow BR, Yates JR. Journal of Separation Science. 2009;32:1175–1188. doi: 10.1002/jssc.200800592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Page JS, Rubakhin SS, Sweedler JV. Analyst. 2000;125:555–561. [Google Scholar]
- 6.Zuberovic A, Ullsten S, Hellman U, Markides KE, Bergquist J. Rapid Communications in Mass Spectrometry. 2004;18:2946–2952. doi: 10.1002/rcm.1712. [DOI] [PubMed] [Google Scholar]
- 7.Snovida SI, Chen VC, Krokhin O, Perreault H. Analytical Chemistry. 2006;78:6556–6563. doi: 10.1021/ac060738k. [DOI] [PubMed] [Google Scholar]
- 8.Williams BJ, Russell WK, Russell DH. Anal Chem. 2007;79:3850–3855. doi: 10.1021/ac062395w. [DOI] [PubMed] [Google Scholar]
- 9.Amantonico A, Urban PL, Zenobi R. Analyst. 2009;134:1536–1540. doi: 10.1039/b907039g. [DOI] [PubMed] [Google Scholar]
- 10.Rubakhin SS, Page JS, Monroe BR, Sweedler JV. Electrophoresis. 2001;22:3752–3758. doi: 10.1002/1522-2683(200109)22:17<3752::AID-ELPS3752>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 11.Lapainis T, Sweedler JV. Journal of Chromatography A. 2008;1184:144–158. doi: 10.1016/j.chroma.2007.10.098. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Ma M, Chen R, Li L. Analytical Chemistry. 2008;80:6168–6177. doi: 10.1021/ac800382t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson T, Bergquist J, Ekman R, Nordhoff E, Schurenberg M, Kloppel KD, Muller M, Lehrach H, Gobom J. Anal Chem. 2001;73:1670–1675. doi: 10.1021/ac0011888. [DOI] [PubMed] [Google Scholar]
- 14.Helmja K, Borissova M, Knjazeva T, Jaanus M, Muinasmaa U, Kaljurand M, Vaher M. Journal of Chromatography A. 2009;1216:3666–3673. doi: 10.1016/j.chroma.2008.12.061. [DOI] [PubMed] [Google Scholar]
- 15.Hanton SD, Hyder IZ, Stets JR, Owens KG, Blair WR, Guttman CM, Giuseppetti AA. Journal of the American Society for Mass Spectrometry. 2004;15:168–179. doi: 10.1016/j.jasms.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Wei H, Nolkrantz K, Powell DH, Woods JH, Ko MC, Kennedy RT. Rapid Communications in Mass Spectrometry. 2004;18:1193–1200. doi: 10.1002/rcm.1458. [DOI] [PubMed] [Google Scholar]
- 17.Jeong KH, Seo J, Yoon HJ, Shin SK. Bulletin of the Korean Chemical Society. 2010;31:2293–2298. [Google Scholar]
- 18.Baluya DL, Garrett TJ, Yost RA. Analytical Chemistry. 2007;79:6862–6867. doi: 10.1021/ac070958d. [DOI] [PubMed] [Google Scholar]
- 19.Vannatta MW, Whitmore CD, Dovichi NJ. Electrophoresis. 2009;30:4071–4074. doi: 10.1002/elps.200900414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojima N, Shingaki T, Yamamoto T, Masujima T. Electrophoresis. 2001;22:3478–3482. doi: 10.1002/1522-2683(200109)22:16<3478::AID-ELPS3478>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 21.Busnel JM, Josserand J, Lion N, Girault HH. Analytical Chemistry. 2009;81:3867–3872. doi: 10.1021/ac900128q. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HY, Caprioli RM. Journal of Mass Spectrometry. 1996;31:1039–1046. doi: 10.1002/(SICI)1096-9888(199609)31:9<1039::AID-JMS398>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Stoeckli M, Andren PE, Caprioli RM. Journal of Mass Spectrometry. 1999;34:377–383. doi: 10.1002/(SICI)1096-9888(199904)34:4<377::AID-JMS778>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 24.Preisler J, Hu P, Rejtar T, Karger BL. Analytical Chemistry. 2000;72:4785–4795. doi: 10.1021/ac0005870. [DOI] [PubMed] [Google Scholar]
- 25.Rejtar T, Hu P, Juhasz P, Campbell JM, Vestal ML, Preisler J, Karger BL. Journal of Proteome Research. 2002;1:171–179. doi: 10.1021/pr015519o. [DOI] [PubMed] [Google Scholar]
- 26.Caprioli RM, Farmer TB, Gile J. Anal Chem. 1997:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 27.Chaurand P, Schwartz SA, Caprioli RM. Current Opinion in Chemical Biology. 2002;6:676–681. doi: 10.1016/s1367-5931(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 28.McDonnell LA, Heeren RMA. Mass Spectrometry Reviews. 2007;26:606–643. doi: 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- 29.Franck J, Arafah K, Elayed M, Bonnel D, Vergara D, Jacquet A, Vinatier D, Wisztorski M, Day R, Fournier I, Salzet M. Molecular & Cellular Proteomics. 2009;8:2023–2033. doi: 10.1074/mcp.R800016-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen RB, Li LJ. Analytical and Bioanalytical Chemistry. 2010;397:3185–3193. doi: 10.1007/s00216-010-3723-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeKeyser SS, Li L. Analyst. 2006;131:281–290. doi: 10.1039/b510831d. [DOI] [PubMed] [Google Scholar]
- 32.Xu QF, Ji XH, Li HG, Liu J, He ZK. Journal of Chromatography A. 2010;1217:5628–5634. doi: 10.1016/j.chroma.2010.06.071. [DOI] [PubMed] [Google Scholar]
- 33.Aerni HR, Cornett DS, Caprioli RM. Anal Chem. 2006;78:827–834. doi: 10.1021/ac051534r. [DOI] [PubMed] [Google Scholar]
- 34.Wang JH, Jiang XY, Sturm RM, Li LJ. Journal of Chromatography A. 2009;1216:8283–8288. doi: 10.1016/j.chroma.2009.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northen TR, Yanes O, Northen MT, Marrinucci D, Uritboonthai W, Apon J, Golledge SL, Nordstrom A, Siuzdak G. Nature. 2007;449:1033–U1033. doi: 10.1038/nature06195. [DOI] [PubMed] [Google Scholar]
- 36.Hsu JL, Huang SY, Chow NH, Chen SH. Analytical Chemistry. 2003;75:6843–6852. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 37.Ji CJ, Guo N, Li L. Journal of Proteome Research. 2005;4:2099–2108. doi: 10.1021/pr050215d. [DOI] [PubMed] [Google Scholar]
- 38.DeKeyser SS, Li LJ. Analyst. 2006;131:281–290. doi: 10.1039/b510831d. [DOI] [PubMed] [Google Scholar]
- 39.Wang JH, Zhang YZ, Xiang F, Zhang ZC, Li LJ. Journal of Chromatography A. 2010;1217:4463–4470. doi: 10.1016/j.chroma.2010.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.