

Abstract
The role of the α-helical domain (MH) of dengue virus (DENV) precursor membrane protein in replication was investigated by site-directed mutagenesis. Proline substitutions of three residues (120, 123 and 127) at the C-terminus, but not those at the N-terminus of MH domain, reduced the virus-like particles of DENV1, DENV2 and DENV4 detected in supernatants. In a DENV2 replicon trans-packaging system, these three mutations suppressed particles detected; two of them (I123P and V127P) also affected viral entry. In the context of DENV2 genome-length RNA, all three mutations reduced virion assembly and virus spreading in cell culture. Analysis of revertants showed that mutation A120P could partially support viral infection cycle; in contrast, mutations I123P and V127P were lethal, and adaptations of I123P→I123L and V127P→ V127L were required to restore the viral infection cycle. These findings demonstrate that the C-terminus of the MH domain is involved in both assembly and entry of DENV.
Keywords: dengue virus, precursor membrane, virus-like particles, assembly, entry
Introduction
Dengue viruses (DENV) are members of the genus Flavivirus in the family Flaviviridae. The four serotypes of DENV (DENV1, DENV2, DENV3, and DENV4) are the leading cause of arboviral diseases in the tropical and subtropical regions. While most DENV infections are asymptomatic, some people present with a debilitating and self-limited disease, dengue fever, or a severe and potentially life-threatening disease, dengue hemorrhagic fever /dengue shock syndrome (Gubler, 2002; Guzman and Kouri, 2002; Halstead, 1988; WHO, 2009). DENV contains a positive-sense, single-stranded RNA genome of about 10.6 kilobases in length. Flanked by the 5’ and 3’ untranslated regions, the single open reading frame encodes a polyprotein precursor, which is cleaved by cellular and viral protease into three structural proteins, capsid (C), precursor membrane (prM) and envelope (E), and seven nonstructural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5 (Lindenbach et al., 2007).
After binding to its cellular receptor, DENV enters the cell through receptor mediated endocytosis (Guirakhoo et al., 1993; Lindenbach et al., 2007; Mukhopadhyay et al., 2005; Randolph and Stollar, 1990). This is followed by uncoating, translation and genome replication. Assembly occurs in the membrane of rough ER, where the immature virions bud into the lumen of ER and transport through the secretary pathway (Lindenbach et al., 2007; Mackenzie and Westaway, 2001; Mukhopadhyay et al., 2005; Welsch et al., 2009). In the trans-Golgi, cleavage of prM protein by furin or furin-like protease results in the formation of mature virions, though the cleavage was inefficient for DENV (Keelapang et al., 2004; Murray et al., 1993; Stadler et al., 1997; Wang et al., 1999; Yu et al., 2008). A unique property of flaviviral replication is the formation of subviral particles, which are smaller and sediment slower than mature virions (Lindenbach et al., 2007; Russell, 1980). Expression of both prM and E proteins can produce recombinant virus-like particles (VLPs). VLPs are similar to the infectious virions in the biophysical and antigenic features, though some studies have shown that VLPs are more heterogeneous in size than virions and not hemagglutinating in certain preparations probably related to the efficiency of prM cleavage in different cells (Allison et al., 2003; Ishikawa and Konishi, 2006; Junjhun et al., 2008; Stadler et al., 1997; Wang et al., 1999). VLPs have been employed as a model system to study the functions of prM/E proteins and assembly of particles (Ferlenhi et al., 2001; Lorenz et al., 2003; Schalich et al., 1996). Moreover, VLPs have been shown to be useful non-infectious serodiagnostic antigens and potential vaccine candidates (Chang et al., 2003; Davis et al., 2001; Hunt et al., 2001; Konishi and Fujii, 2002; Kroeger and McMinn, 2002; Martin et al., 2007; Purdy et al., 2004).
The E protein is the major determinant of cellular tropism and virulence, and the major target of neutralizing and enhancing antibodies of DENV (Bray et al., 1998; Halstead, 1988; Lindenbach et al., 2007; Mukhopadhyay et al., 2005). In the N-terminal ectodomain of E protein, there are three well characterized domains (domains I, II and III) based on X-ray crystallographic studies (Modis et al., 2003; Modis et al., 2004; Modis et al., 2005). The C-terminus of E protein contains two α-helices (EH1 and EH2) in the stem region and two transmembrane domains (ET1 and ET2) in the anchor region, which crosses the two leaflets of the lipid bilayer (Allison et al., 1999; Zhang et al., 2003) (Fig. 1A). Based on the studies of the tick-borne encephalitis virus (TBEV), both ET2 and ET1 were required for the assembly of E protein into particles. EH2 can stabilize the prM-E heterodimer, whereas EH1 is involved in the irreversible trimerization of soluble E protein in low pH environment (Allison et al., 1999; Orlinger et al., 2006; Stiasny et al., 1996). In addition, a study of the yellow fever virus reported that transmembrane domains of prM and E proteins are involved in the formation of VLPs (Op De Beeck et al., 2003).
Fig. 1.

Schematic drawing of the stem region of DENV4 prM protein, MH domain mutants, production of VLPs and prM-E heterodimerization. (A) The C-terminus of prM protein contains an α-helical domain (MH) (residues 113 to 128) in the stem region, followed by two transmembrane domains (MT1 and MT2) (Zhang et al., 2003). Single letter designations of amino acids are shown with numbers above indicating the amino acid positions. The proline or alanine substitution mutants in the MH domain of pCB-D4 are shown. (B) Helical wheel analysis of the MH domain revealed an amphipathic helix with hydrophobic residues at the a or d positions facing one side and charged or polar residues mainly facing the other side. (C, D) At 48 h post-transfection, 293T cell lysates and pellets derived from culture supernatants of WT pCB-D4 or mutants were subjected to Western blot analysis using human serum of a dengue case (Wang et al., 2006). (E) Transfected 293T cells were labeled with 35S-Met, immunoprecipitated with anti-E Mab FL0232 and subjected to SDS-12% PAGE. The size of molecular weight markers is shown in kDa. One representative experiment of three is shown.
The prM protein contains 166 amino acids, of which the cleavage occurs at position 91 to produce the pr peptide and M protein. At the C-terminus of prM protein, there is an α-helical domain (MH), followed by two transmembrane domains (MT1 and MT2) (Fig. 1A) (Zhang et al., 2003). After synthesis in the rough ER, prM protein formed a heterodimer with E protein and was reported to function as a chaperone for proper folding of E protein of TBEV and Japanese encephalitis virus (JEV) (Konishi and Mason, 1993; Lorenz et al., 2002). Recent cryo-electronic microscopic (EM) study of immature DENV at low pH has provided new insights into the maturation of DENV particles along the secretory pathway (Yu et al., 2008). Moreover, a study of the crystal structure of recombinant prM-E proteins complex revealed the heterodimeric interaction between the N-terminus of prM protein (pr peptide, residues 1 to 81) and the E protein ectodomain at high resolution, and predicted that the extended part of M protein (residues 82 to 112) undergoes a large conformational change during the maturation process (Li et al., 2008).
Previously, a histidine residue at position 99 of prM protein of JEV was reported to be involved in the prM-E interaction (Lin and Wu, 2005). Moreover, a histidine residue at position 39 of M protein (position 130 of prM protein) of DENV2 and a tyrosine residue at position 78 of prM protein of West Nile virus were reported to be important for virus assembly (Pryor et al., 2004; Tan et al., 2009). However, the functional roles of the MH domain (residues 113 to 128) in the replication cycle of DENV remains unknown. In this study, we investigated the involvement of the MH domain of prM protein in the assembly of VLPs by site-directed mutagenesis in the prM/E expression constructs of DENV4, DENV1 and DENV2, and found that three proline mutants at the C-terminus of MH domain severely impair the production of VLPs. This was further verified by examining the assembly of DENV2 replicon particles and virions derived from a full-length infectious clone. Moreover, two proline mutants were found to greatly affect virus entry, suggesting that the C-terminus of MH domain is involved in both assembly and entry of the DENV replication cycle.
Results
C-terminus of MH domain is involved in the production of DENV4 VLPs
Helical wheel analysis of the MH domain revealed an amphipathic helix, in which hydrophobic residues at the a or d positions of the wheel faced one side and charged or polar residues mainly faced the other side (Fig. 1B). To investigate the roles of the MH helical domain in the assembly of VLPs, site-directed mutagenesis was carried out to replace each of the residues at the a or d positions (residues 113, 116, 120, 123 and 127) with a proline or alanine in a DENV4 prM/E expression construct, pCBD4 (Fig. 1A). Substitutions with proline, an α-helix breaker, and alanine are commonly employed to study the effect of disrupting helical structure and the effect of removing side chain with helical structure preserved, respectively.
As shown in Fig. 1C, the amounts of prM/E proteins of the proline mutants in cell lysates were generally comparable to those of wild type (WT) pCB-D4, suggesting that substitutions of proline do not greatly affect the expression of prM/E proteins. In contrast, the amounts of prM/E proteins in pellets, compared to those of the WT, were greatly reduced in mutants A120P, V123P and I127P and slightly reduced in mutant A116P, suggesting that proline substitutions introduced to residues 120, 123 and 127 at the C-terminus of the MH domain, but not those at the N-terminus, greatly affect the production of VLPs (Fig. 1C, long exposure for prM bands in pellets not shown). Alanine substitutions introduced to two of these residues, V123A and I127A, resulted in slightly reduced VLP production (Fig. 1D). Of note, residues 116 and 120 are alanine themselves. Other two alanine substitutions including S113A and H98A (which was included in the analysis because its corresponding residue at position 99 of JEV prM protein has been reported to be important for prM-E interaction) did not affect VLPs production (Lin and Wu, 2005). Together, these findings suggested that the C-terminus of the helical structure is involved in production of VLPs of DENV4. To investigate whether these mutations affect the prM-E interaction, lysates of 293T cells transfected with each of these mutants were subjected to a radioimmunoprecipitation assay using anti-E Mab (Hsieh et al., 2008). As shown in Fig. 1E, the amounts of prM protein relative to E protein of these mutants, though slightly reduced for mutants V123P and I127P, were generally comparable to that of the WT, suggesting that alanine or proline substitutions introduced to the MH domain of DENV4 do not greatly affect the prM-E heterodimeric interaction.
C-terminus of MH domain is involved in the production and release of VLPs from the membrane to the lumen of ER
To further investigate whether the intracellular prM/E proteins of these proline mutants retain in the membrane-bound fraction or form VLPs in the soluble fraction of ER or other compartments, 293T cells transfected with each of these proline mutants were subjected to a previously described subcellular fractionation experiment (Hsieh et al., 2008; Xu et al., 1997). As shown in Fig. 2A, the amounts of prM/E proteins in pellets of the soluble fraction relative to those in the membrane fraction were reduced for the proline mutants (A120P, V123P and I127P) compared with those of the WT. Moreover, the amounts of prM/E proteins in pellets of supernatants were much less in these mutants than those in the WT. As a control, calnexin, an integral ER membrane protein, was found in the membrane fraction but not in the soluble fraction or supernatants. Enzyme digestion with endo H or PNGase F of the membrane fraction and pellets of soluble fraction revealed a predominantly endo-H sensitive pattern for both WT and mutants (Fig. 2B), suggesting that intracellular prM/E proteins of WT and mutants, either in the membrane bound fraction (before assembly to form VLPs) or in the soluble fraction (after assembly), were mainly present in a compartment prior to trans-Golgi, most likely ER. Collectively, these findings suggest that proline substitutions introduced to the C-terminus of the MH domain result in impairment in the production of VLPs from prM/E proteins in the membrane of ER and release to the lumen and culture supernatants.
Fig. 2.

Subcellular fractionation experiment of the MH domain mutants of DENV4 prM/E construct. (A) 293T cells transfected with mock, WT or mutants were washed with PBS, resuspended in modified buffer B, and frozen-thawed 8 times (Hsieh et al., 2008; Xu et al., 1997). After clearing the nuclei and debris, the membrane fraction, pellets derived from the soluble fraction by ultracentrifugation and pellets derived from culture supernatants were subjected to Western blot analysis using human serum of a dengue case (upper gels) (Wang et al., 2006), followed by reprobing with anti-calnexin Mab (lower gels). The long exposure gels for prM bands in pellets were shown (middle gels). (B) Membrane fraction and pellets of the soluble fraction of WT and mutants were treated with endo H (H) or PNGase F (F), and subjected to Western blot analysis as in panel A. Arrow heads indicate E or deglycosylated E protein (Edg). The size of molecular weight markers is shown in kDa. One representative experiment of three is shown.
C-terminus of MH domain is involved in the production of VLPs of other serotypes
To examine whether the C-terminus of the MH domain is also involved in the production of VLPs of other DENV serotypes, proline substitutions were introduced to residues 120I, 123V and 127A of the DENV1 prM/E construct, as well as to residues 120V, 123I and 127I of the DENV2 prM/E construct. In agreement with the findings in DENV4 mutants, the amounts of prM/E proteins in pellets relative to those in cell lysates were greatly reduced in both DENV1 mutants (D1-I120P, V123P and A127P) and DENV2 mutants (D2-V120P, I123P and I127P), when compared with the WT pCB-D1 and pCB-D2, respectively (Figs. 3A and 3B). Radioimmunoprecipitation assay revealed that amounts of prM protein relative to E protein of these mutants were generally comparable to those of the WT (Fig. 3C). These findings suggest that the C-terminus of the MH domain of DENV1 and DENV2 is also involved in the production of VLPs but not in the prM-E heterodimerization.
Fig. 3.

Production of VLPs and prM-E heterodimerization of the MH domain mutants of DENV1 and DENV2 prM/E constructs. (A, B) At 48 h post-transfection, 293T cell lysates and pellets derived from culture supernatants of WT or mutants were subjected to Western blot analysis using human serum of a dengue case (Wang et al., 2006). The long exposure gels for DENV1 prM bands in pellets were shown below. (C) Transfected 293T cells were labeled with 35S-Met, immunoprecipitated with anti-E Mab FL0232 and subjected to SDS-12% PAGE. The size of molecular weight markers is shown in kDa. One representative experiment of two is shown.
C-terminal MH domain mutations affect the assembly and entry of replicon particles
To further investigate the effects of the C-terminal MH domain mutations on the assembly of replicon particles and infectivity, we employed a replicon particle packaging system, in which a DENV2 luciferase-reporting replicon was trans supplied with DENV2 WT or mutant structural proteins (CprME) by sequential transfections (Fig. 4A). Three mutant CprME constructs, of which each contained a proline substitution at the C-terminus of the MH domain (A120P, I123P and V127P) were generated. After the second transfection, Western blot analysis revealed comparable amounts of E proteins in the WT and mutants’ packaging cells, suggesting that these MH domain mutations do not affect the expression of E protein (Fig. 4B). The luciferase activities detected in the mutants’ packaging cells were similar to that in the WT packaging cells, suggesting that these mutations do not affect the replication of replicon RNA (Fig. 4C). To monitor the amounts of packaged replicon particles in the culture supernatants, we performed a real-time RT-PCR assay to quantify the extracellular replicon RNA at 48 and 72 h post-transfection. As shown in Fig. 4D, all three mutations significantly reduced the amounts of replicon RNA in the culture supernatants. Compared with that from the WT packaging cells, the extracellular replicon RNA from mutants A120A-, I123P- and V127P-packaging cells reduced by 87.3%, 96.7% and 97.6%, respectively, at 72 h post-transfection. These findings suggest that the C-terminal MH domain mutations affect the assembly of replicon particles.
Fig. 4.

Effects of MH domain mutations on the assembly and infectivity of replicon particles. (A) DENV2 replicon particles were prepared by sequential transfections of BHK-21 cells with replicon RNA and WT or mutant CprME RNA. (B) At 30 h after the first transfection, cells were examined for E protein expression by Western blot analysis using anti-E Mab 4G2. Host γ-tubulin protein was used as a loading control. (C) At 32, 48 and 72 h post-transfection, the luciferase activities in the transfected cells were measured. (D) Viral RNA extracted from the culture supernatants (containing replicon particles) were quantified by real-time RT-PCR; the relative amounts of replicon RNA to that of the WT replicon RNA at 72 h post-transfection (100%) are presented. (E) Equal amounts of WT and mutant replicon particles (normalized by RNA amounts) were inoculated to Vero cells. At 48 h, the luciferase activities in the mutant-infected cells were measured; the luciferase signals relative to that of the WT-infected cells (100%) are shown. Data are mean and standard errors from three independent experiments.
To examine the effect of these mutations on the infectivity of replicon particles, which have been shown to be able to complete one-round of replication (Jones et al., 2005; Pierson et al., 2006; Puig-Basagoiti et al., 2005), we infected Vero cells with equal amounts of WT and mutant replicon particles normalized by the amounts of replicon RNA, and measured the luciferase activities in the target cells at 48 h post-infection. As shown in Fig. 4E, the luciferase signals in the mutant replicon particle A120P-, I123P-, and V127P-infected cells were 29.7%, 0.16%, and 0.03%, respectively, of those of the WT replicon particle-infected cells. Since these mutations were present on the surface of replicon particles and did not affect the replication of replicon RNA (Fig. 4C), the reduction of luciferase signals in the target cells was most likely due to a defect in the entry of these MH domain mutant particles.
C-terminal MH domain mutations affect the assembly and infectivity of virions
To further investigate the involvement of the C-terminal MH domain in the assembly and infectivity of virions, the three mutations (A120P, I123P and V127P) were introduced to an infectious clone of DENV2. After transfection to BHK-21 cells with equal amounts of RNA, the numbers of positive cells detected by immunofluorescence assay (IFA) increased from 24 to 72 h in the cells transfected with WT RNA (Fig. 5A). In contrast, the numbers of IFA-positive cells remained similar from 24 to 72 h in the mutant I123P RNA- or V127P RNA-transfected cells, suggesting no or limited spreading of virus in the cultures. Mutant A120P RNA-transfected cells exhibited a slight increase in IFA-positive cells over time. At 72 h, the IFA-positive cells in the cells transfected with WT, A120P, I123P and V127P RNA were 80%, 20%, 13% and 14%, respectively (calculated from three random views under microscope).
Fig. 5.

Effects of MH domain mutations on the assembly and infectivity of virions. (A) BHK-21 cells were transfected with equal amounts (10 µg) of WT and mutant genome-length RNA derived from a DENV2 infectious cDNA clone, and viral protein synthesis was analyzed by IFA at 24, 48 and 72 h post-transfection. Anti-E Mab 4G2 and Alexa Fluor 488 goat anti-mouse IgG were used as primary and secondary antibodies, respectively. (B) Pellets (containing virions) derived from ultracentrifugation of culture supernatants on day 5 post-transfection were examined by Western blot analysis using anti-E Mab 4G2 (upper panel). The size of molecular weight markers is shown in kDa. Viral RNA extracted from the culture supernatants (containing virions) were quantified by real-time RT-PCR; the relative amount of viral RNA of each mutant to that of WT virus (100%) is shown (lower panel). (C) Plaque morphologies of WT and mutant viruses. Viruses collected on day 5 post-transfection (passage 0) were subjected to plaque assay in BHK-21 cells as described previously (Poh et al., 2009). After five rounds of continuous culture on Vero cells, the passage 5 viruses were also subjected to plaque assay. (D) Sequencing chromatograms of viral RNA extracted from culture supernatants of passages 0 and 5 viruses. The nucleotides and corresponding amino acids are indicated above the chromatogram peaks.
To examine the production of virions, pellets derived from culture supernatants post-transfection were subjected to Western blot analysis using anti-E Mab. Compared with that in the WT pellets, the amount of E protein in pellets derived from these mutants was greatly reduced with very faint band detected for mutants I123P and V127P (Fig. 5B, upper panel). Consistent with this, the amount of viral RNA in the culture supernatants (containing virions) of mutants A120P, I123P and V127P were 1.8, 4.3 and 4.2 logs, respectively, lower than that of the WT (Fig. 5B, lower panel). Together, these results suggest that all three MH domain mutants affect the assembly of virions with greater impairment in mutants I123P and V127P than in mutant A120P.
Plaque assay was also carried out to examine the culture supernatants collected from transfected cells (passage 0) and revealed similar plaque sizes for WT, mutants I123P and V127P (Fig. 5C); the plaque sizes of these viruses did not change after five rounds of passages on Vero cells. Sequencing the entire CprME genes of the passage 0 viruses of mutants I123P and V127P showed that the engineered proline had been predominantly reverted to leucine and low levels of the engineered proline were detected in both mutants, possibly derived from the residual undegraded RNA after trsnfection. The original proline mutations (I123P and V127P) were completely reverted to leucine substitutions (I123L and V127L, respectively) in the passage 5 viruses (Fig. 5D, middle and lower panels). These results suggest that a reversion of proline to leucine at positions 123 and 127 is required to restore viral replication/infection.
In contrast to I123P and V127P mutants, the passage 0 virus of A120P had heterogeneous plaque sizes with more small plaques than large plaques; the passage 5 virus of this mutant showed more homogeneous plaque sizes with predominant large plaques (Fig. 5C). Sequencing the CprME genes of the passage 0 and 5 viruses of mutant A120P revealed that the passage 0 virus retained the original mutation (proline at position 120), whereas the passage 5 virus displayed a mixed population (proline and leucine at position 120; tryptophan and glycine at position 115) (Fig. 5D, upper panels), suggesting that the original A120P mutation could be tolerated for productive infection (though with reduced efficiency) and adaptive mutations accumulated gradually.
Validation of V127L adaptation
Since only the CprME genes rather than complete genome were sequenced, it is possible that in addition to the observed leucine substitutions (123L and 127L) in the passage 5 viruses of mutants I123P and V127P (Fig. 5D), other mutations outside the structural genes may also contribute to the restored viral replication. To exclude this possibility, we engineered V127L substitution into the WT infectious cDNA clone. After transfection to BHK-21 cells with equal amounts of RNA, the numbers of IFA-positive cells increased from 24 to 72 h in both WT and V127L RNA-transfected cells (Fig. 6A), suggesting rapid virus spreading in both cultures. This was in stark contrast to the V127P RNA-transfected cells, which showed no virus spreading (Fig. 5A). The mutant V127L virus exhibited plaque size and morphology similar to those of the WT virus (Fig. 6B). Sequencing the CprME genes revealed that the engineered mutation retained in mutant virus V127L (Fig. 6C). Comparable viral titers were detected for WT and mutant V127L viruses at various time points post-transfection (Fig. 6D). These results suggest that a single amino acid substitution from proline to leucine at position 127 of prM protein can restore viral replication of mutant V127P.
Fig. 6.

Characterization of 127VL adaptation. (A) Analysis of viral protein synthesis in BHK-21 cells, which were transfected with equal amounts (10 µg) of WT and mutant V127L genome-length RNA, by IFA at 24, 48 and 72 h post-transfection. Data are presented as in Fig. 5A. (B) Plaque morphologies of the WT and mutant V127L viruses collected on day 5 post-transfection (Poh et al., 2009). (C) Sequencing chromatograms of viral RNA extracted from culture supernatants of WT and mutant V127L viruses on day 5 post-transfection. (D) Production of the WT and mutant V127L viruses after RNA transfection. Viral titers (PFU/ml) in the culture supernatants collected every 24 h from day 1 to 5 post-transfection were determined by plaque assay (Poh et al., 2009).
Discussion
In this study, we used the approach of site-directed mutagenesis and different functional assays to investigate the roles of the MH domain of prM protein in the DENV replication cycle. Analysis of the assembly of VLPs of DENV1, DENV2 and DENV4 suggested that proline substitutions of three residues (120, 123 and 127) at the C-terminus of MH domain greatly affected the amounts of VLPs detected in the lumen of ER and in the culture supernatants. Due to the size heterogeneity of VLPs, it is possible that some small VLPs might not be sedimented through 20% sucrose cushion after ultracentrifugation and thus be detected in the pellets. However, it has been shown in a study of TBEV prM/E constructs that a sensitive four-layer ELISA detected all the E protein in the pellets (presumably containing both large and small sized VLPs) but not in the supernatants after ultracentrifugation of the culture medium (Allison et al., 1999), suggesting that the pelleting efficiency of heterogenous VLPs is unlikely to account for the less amounts of VLPs detected in the pellets of mutant VLPs (presumably containing large and small sized VLPs as well). The possibility that these mutations may affect the stability of VLPs and thus the amounts of VLPs detected in the ER lumen and culture supernatants could not be completely ruled out; it is worth noting that the amounts of mutant prM/E proteins in the total cell lysates were generally comparable to those of the WT (Figs. 1D, 3A and 3B), suggesting that the expression and stability of mutant prM/E proteins in the cells were not greatly affected. Further pulse-chase experiment is needed to address the stability of these mutant VLPs in different fractions. Introducing these mutations to a DENV2 replicon packaging system revealed that all three mutants affect the assembly of replicon particles and two mutants greatly affect the entry. Moreover, all three mutants affect the assembly and infectivity of virions from a DENV2 infectious clone. To our knowledge, this is the first report demonstrating that the C-terminus of the MH domain of prM protein plays an important role in both assembly and entry of DENV, two important steps of the replication cycle.
Characterization of the adaptive changes of these mutant viruses provided interesting information regarding the structure and function of the MH domain. After sequencing the entire CprME gene, all the adaptive changes were found in the MH domain rather than in other domains of the CprME proteins (Fig. 5D), suggesting that the MH domain is probably structurally distinct. This is consistent with the cryo-EM picture of the MH domain, which is close to and partially buried in the membrane rather than interacting with the ectodomain of E protein or other part of prM protein (Zhang et al., 2003). That all adaptive changes contained substitutions to a hydrophobic but not a non-hydrophobic residue suggests the importance of hydrophobic residues to maintain the helical structure of MH domain. Interestingly, the adaptive changes involved substitutions to either leucine or tryptophan rather than to other hydrophobic residues such as isoleucine, valine, methionine and phenylalanine. Analysis of the codons revealed that the substitutions from proline to leucine (CCA to CTA of residue 120, CCT to CTT of residue 123, and CCC to CTC of residue 127) and from glycine to tryptophan (GGG to TGG of residue 115) involve only one nucleotide change, presumably the easiest and quickest way to acquire viable adaptation, whereas substitutions to other hydrophobic residues generally involve two or more nucleotide changes (Table 1). Notably, the adaptive changes for mutants I123P and V127P were found in the passage 0 viruses, suggesting that the original proline mutations at these positions were not tolerated and the adaptation occurred very rapidly (Fig. 5D). The observations that introducing one substitution (V127L) to the WT infectious clone completely restore virus replication of V127P (Figs. 6A and 6D) suggest a single amino acid adaptation from proline to leucine at position 127 is sufficient. Compared with the severe impairment of VLPs assembly by DENV4 mutant I127P, introducing leucine substitution at position 127 in the DENV4 prM/E expression construct completely restore the assembly of VLPs (data not shown).
Table 1.
Adaptive codon changes in the prM gene of passage 0 and 5 viruses of mutants A120P, I123P and V127P
| Passage 0 virusesa | Passage 5 virusesa | Possible codon changes of the adapted residues in these mutants to other hydrophobic residuesb |
|---|---|---|
| Mutant A120P: | 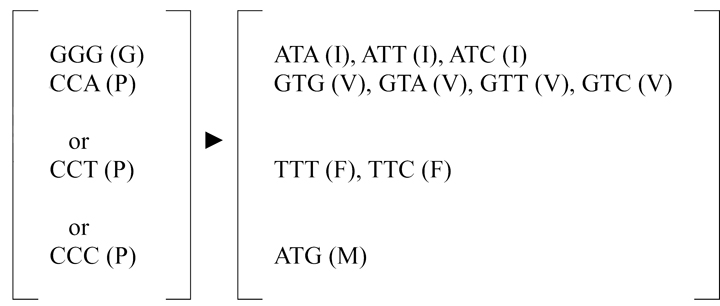 |
|
| residue 115, GGG (G)* ►TGG (W) | ||
| residue 120, CCA (P) ►CTA (L)* | ||
| Mutant I123P: | Mutant I123P | |
| residue 123, CCT (P)* ►CTT (L) | residue 123, CCT (P) ►CTT (L) | |
| Mutant V127P: | Mutant V127P: | |
| residue 127, CCC (P)* ►CTC (L) | residue 127, CCC (P) ►CTC (L) |
BHK-21 cells were transfected with equal amounts of WT and each mutant genome-length RNA derived from a DENV2 infectious cDNA clone, and culture supernatants collected on day 5 post-transfection of each mutant (passage 0 viruses) were continuously cultured on Vero cells for 5 rounds (5 days per round) to obtain the passage 5 viruses. Passage 0 and 5 viruses of each mutant (A120P, I123P and V127P) were subjected to RNA extraction, RT-PCR and sequencing of the entire CprME genes. Adaptive codon changes were only found in the prM gene and are presented. Parentheses indicate the amino acids with single letter designations, and single nucleotide changes are underlined. Asters indicate a minor proportion based on the sequencing chromatograms in Fig. 5D.
As a comparison, all the possible codon changes of the adapted residues in these mutants to other hydrophobic residues, including isoleucine, valine, phenylalanine, and methionine are shown.
On the other hand, the passage 0 virus of mutant A120P retained the original proline mutation and the adaptive changes were found in the passage 5 virus, suggesting that the original mutation was tolerated for a productive but probably inefficient replication, as was evidenced by the predominant small sized plaques in the passage 0 virus (Fig. 5C). The appearance of predominant large sized plaques in the passage 5 virus, which coincided with the adaptive changes of tryptophan over glycine at position 115, may suggest gradual adaptation to more efficient replication. Whether the large plaques in the passage 5 virus contain tryptophan substitution at position 115 and the small plaques in the passage 0 virus contain the original proline mutation at position 120 remain to be verified by sequencing each plaque-purified virus.
By studying VLPs, packaging of replicon particles and virions production, we showed that the C-terminus rather than the N-terminus of the MH domain is involved in the assembly. Cryo-EM study of DENV2 virions revealed that the C-terminus of the MH domain as well as the EH1 and EH2 domains of E protein are partially buried in the outer leaflet of viral membrane (Zhang et al., 2003), it is conceivable that mutations introduced to the MH domain might affect its association with the membrane in ER after biosynthesis, and thus block the curving and bending of the membrane during assembly. While the assembly and budding of flaviviral particles or VLPs from the ER membrane to the lumen is believed to be a rapid process due to the lack of budding intermediates clearly observed thus far (Ishak et al., 1988; Lindenbach et al., 2007; Mackenzie and Westaway, 2001), future EM studies in combination with blocking of vesicles budding from ER or trans-Golgi by low temperature might provide morphological information regarding how the MH domain is involved in the assembly (Beckers and Balch, 1989; Greenfield et al., 1999).
In the infectivity assay of replicon particles, two of the three proline mutants (I123P, V127P) at the C-terminus of the MH domain affected entry. After the entry of DENV through receptor mediated endocytosis, the low-pH environment in the endosome triggers a series of conformational change of E protein and fusion process, including the hinge outward of domain II of E protein, lateral rearrangement of E protein, exposure and insertion of the fusion loop of domain II into endosomal membrane, trimerization of E protein, folding back of domain III and the stem-anchor toward domain II, approximation of membranes, and formation of fusion pore (Bressanelli et al., 2004; Liao and Kielian, 2005; Lindenbach et al., 2007; Modis et al., 2004; Mukhopadhyay et al., 2005; Zhang et al., 2004). The presence of a pre-fusion intermediate with exposed EH1 and EH2 domains was supported by the observations that peptides derived from the stem as well as recombinant domain III, which bound fusion intermediate, can block the fusion and by the recent capture of such intermediate using cryo-EM (Hrobowski et al., 2005; Kaufmann et al., 2009; Liao and Kielian, 2005; Schmidt et al., 2010). Although how the M protein, which is believed to locate in the “hole” between two E monomers with the MH domain partially buried in the membrane, changes during the fusion process remains unclear, it is likely that the MH domain also undergoes considerable conformational change given that dramatic conformational changes of the EH1 and EH2 domains occur from the membrane-associated position to outward extension in the pre-fusion intermediate and finally folding back to domain II. In this regard, our findings suggest that disrupting the C-terminal helical structure of the MH domain could affect the fusion process during virus entry. Alternatively, these two mutants might affect entry by reducing the efficiency of prM cleavage, though only sequences proximal to the cleavage site (residue 91) have been reported to affect prM cleavage thus far (Junjhon et al., 2008; Keelapang et al., 2004).
Comparison of the amino acid sequences of the MH domain among different flaviviruses revealed that the three residues, 120, 123 and 127, at the C-terminus of the MH domain are not absolutely conserved by particular amino acids but are conserved by hydrophobic amino acids, which is consistent with their location at the hydrophobic face (a or d position) of the MH helical domain (Table 2). This feature is also in agreement with the observed phenotypes of the mutants, in which substitutions of alanine but not proline were somewhat tolerated (Fig. 5A), and consistent with the adaptive changes, in which substitutions to leucine, one of the hydrophobic residues, rather than the original hydrophobic residue were found (Table 1). Interestingly, there are nine residues in this region, which are adjacent to the residues at the a or d position and are highly conserved by all four DENV serotypes or all flaviviruses, suggesting the structural or functional importance of this domain. It should be noted that since proline substitution greatly disrupted the helical structure, the phenotypes of proline mutants in this study were attributed to the effect of structure rather than the side chains of particular amino acids. Further studies involving substitutions to alanine and other amino acids on these highly conserved residues would better delineate their functional roles and lead to the identification of potential targets of anti-virals in the future.
Table 2.
Comparison of amino acid sequences of the MH domain of prM protein of different flavivirusesa
 |
The MH domain of prM protein is an α-helical domain (Zhang et al., 2003). sequences of the MH domain of multipile strain of flaviviruses were obtained from GenBank and analyzed. single letter designations of amino acid are shown with the numbers above indicating amino acid positions and the numbers beneath indicating the numbers of strains containing such residue. The residues at the a or d position of the helical wheel are bolded. Asters indicate residues highly conserved among the four DENV serotypes (*) or flaviviruses (**) analyzed.
Materials and Methods
Plasmid constructs of VLPs
The prM/E expression constructs of DENV1 (pCB-D1), DENV2 (pCB-D2) and DENV4 (pCB-CD4) were described previously (Hu et al., 2007). To generate the MH mutants of DENV4, a two-step PCR mutagenesis was performed using pCB-D4 as template and the primers (d4MS113PA2, d4MS113PB1, d4MA116PA2, d4MA116PB1, d4MA120PA2, d4MA120PB1, d4MV123PA2, d4MV123PB1, d4MI127PA2, d4MI127PB1, d4MS113AA2, d4MS113AB1, d4MV123AA2, d4MV123AB1, d4MI127AA2, d4MI127AB1, d4KpnSS, d4EBstEIIB2) as described previously (Hu et al., 2007). After the second round PCR, the 950-bp product containing the MH mutation was digested with KpnI/BstEII, and cloned into pCB-D4. To generate the MH mutants of DENV1 and DENV2, a two-step PCR mutagenesis was performed using pCB-D1 and pCB-D2 as templates, respectively, and the primers (d1MI120PA2, d1MI120PB1, d1MV123PA2, d1MV123PB1, d1MA127PA2, d1MA127PB1, d2MV120PA2, d2MV120PB1, d2MI123PA2, d2MI123PB1, d2MI127PA2, d2MI127PB1). After the second round PCR, the 997-bp and 722-bp products containing the MH mutations were digested with KpnI/DraIII and KpnI/Bsu36I, and cloned into pCB-D1 and pCB-D2, respectively. All the constructs were confirmed by sequencing the entire inserts to rule out second site mutations. The sequences of all primers for mutagenesis will be provided upon request.
Cell lysates, VLPs and Western blot analysis
293T cells prepared in a 10 cm-culture dish at 5×105 cells per dish one day earlier were transfected with 10 µg of plasmid DNA by calcium phosphate method (Hsieh et al., 2008). At 48 h post-transfection, culture supernatants were collected (see below), and cells were washed with PBS and treated with 1% NP40 lysis buffer containing protease inhibitors (Roche Diagnostics), followed by centrifugation at 20,000 × g at 4°C for 30 min to obtain cell lystates (Hsieh et al., 2008). Culture supernatants were clarified by centrifugation at 1,250 × g for 20 min, filtered through a 0.22 µm pore-sized membrane (Sartorius), layered over a 20 % sucrose buffer, and ultracentrifuged at 65,000 × g at 4°C for 5 h to obtain pellets, which were resuspended in 30 µl TNE buffer (Hsieh et al., 2008). For Western blot analysis, cell lysates and pellets were subjected to SDS-12% polyacrylaminde gel electrophoresis (PAGE), followed by transfer to nitrocellulose membrane, blocking and incubation with primary antibody, human serum from confirmed dengue case or anti-calnexin mouse Mab E-10 (Santa Cruz), and secondary antibody (Hsieh et al., 2008; Wang et al., 2006). After final washing, the signals were detected by enhanced chemiluminescence reagents (Perkin Elmer life sciences).
Radioimmunoprecipitation
293T cells prepared in 6-well plate were transfected with plasmid DNA by calcium phosphate method. At 20 h, the cells were washed, incubated with methionine-free medium, followed by 50 Ci [35S] methonine (Amersham Biosciences) at 37°C for 6 h, and collected to obtain cell lysates (Hsieh et al., 2008). Following pre-clear, cell lysates were incubated with anti-E mouse Mab FL0232 (Chance Biotechnology) at 4°C overnight and then with protein A sepharose beads (Amersham Biosciences) at 4°C for 6 h. After washing, the beads were mixed with 2X sample buffer and heated, and the solubilized fraction was subjected to SDS-12% PAGE (Hsieh et al., 2008).
Subcellular fractionation and enzyme digestion
293T cells transfected with plasmid DNA were washed 3 times with PBS at 48 h, resuspended in modified buffer B (10% sucrose, 20mM Tris, 150mM NaCl, 10mM Mg acetate, 1mM EGTA [pH7.6]), and frozen-thawed 8 times as described previously (Hsieh et al., 2008; Xu et al., 1997). After clearing the nuclei and debris by centrifugation at 1,000 × g for 5 min, the membrane fraction was obtained by centrifugation at 20,000 × g for 30 min at 4°C. The resulting supernatants were layered over a 20% sucrose buffer and ultracentrifuged at 246,000 × g at 4°C for 1 h to obtain the pellets of soluble fraction, which were resuspended in 30 µl TNE buffer (Hsieh et al., 2008). Membrane fraction and pellets of soluble fraction were subjected to Western blot analysis. Aliquots from above were treated with 500 U of endo-β-N-acetylglucosaminidase H (endo H) or peptide N-glycosidase F (PNGase F) at 37°C for 1 h according to the manufacture’s instructions (New England Biolabs), and subjected to Western blot analysis.
Infectious clone, replicon and CprME constructs
An infectious cDNA clone of DENV2 (pACYC FLTSV; strain TSV01) and a subclone pACYC TSV-E were used to engineer mutations of the MH domain in the context of genome-length RNA (Qing et al., 2010b). The subclone pACYC TSV-E contains the SacII-XhoI fragment from the pACYC FLTSV (spanning from T7 promoter to nucleotide position 5,426 of the viral genome; GenBank accession number AY037116). A QuikChange II XL site-directed mutagenesis Kit (Stratagene) was used to engineer MH mutations into the pACYC TSV-E. The mutated DNA fragment was cut and cloned back into the pACYC FLTSV by the SacII and BsrGI sites (nucleotide position 1,840 of the genome). The DENV2 TSV01 Rluc2A replicon was constructed by replacing the structural genes (CprME) with renilla luciferase gene followed by FMDV 2A; the coding sequences of 38 amino acids at the N-terminus of C protein and 31 amino acids at the C-terminus of E protein was retained to preserve correct processing and translocation of NS1 as well as correct topology of the remaining nonstructural polyprotein across the membrane of ER. For replicon packaging constructs, the CprME-coding fragment with Kozak sequence at the 5’-end and stop codon at the 3’-end was cloned into pCR2.1-TOPO (Invitrogen); the resulting plasmid was used to engineer MH mutations by QuikChange II XL site-directed mutagenesis Kit; the mutated fragments were then cloned into a Semliki forest virus (SFV) expression vector (Puig-Basagoiti et al., 2005) through a unique XmaI site, resulting in the SFV-DENV2- prME cDNA plasmid. All the constructs were verified by sequencing the inserts and junctions.
In vitro transcription, RNA transfection, and immunofluorescence assay
The genome-length RNA and replicon RNA were in vitro transcribed from corresponding cDNA plasmids that were linealized with ClaI. A T7 mMESSAGE mMACHINE kit (Ambion) was used for RNA synthesis as described previously (Shi et al., 2002). The SFV-DENV2-CprME RNA was in vitro transcribed from a linealized DNA (predigested with the SapI) using an SP6 mMESSAGE mMACHINE kit (Ambion). Both genome-length and replicon RNA were electroporated into BHK-21 cells (Shi et al., 2002). After transfection of genome-length RNA, the cells were cultured at 37°C for 24 h and then at 30°C; culture supernatants containing viruses were collected every 24 h until day 5, and were aliquoted and stored at −80°C.
For the IFA, BHK-21 cells transfected with genome-length RNA were seeded on a chamber slide (Nalge Nunc). At the indicated time points, the cells were fixed in 100% methanol at 4°C for 30 min, washed with PBS, and incubated with mouse anti-E Mab 4G2 at room temperature for 1 h, followed by three washings with PBS and incubation with Alexa Fluor 488 goat anti-mouse IgG at room temperature for 1 h. After three washings with PBS and mounting with DAPI, the slides were analyzed under a fluorescence microscope (Leica).
Replicon particle assays
Replicon particles of DENV2 were prepared by trans-supplying of CprME proteins to replicon RNA as reported previously (Qing et al., 2010a). Briefly, 8×106 BHK-21 cells were electroporated with 10 µg of DENV2 luciferase-reporting replicon RNA in a 0.4-cm cuvette with the GenePulser apparatus (Bio-Rad) using the settings of 0.85 kV, 25 µF and three pulsings at 3-s intervals. The transfected cells were resuspended in DMEM with 10% FBS, incubated at 37°C for 24 h, and electroporated again with 10 µg of SFV replicon RNA expressing WT or mutant CprME using the settings described above. After incubation at 30°C for 48 h and 72 h post-first transfection, culture supernatants were centrifuged to remove cellular debris, and the supernatants containing replicon particles were aliquoted and stored at −80°C. To determine the RNA copy numbers in the replicon particles, replicon RNA was extracted from 140 µl of supernatants and dissolved in 50 µl RNase free water, of which 5 µl was quantified by a real-time RT-PCR assay using primers targeting the NS5 gene (5'-GAAGGAGAAGGGCTGCACAAAC-3' and 5'-GCACACGCACCACCTTGTTTTG-3') and the iScriptTM one-step RT-PCR kit with SYBR Green (Bio-Rad). For replicon particle infection assay, Vero cells (4×104 cells per well in 96-well plate) was infected with equal amounts of WT or mutant replicon particles (normalized by viral RNA copy numbers). At the indicated time points, the cells were washed once with cold PBS, lysed in 20 µl of 1× lysis buffer for 20 min, and assayed for luciferase activities by using a Renilla luciferase assay kit (Promega) and a Clarity luminescence microplate reader (BioTek).
Virion production from infectious clones
Twenty microliters of culture supernatants from genome-length RNA-transfected BHK-21 cells on day 5 were centrifuged at 4,000 × g and 4°C for 30 min to remove the cell debris. The supernatants (containing virions) were transferred to sterile Sorvall SS-34 tubes and ultracentrifuged at 45,000 × g and 4°C for 1 h. After careful removal of the supernatants, the pellets containing virions were resuspended in 500 µl PBS; aliquots were dissolved in sample buffer, boiled for 5 min, and subjected to SDS 5–12% PAGE and Western blot analysis using mouse anti-E Mab 4G2 as described above. Viral RNA in the pellets was quantified by real-time RT-PCR as described above.
Plaque assay, continuous passaging of mutants and adaptive mutations
Plaque assay in BHK-21 cells was performed as described previously (Poh et al., 2009). Viruses collected on day 5 post-transfection (passage 0) of each mutant were continuously cultured on Vero cells for 5 rounds (5 days per round) to obtain the passage 5 viruses. Viral RNA extracted from culture supernatants of passages 0 and 5 viruses was subjected to RT-PCR and sequencing of the entire CprME genes to examine the adaptive mutations.
Acknowledgment
This work was supported by the National Science Council Taiwan (NSC95-2320-B-002-084-MY3) and start-up fund from JABSOM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allison SL, Stiasny K, Stadler K, Mandl CW, Heinz FX. Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J.Virol. 1999;73:5605–5612. doi: 10.1128/jvi.73.7.5605-5612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison SL, Tao YJ, O'Riordain G, Mandl CW, Harrison SC, Heinz FX. Two distinct size classes of immature and mature subviral particles from tick-borne encephalitis virus. J. Virol. 2003;77:11357–11366. doi: 10.1128/JVI.77.21.11357-11366.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CJ, Balch WE. Calcium and GTP: essential components in vesicular trafficking between the endoplasmic reticulum and Golgi apparatus. J Cell Biol. 1989;108:1245–1256. doi: 10.1083/jcb.108.4.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Men R, Tokimatsu I, Lai CJ. Genetic determinants responsible for acquisition of dengue type 2 virus mouse neurovirulence. J. Virol. 1998;72:1647–1651. doi: 10.1128/jvi.72.2.1647-1651.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GJ, Hunt AR, Holmes DA, Springfield T, Chiueh TS, Roehrig JT, Gubler DJ. Enhancing biosynthesis and secretion of premembrane and envelope proteins by the chimeric plasmid of dengue virus type 2 and Japanese encephalitis virus. Virology. 2003;306:170–180. doi: 10.1016/s0042-6822(02)00028-4. [DOI] [PubMed] [Google Scholar]
- Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, Bowen R, Bunning ML. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J.Virol. 2001;75:4040–4047. doi: 10.1128/JVI.75.9.4040-4047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol.Cell. 2001;7:593–602. doi: 10.1016/s1097-2765(01)00206-4. [DOI] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U. S. A. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends. Microbiol. 2002;10:100–103. doi: 10.1016/s0966-842x(01)02288-0. [DOI] [PubMed] [Google Scholar]
- Guirakhoo F, Hunt AR, Lewis JG, Roehrig JT. Selection and partial characterization of dengue 2 virus mutants that induce fusion at elevated pH. Virology. 1993;194:219–223. doi: 10.1006/viro.1993.1252. [DOI] [PubMed] [Google Scholar]
- Guzman MG, Kouri G. Dengue: an update. Lancet Infect. Dis. 2002;2:33–42. doi: 10.1016/s1473-3099(01)00171-2. [DOI] [PubMed] [Google Scholar]
- Halstead SB. Pathogenesis of dengue: challenges to molecular biology. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol. J. 2005;2:49. doi: 10.1186/1743-422X-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh SC, Liu IJ, King CC, Chang GJ, Wang WK. A strong endoplasmic reticulum retention signal in the stem-anchor region of envelope glycoprotein of dengue virus type 2 affects the production of virus-like particles. Virology. 2008;374:338–350. doi: 10.1016/j.virol.2007.12.041. [DOI] [PubMed] [Google Scholar]
- Hu HP, Hsieh SC, King CC, Wang WK. Characterization of retrovirus-based reporter viruses pseudotyped with the precursor membrane and envelope glycoproteins of four serotypes of dengue viruses. Virology. 2007;368:376–387. doi: 10.1016/j.virol.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AR, Cropp CB, Chang GJ. A recombinant particulate antigen of Japanese encephalitis virus produced in stably-transformed cells is an effective noninfectious antigen and subunit immunogen. J Virol. Methods. 2001;97:133–149. doi: 10.1016/s0166-0934(01)00346-9. [DOI] [PubMed] [Google Scholar]
- Ishak R, Tovey DG, Howard CR. Morphogenesis of yellow fever virus 17D in infected cell cultures. J. Gen.Virol. 1988;69:325–335. doi: 10.1099/0022-1317-69-2-325. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Konishi E. Mosquito cells infected with Japanese encephalitis virus release slowly-sedimenting hemagglutinin particles in association with intracellular formation of smooth membrane structures. Microbiol. Immunol. 2006;50:211–223. doi: 10.1111/j.1348-0421.2006.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Jones CT, Patkar CG, Kuhn RJ. Construction and applications of yellow fever virus replicons. Virology. 2005;331:247–259. doi: 10.1016/j.virol.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Junjhon J, Lausumpao M, Supasa S, Noisakran S, Songjaeng A, Saraithong P, Chaichoun K, Utaipat U, Keelapang P, Kanjanahaluethai A, Puttikhunt C, Kasinrerk W, Malasit P, Sittisombut N. Differential modulation of prM cleavage, extracellular particle distribution, and virus infectivity by conserved residues at nonfurin consensus positions of the dengue virus pr-M junction. J. Virol. 2008;82:10776–10791. doi: 10.1128/JVI.01180-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann B, Chipman PR, Holdaway HA, Johnson S, Fremont DH, Kuhn RJ, Diamond MS, Rossmann MG. Capturing a flavivirus pre-fusion intermediate. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000672. e1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelapang P, Sriburi R, Supasa S, Panyadee N, Songjaeng A, Jairungsri A, Puttikhunt C, Kasinrerk W, Malasit P, Sittisombut N. Alterations of pr-M cleavage and virus export in pr-M junction chimeric dengue viruses. J.Virol. 2004;78:2367–2381. doi: 10.1128/JVI.78.5.2367-2381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi E, Fujii A. Dengue type 2 virus subviral extracellular particles produced by a stably transfected mammalian cell line and their evaluation for a subunit vaccine. Vaccine. 2002;20:1058–1067. doi: 10.1016/s0264-410x(01)00446-7. [DOI] [PubMed] [Google Scholar]
- Konishi E, Mason PW. Proper maturation of the Japanese encephalitis virus envelope glycoprotein requires cosynthesis with the premembrane protein. J. Virol. 1993;67:1672–1675. doi: 10.1128/jvi.67.3.1672-1675.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger MA, McMinn PC. Murray Valley encephalitis virus recombinant subviral particles protect mice from lethal challenge with virulent wild-type virus. Arch.Virol. 2002;147:1155–1172. doi: 10.1007/s00705-002-0809-3. [DOI] [PubMed] [Google Scholar]
- Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830–1834. doi: 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- Liao M, Kielian M. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J. Cell Biol. 2005;171:111–120. doi: 10.1083/jcb.200507075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YJ, Wu SC. Histidine at residue 99 and the transmembrane region of the precursor membrane PrM protein are important for the PrM-E heterodimeric complex formation of Japanese encephalitis virus. J.Virol. 2005;79:8535–8544. doi: 10.1128/JVI.79.13.8535-8544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Thiel HJ, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott William & Wilkins; 2007. pp. 1101–1152. [Google Scholar]
- Lorenz IC, Allison SL, Heinz FX, Helenius A. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J. Virol. 2002;76:5480–5491. doi: 10.1128/JVI.76.11.5480-5491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz IC, Kartenbeck J, Mezzacasa A, Allison SL, Heinz FX, Helenius A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J. Virol. 2003;77:4370–4382. doi: 10.1128/JVI.77.7.4370-4382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J.Virol. 2001;75:10787–10799. doi: 10.1128/JVI.75.22.10787-10799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JE, Pierson TC, Hubka S, Rucker S, Gordon IJ, Enama ME, Andrews CA, Xu Q, Davis BS, Nason M, Fay M, Koup RA, Roederer M, Bailer RT, Gomez PL, Mascola JR, Chang GJ, Nabel GJ, Graham BS. A West Nile virus DNA vaccine induces neutralizing antibody in healthy adults during a phase 1 clinical trial. J..Infect. Dis. 2007;196:1732–1740. doi: 10.1086/523650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc.Natl.Acad.Sci.U. S. A. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 2005;79:1223–1231. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- Murray JM, Aaskov JG, Wright PJ. Processing of the dengue virus type 2 proteins prM and C-prM. J. Gen. Virol. 1993;74:175–182. doi: 10.1099/0022-1317-74-2-175. [DOI] [PubMed] [Google Scholar]
- Op De Beeck A, Molenkamp R, Caron M, Ben Younes A, Bredenbeek P, Dubuisson J. Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J. Virol. 2003;77:813–820. doi: 10.1128/JVI.77.2.813-820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlinger KK, Hoenninger VM, Kofler RM, Mandl CW. Construction and mutagenesis of an artificial bicistronic tick-borne encephalitis virus genome reveals an essential function of the second transmembrane region of protein e in flavivirus assembly. J. Virol. 2006;80:12197–12208. doi: 10.1128/JVI.01540-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology. 2006;346:53–65. doi: 10.1016/j.virol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Poh MK, Yip A, Zhang S, Priestle JP, Ma NL, Smit JM, Wilschut J, Shi PY, Wenk MR, Schul W. A small molecule fusion inhibitor of dengue virus. Antiviral Res. 2009;84:260–266. doi: 10.1016/j.antiviral.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Pryor MJ, Azzola L, Wright PJ, Davidson AD. Histidine 39 in the dengue virus type 2 M protein has an important role in virus assembly. J. Gen. Virol. 2004;85:3627–3636. doi: 10.1099/vir.0.80283-0. [DOI] [PubMed] [Google Scholar]
- Puig-Basagoiti F, Deas TS, Ren P, Tilgner M, Ferguson DM, Shi PY. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 2005;49:4980–4988. doi: 10.1128/AAC.49.12.4980-4988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy DE, Noga AJ, Chang GJ. Noninfectious recombinant antigen for detection of St. Louis encephalitis virus-specific antibodies in serum by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 2004;42:4709–4717. doi: 10.1128/JCM.42.10.4709-4717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing M, Liu W, Yuan Z, Gu F, Shi PY. A high-throughput assay using dengue-1 virus-like particles for drug discovery. Antiviral Res. 2010a;86:163–171. doi: 10.1016/j.antiviral.2010.02.313. [DOI] [PubMed] [Google Scholar]
- Qing M, Zou G, Wang QY, Xu HY, Dong H, Yuan Z, Shi PY. Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob. Agents Chemother. 2010b July 6; doi: 10.1128/AAC.00561-10. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph VB, Stollar V. Low pH-induced cell fusion in flavivirus-infected Aedes albopictus cell cultures. J. Gen. Virol. 1990;71:1845–1850. doi: 10.1099/0022-1317-71-8-1845. [DOI] [PubMed] [Google Scholar]
- Russell PK, Brandt WE, Dalrymple JM. Chemical and antigenic structure of flaviviruses. In: Schlesinger RW, editor. The Togaviruses. New York: Academic Press; 1980. pp. 503–529. [Google Scholar]
- Schalich J, Allison SL, Stiasny K, Mandl CW, Kunz C, Heinz FX. Recombinant subviral particles from tick-borne encephalitis virus are fusogenic and provide a model system for studying flavivirus envelope glycoprotein functions. J. Virol. 1996;70:4549–4557. doi: 10.1128/jvi.70.7.4549-4557.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000851. e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi PY, Tilgner M, Lo MK, Kent KA, Bernard KA. Infectious cDNA clone of the epidemic west nile virus from New York City. J. Virol. 2002;76:5847–5856. doi: 10.1128/JVI.76.12.5847-5856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J.Virol. 1997;71(11):8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiasny K, Allison SL, Marchler-Bauer A, Kunz C, Heinz FX. Structural requirements for low-pH-induced rearrangements in the envelope glycoprotein of tick-borne encephalitis virus. J. Virol. 1996;70:8142–8147. doi: 10.1128/jvi.70.11.8142-8147.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TT, Bhuvanakantham R, Li J, Howe J, Ng ML. Tyrosine 78 of premembrane protein is essential for assembly of West Nile virus. J. Gen. Virol. 2009;90:1081–1092. doi: 10.1099/vir.0.007872-0. [DOI] [PubMed] [Google Scholar]
- Wang S, He R, Anderson R. PrM- and cell-binding domains of the dengue virus E protein. J. Virol. 1999;73:2547–2551. doi: 10.1128/jvi.73.3.2547-2551.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WK, Chen HL, Yang CF, Hsieh SC, Juan CC, Chang SM, Yu CC, Lin LH, Huang JH, King CC. Slower rates of clearance of viral load and virus-containing immune complexes in patients with dengue hemorrhagic fever. Clin. Infect. Dis. 2006;43:1023–1030. doi: 10.1086/507635. [DOI] [PubMed] [Google Scholar]
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Dengue and dengue hemorrhagic fever. Fact sheet no. 117. 2009 http://www.who.int/mediacentre/factsheets/fs117/en.
- Xu Z, Bruss V, Yen TS. Formation of intracellular particles by hepatitis B virus large surface protein. J. Virol. 1997;71:5487–5494. doi: 10.1128/jvi.71.7.5487-5494.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 2003;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]