

Abstract
Lys120 in the DNA-binding domain (DBD) of p53 becomes acetylated in response to DNA damage. But, the role and effects of acetylation are obscure. We prepared p53 specifically acetylated at Lys120, AcK120p53, by in vivo incorporation of acetylated lysine to study biophysical and structural consequences of acetylation that may shed light on its biological role. Acetylation had no affect on the overall crystal structure of the DBD at 1.9-Å resolution, but significantly altered the effects of salt concentration on specificity of DNA binding. p53 binds DNA randomly in vitro at effective physiological salt concentration and does not bind specifically to DNA or distinguish among its different response elements until higher salt concentrations. But, on acetylation, AcK120p53 exhibited specific DNA binding and discriminated among response elements at effective physiological salt concentration. AcK120p53 and p53 had the highest affinity to the same DNA sequence, although acetylation reduced the importance of the consensus C and G at positions 4 and 7, respectively. Mass spectrometry of p53 and AcK120p53 DBDs bound to DNA showed they preferentially segregated into complexes that were either DNA(p53DBD)4 or DNA(AcK120DBD)4, indicating that the different DBDs prefer different quaternary structures. These results are consistent with electron microscopy observations that p53 binds to nonspecific DNA in different, relaxed, quaternary states from those bound to specific sequences. Evidence is accumulating that p53 can be sequestered by random DNA, and target search requires acetylation of Lys120 and/or interaction with other factors to impose specificity of binding via modulating changes in quaternary structure.
Keywords: electrostatic interactions, transcription, genetic code expansion, posttranslational modifications
The tumor suppressor p53 is a key component in the signaling network activated in response to oncogenic and other cellular stresses. It is pivotal in the prevention of cancer development, by promoting cell cycle arrest, senescence, or apoptosis (1). As a transcription factor, p53 is active as a homotetramer of multidomain monomers, each composed of the tetramerization (Tet) and DNA-binding domains (DBD) and intrinsically disordered regions (2). Tetrameric p53 binds to DNA response elements (REs) that consist of two decameric motifs or half sites of the general form RRRCWWGYYY (R = A, G; W = A, T; Y = C, T) separated by 0–14 base pairs, although more than two spacing base pairs is rare (3). Although once thought essential, the C at position 4 and the G at 7 are now known to be replaced in some REs, and there is a huge range of targets (4).
The complex activities of p53 are tightly regulated by an assortment of protein–protein interactions and posttranslational modifications (5, 6). More than 35 different amino acids in p53 are posttranslationally modified during normal homeostasis and under stress. In many cases, the exact role of individual posttranslational modification is not clear. Here, we study the acetylation of Lys120 in the DBD. In response to DNA damage, Lys120 is acetylated by TIP60 and hMOF acetyltransferases of the MYST family (7, 8). There is evidence that the acetylation of Lys120 is important for p53-induced apoptosis, and not cell cycle arrest (7, 8). In contrast to wild-type p53, the K120R mutant is incapable of promoting the transcription of proapoptotic genes BAX or PUMA and inducing p53-mediated apoptosis. This discrimination was explained by the selective accumulation of the Lys120-acetylated form of p53, Ac120Kp53, at proapoptotic BAX and PUMA promoters, seen by ChIP of camptothecin-treated LNCaP and MCF-7 cell lines (7). It was also suggested that Lys120 acetylation is important for p53-induced transcription-independent apoptosis, mainly via the mitochondria (9). The population of p53 in the mitochondria is enriched with the acetylated Lys120 variant, although the acetylation is not required for the localization of p53. Further, Lys120 acetylation is essential for effective displacement of the antiapoptotic protein MCL-1 from the complex with proapoptotic BAK (9). Hence, it is assumed that Lys120 acetylation differentiates between p53-mediated cell cycle arrest and apoptosis.
The exact mechanism by which Lys120 acetylation promotes apoptosis is not clear, especially its correlation with the acetylation of six lysine residues at the C terminus. For example, homozygous mutant mice in which the C-terminal lysine residues are mutated to arginine show no significantly different p53 response from wild-type mice, except for partially impaired transcriptional activation upon DNA damage (10). There are similar results for the same mutations introduced into endogenous p53 in mouse embryonic stem cells (11). However, when all eight lysines that are known to be acetylated are mutated to arginine (six at the C terminus and two within the DNA-binding domain, including K120), p53 is incapable of inducing growth arrest and apoptosis, but at the same time is able to retain its DNA-binding capacity as a transcription factor and induce the p53-Mdm2 feedback loop (12). Although Lys120 directly interacts with DNA (13, 14), the eight lysine-to-arginine mutations have no effect on DNA binding, as measured by electrophoretic mobility shift assay. In contrast, a different study showed that the mutant K120A expressed at low concentration in H1299 cells has lower affinity for DNA than does the wild-type protein (15).
In order to study the effect of Lys120 acetylation on p53 structure and DNA-binding properties, we used an evolved Methanosarcina barkeri pyrrolysyl-tRNA synthetase/tRNACUA pair to encode genetically the incorporation of acetylated lysine in response to an amber stop codon in Escherichia coli (16). This system allowed us to express and purify a superstable quadruple mutant p53 (17) and the wild-type DBD, acetylated at position Lys120 (AcK120p53 and AcK120DBD, respectively). We were able to measure the binding affinity of AcK120p53 to different DNA REs, solve the crystal structure of the AcK120DBD, and perform mass spectrometry studies to search for biophysical and structural effects of acetylation that may be relevant to its biological role.
Results
Expression and Purification of Site-Specifically Acetylated p53.
We expressed site-specifically acetylated p53 in E. coli cells by genetically encoding the incorporation of Nε-acetyl-lysine in response to the amber stop codon. The quadruple-mutant version of full-length p53 (p53) (17, 18) or the wild-type DBD (residues 94–293) carrying a TAG stop codon at position 120, were coexpressed with an orthogonal Nε-acetyllysyl-tRNA synthetase/tRNACUA pair, in the presence of Nε-acetyl-lysine. Incorporation of acetylated lysine was verified by Western blot, using an antibody against acetylated lysine (Fig. 1, lanes 3–6), and by electrospray ionization mass spectroscopy. Typical yields of pure protein were about 20% to 30% of that obtained for the nonacetylated version due, mainly, to the expression of the truncated version (residues 1–119, Fig. 1, lane 9). When protein was expressed without Nϵ-acetyl-lysine in the medium (Fig. 1, lane 10), only the truncated version could be detected by Western blot using an antibody against residues 44–56 of p53. We found that in vitro, Lys120 acetylation has no effect on the thermodynamic and kinetic stability of the DBD (Fig. S1). In addition, our data suggest that in vitro, AcK120DBD does not bind to Mcl-1 (residues 183–330) with a Kd in the micromolar range (Fig. S2).
Fig. 1.

Expression of site-specifically acetylated p53. Coomassie blue staining of site-specifically acetylated and nonacetylated p53 (lanes 1, 2). Only proteins expressed in the presence of acetylated lysine were detected (lanes 3–6) on analysis by Western blot using an antibody against Nε-acetyl-lysine. Using an antibody against p53 (residues 44–56), we detected both acetylated and nonacetylated proteins (lanes 7–10). The presence of the amber stop codon in position 120 gives rise to the expression of a truncated version (lane 9, middle band, lipoyl domain and residues 1–119) together with the full-length protein (lane 9, upper band, lipoyl domain and full-length p53). Specificity of the Nε-acetyllysyl-tRNA synthetase/tRNACUA pair is demonstrated by only the truncated version being expressed when cells were grown in media not supplemented with Nε-acetyl-lysine (lane 10).
DNA-Binding Affinity and Specificity.
We used fluorescence anisotropy in order to study the effect of Lys120 acetylation on DNA-binding affinity and specificity (Table 1) (19, 20). We compared the binding of p53 and AcK120p53 to REs with different affinities: (i) high affinity sequences—a 20 base-pair consensus-sequence response element (20 bp RE) (21) and the p21 RE; (ii) moderate affinity sequence, PUMA2; (iii) low affinity sequences, BAX and PUMA1 REs; and (iv) as controls for nonspecific binding, a 20-bp random sequence and the 30-bp NFκB RE (Table 1).
Table 1.
DNA-binding affinities determined by fluorescence anisotropy titration
|
I = 150 mM |
I = 211 mM |
I = 286 mM |
I = 225 mM |
||||
| DNA* | p53 WT Kd/nM (h) | AcK120 Kd/nM (h) | p53 wt Kd/nM (h) | AcK120 Kd/nM (h) | p53 Kd/nM (h) | AcK120 Kd/nM (h) | CT†Kd/nM (h) |
| 20-bp random | 59 (1.7) | 140 (1.2) | 290 (1) | 1,100 (1) | ≫1,000 | ≫1,000 | |
| 20-bp random + 2 mM MgCl2 | 380 (1) | ||||||
| 20-bp random KCl buffer | 310 (1) | ||||||
| 30-bp NFκB | 25 (2.1) | 35 (2.0) | 41 (1.4) | 85 (1.2) | 500 (1) | ≫1,000 | |
| 20-bp RE | 22 (1.5) | 17 (1.4) | 18 (1.3) | 22 (1.3) | 51 (1.3) | 70 (1) | |
| 20-bp RE + 2 mM MgCl2 | 18 (1.6) | 25 (1.4) | |||||
| P21 | 40 (1.8) | 29 (1.8) | 26 (1.9) | 33 (1.3) | 55 (1.2) | 180 (1) | 10 |
| P21 KCl buffer | 38 (1.7) | ||||||
| PUMA2 | 39 (1.5) | 30 (1.5) | 24 (1.9) | 66 (1) | 69 (1) | 1,000 (1) | 14 |
| PUMA2 + 2 mM MgCl2 | 36 (1.4) | 120 (1) | |||||
| BAX | 46 (1.6) | 45 (1.3) | 33 (1.4) | 390 (1) | 570 (1) | ≫1,000 | 150 |
| PUMA1 | 35 (1.6) | 43 (1.4) | 32 (1.5) | 350 (1) | ≫1,000 | ≫1,000 | 520 |
| PUMA1 + 2 mM MgCl2 | 80 (1.1) | ≫1,000 | |||||
| PUMA1 KCl buffer | 45 (1.5) | ||||||
Data fitted to Hill equation, with variable Hill constant (h) (20). Tabulated values of Kd are concentrations of p53 in terms of monomer required for 50% binding of labeled DNA. The apparent cooperativity, h, of binding results from the association of p53 dimers into tetramers with a Kd of 20 nM (45, 46). Thus, the predominant form of p53 during the measurement of weak binders (that is around the concentration for 50% binding) is the tetramer, and the binding is not cooperative (20).
*20-bp random = 5′-GGAAATTTCCGGAAATTTCC-3′.
30-bp NFκB = 5′-TCGACAGAGGGGACTTTCCGAGAGGCTCGA-3′.
20-bp RE = 5′-GGACATGTCCGGACATGTCC-3′.
p21 = 5′-ATCAGGAACATGTCCCAACATGTTGAGCTC-3′.
PUMA2 = 5′-CGCGCCTGCAAGTCCTGACTTGTCCGCGGC-3′.
Bax = 5′-TGGGCTCACAAGTTAGAGACAAGCCTGGGCG-3′.
PUMA1 = 5′-GGGTCCTCCTTGCCTTGGGCTAGGCCCTGCC-3′.
Labeled DNA sequences were 5′ fluorescein- or Alexa488-labelled (see Materials and Methods).
†Data for the core plus Tet domain construct (CT) are taken from Weinberg et al. (19).
We used three major buffers containing 90, 150, or 225 mM NaCl as salts plus 25 mM phosphate buffer pH 7.2. These have calculated ionic strengths, I, of 150 mM, 211 mM, and 286 mM, respectively. Strictly speaking, the classical concept of ionic strength breaks down because of ion condensation effects, but for convenience we refer to these buffers in terms of ionic strength. The middle buffer 211 mM I is approximately equal to the effective salt concentration in vivo. At 150 mM I, p53 bound all REs within an affinity range of 22–59 nM. Ac120Kp53 had similar affinities to p53, apart from weaker binding to the 20-bp random sequence. In effective physiological salt concentration buffer (211 mM I), p53 began to discriminate weakly against the 20-bp random sequence, but not among the others. In contrast, AcK120p53 showed strong discrimination against the 20-bp random sequence and significant discrimination among the REs. In 286 mM I buffer, p53 showed strong discrimination against the 20-bp random sequence and the 30-bp NFκB, as well as BAX and PUMA REs. AcK120p53 showed even stronger discrimination. Previous experiments with a construct of p53 containing just the DBD and the tetramerization domain (CT) showed a similar loss of specificity at 150 mM I, but specificity at 225 mM I (19, 20). For comparison in Table 1, CT has a similar specificity at 225 mM I buffer to AcK120p53 at 211 mM I buffer.
The addition of 2 mM Mg2+, which is above mammalian cellular levels of 0.2–0.6 mM (22–24), to the effective physiological salt concentration buffer (211 mM I) did not affect the binding of strongly binding REs but did weaken the binding of a weak binder (Table 1) so that AcK120p53 reached its full selectivity, which was still much higher than that for nonacetylated p53, measured under the same Mg2+ concentrations. Substitution of KCl for NaCl had a negligible effect on binding (Table 1).
We scanned the more general specificity of AcK120p53 by synthesizing 30 different competitor DNA sequences, systematically mutating each position within one half-site of the reference sequence (positions 1–10 in Fig. 2) and measuring their affinity by competition with a fluorescently labeled standard sequence (Table S1) (21). AcK120p53 had the highest affinity to the same DNA sequence as p53, measured at 286 mM I (Fig. 2). But, the acetylation of Lys120 lowers the importance of positions 4, the canonical C, and 7, the canonical G (equivalent to positions 14 and 17 in our longer sequences) in defining the preferred DNA-binding site. Compared with nonacetylated p53, the importance of other positions in the consensus sequence was not affected by the acetylation of Lys120 (Table S1), including positions 3 and 8 (equivalent to positions 13 and 18), which are known to form hydrogen bonds with the Lys120 side chain (13, 14).
Fig. 2.

Sequence logo representation of DNA binding. Fluorescently labeled DNA was allowed to form a complex with AcK120p53 (A) or p53 (21) (B) and the complex was titrated with unlabeled competitor DNA. Bit values (48) are plotted against the position in sequence. Bar heights indicate the relative importance of different nucleotides at each position. Both proteins had the highest affinity to the same consensus sequence written at the bottom.
Structure of the Acetylated Core Domain.
We solved the crystal structure of AcK120 DBD (residues 94–293) at 1.9-Å resolution (Table 2). The crystals used for structure solution belonged to space group P21 with four molecules in the asymmetric unit and were isomorphous to those reported for the nonacetylated wild-type DBD (25) and a cancer-related mutant (26). The overall structure of the β-sandwich and the DNA-binding surface of AcK120 DBD was virtually identical to that of the nonacetylated DBD [Fig. 3, PDB ID code 2OCJ (25)]. The different molecules of AcK120 DBD and nonacetylated DBD can be superimposed with a rmsd of 0.3–0.8 Å, which is in the same range as the rmsd when superimposing the molecules within each asymmetric unit. As with all structures of unbound DBD, the side chain of Lys120 showed a high degree of flexibility, and the lack of defined electron density prevented unambiguous modeling of the acetylated lysine side chain. The backbone conformation of the L1 loop, however, was well defined and similar to that of the unmodified protein, indicating that K120 acetylation does not alter the conformation of this loop in its DNA-free state.
Table 2.
X-ray data collection and refinement statistics
| A. Data Collection | |
| Space group | P21 |
| Cell, a, b, c, Å | 68.93, 69.58, 83.49 |
| Cell, α, β, γ, ° | 90.00, 90.12, 90.00 |
| Molecules/AU | 4 |
| Resolution, Å* | 49.0–1.9 (2.0–1.9) |
| Unique reflections | 60,047 |
| Completeness, %* | 96.6 (98.6) |
| Multiplicity* | 2.0 (2.0) |
| Rmerge, %*,† | 8.0 (34.1) |
| 〈I/σI〉* | 7.5 (2.4) |
| Wilson B value, Å2 | 17.9 |
| B. Refinement | |
| Number of atoms | |
| Protein‡ | 6,077 |
| Water | 644 |
| Zinc | 4 |
| Rcryst, %§ | 17.4 |
| Rfree, %§ | 22.6 |
| rmsd bonds, Å | 0.006 |
| rmsd angles, ° | 0.97 |
| Mean B value, Å2 | 20.9 |
| Ramachandran favored, %¶ | 98.2 |
| Ramachandran outliers, %¶ | 0.0 |
*Values in parentheses are for the highest resolution shell.
†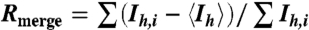
‡Number includes alternative conformations.
§Rcryst and 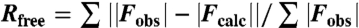 , where Rfree was calculated over 5% of the amplitudes chosen at random and not used in the refinement.
, where Rfree was calculated over 5% of the amplitudes chosen at random and not used in the refinement.
¶Determined using MOLPROBITY (47).
Fig. 3.

Structural effect of Lys120 acetylation. Superposition of the crystal structures of AcK120DBD (gray; PDB ID code 2YBG, this work) and nonacetylated DBD (blue; PDB ID code 2OCJ) in their DNA-free form shows that acetylation of Lys120 has no significant effect on the conformation of the L1 loop in the unbound DBD. Both structures are shown as cartoon representation, with the position of residue 120 highlighted in red. The figure was generated using PyMOL (www.pymol.org).
Composition of Acetylated and Nonacetylated p53DBDs in Complex with DNA.
We studied the composition of complexes formed by competition of excess concentrations of p53DBD and AcK120DBD for the 20-bp RE, for which they have equal affinity. We incubated different ratios of 13C-15N-labeled nonacetylated DBD and AcK120DBD with the consensus 20-bp RE and used soft ionization electrospray mass spectrometry (ESI-MS) to analyze the composition of the resultant complexes (Fig. 4) (27–29). The relative abundance of a particular species was calculated from the ratio between the sum of intensities associated with that species and the sum of all intensities. Completely random formation could theoretically result in five different species, with DBD∶AcK120DBD stoichiometries of 0∶4, 1∶3, 2∶2, 3∶1, or 4∶0. A binomial distribution of equal amounts of both species should yield a ratio of 1∶4∶6∶4∶1.
Fig. 4.

ESI-MS of complexes formed between AcK120-DBD and/or 13C-15N-labeled nonacetylated DBD and DNA. Nonacetylated DBD is marked by blue dots, whereas AcK120DBD is marked by red dots. Molar ratio of 13C-15N-DBD∶AcK120DBD∶DNA gradually changed from 8∶0∶1 (Bottom) to 8∶8∶1 (Top). The composition of the complex associated with each peak is marked at the top (i.e., the ratio between 13C-15N-DBD and AcK120DBD). The intensity of each peak is proportional to the relative abundance of each species. Dashed lines represent the expected m/z value for the DNA(p53DBD)2(AcK120DBD)2 complex (m/z = 6,461, 6,865, and 7,322). Only the peak at m/z = 6,865 was detected, due to low occurrence of the DNA(p53DBD)2(AcK120DBD)2 complex.
The spectra in Fig. 4 were obtained when 13C-15N-labeled nonacetylated DBD and AcK120DBD were mixed at different molar ratios with DNA. As expected, when only 13C-15N-labeled nonacetylated DBD was mixed with DNA at a molar ratio of 8∶1 (monomer to double stranded DNA), only homotetramers of nonacetylated DBD and DNA were observed (bottom spectrum). When equal amounts of 13C-15N-labeled nonacetylated DBD and AcK120DBD were mixed with DNA at molar ratios of 8∶8∶1, both homotetramers and heterotetramers formed in solution (top spectrum). The hetero-tetramers were composed of different ratios between acetylated (red dots) and nonacetylated p53 DBDs (blue dots), as marked in Fig. 4 (Top) according to the composition of the tetramer associated with each peak.
All theoretically possible species were observed (Fig. 5), but importantly the ratio of these complexes significantly deviated from the values expected for a binomial distribution. Of the total population of complexes monitored, 20% were composed of DNA and four nonacetylated DBDs, whereas 17% were composed of DNA and four AcK120DBDs (Fig. 5), three times higher than expected from a random binomial distribution, of about 6%. The deviation from binomial distribution was even more significant for the 2∶2 heterotetramers: 7% observed versus an expected 38%. AcK120DBDs and p53DBDs prefer to form homocomplexes with DNA and have the least preference for DNA(AcK120DBD)2(p53DBD)2 heterocomplexes.
Fig. 5.

Distribution of AcK120DBD and nonacetylated DBD in tetrameric complex with DNA. Equal amounts of 13C-15N-nonacetylated DBD and AcK120DBD were mixed with DNA at molar ratios of 8∶8∶1 and samples were analyzed by ESI-MS. Results are presented as function of number of AcK120DBDs in each complex: 0—a homotetramer composed of four nonacetylated DBDs; 4—a homotetramer composed of four AcK120DBDs. Bars represent the distribution of complexes relative to the total amount of formed complexes. For example, 17% of the complexes were in the form of DNA(AcK120DBD)4 complex, whereas 7% were in the form of DNA(AcK120DBD)2(p53DBD)2 complex. Experimental data are compared to results expected in a nonbiased experiment, assuming binomial distribution.
Discussion
Using an orthogonal pyrrolysyl-tRNA synthetase/tRNA pair evolved to incorporate Nε-acetyllysine in response to the amber stop codon (16), we were able to express in vivo and purify p53 and its DBD that were site-specifically acetylated at position Lys120 and then study in vitro the effect of Lys120 acetylation on the structure and activity of p53. Acetylation did not significantly alter the crystal structure of the DBD, although the acetylated side chain of Lys120 was too mobile to be located. The thermodynamic and kinetic stability of the DBD were unaffected (Fig. S1). But, there were unexpected consequences on the specificity of binding to DNA.
Acetylation of Lys120 and DNA Binding.
Salt concentration.
The most significant effect of acetylation of Lys120 on the activity of p53 is to modulate its sensitivity to salt concentration. Previous studies on measuring the specificity of binding of p53 in vitro (using a simplified construct, CT, lacking the N and C termini) note that p53 binds indiscriminately to DNA at lower salt concentrations, not distinguishing between different response elements or even random sequences. So, measurements were made at an ionic strength of 225 mM (19) where discrimination is observed. Subsequent systematic surveys of full-length p53 in vitro have needed to use higher salt concentrations still, equivalent to 286 mM I (21).
The value of intracellular salt concentration is often considered to be equivalent to a calculated ionic strength of 250 mM. Allowance for formation of complexes between ions reduces the value to 200 mM (23). An effective physiological ionic strength of 215 mM I is also calculated (30). However, the classical Debye–Hückel theory of electrostatic effects of ions breaks down at these high concentrations, and it is better to use a term such as effective physiological salt concentration (31). Here, at 150 mM I, both p53 and AcK120p53 bound DNA near indiscriminately, with equal affinity (Table 1). But, at 211 mM effective salt concentration, close to physiological effective salt concentrations, AcK120p53 achieved > 10-fold discrimination between p21 and Bax response elements, whereas p53 still did not discriminate between different DNA response elements and bound well to a random 30-mer. Nonacetylated p53 exhibited specificity at 286 mM I, as expected (21).
Mg2+ ions can affect DNA–protein interactions by competing for DNA sites (31, 32). The concentration of Mg2+ in mammalian cells is about 0.2–0.6 mM (22–24). The addition of 2 mM Mg2+ to the effective physiological salt buffer (Table 1) had little effect on the binding of either AcK120p53 or nonacetylated p53 to strong-binding REs, but it weakened the binding to weak-binding REs. The affinity of nonacetylated p53 to Puma1 was reduced by a factor of approximately 2 (from 32 to 80 nM) and the affinity of AcK120p53 was reduced from 350 nM to > 1 μM. The specificity of AcK120p53 for Puma1 was thus increased to the level found at 286 mM I. Perhaps, other divalent ions could also enhance selectivity. A major effect of acetylation of Lys120 is to endow p53 with selectivity at salt concentrations close to the effective physiological values where nonacetylated p53 does not exhibit selectivity.
General specificity of AcK120p53.
The affinity of p53 for various REs is mostly affected by mutations of the C at position 4 or G at position 7 (Fig. 2), but acetylation of Lys120 relaxed somewhat the stringent requirements for C and G at those positions (Table S1). It implies that acetylation of Lys120 shifts the equilibrium toward binding to REs with lower binding affinities, in addition to reducing nonspecific DNA binding. Competition between binding to specific and nonspecific DNA sequence has previously suggested as part of the lactose (lac) operon regulatory system (33). According to this model, the binding of lac repressor to specific DNA relative to nonspecific DNA is a crucial parameter in the regulatory mechanism of the lac operon. According to in vitro measurements presented here, at effective physiological salt concentration the ratio between specific (p21 RE) to nonspecific DNA-binding affinities increases from 11 to 33 upon acetylation (Table 1). Assuming similar in vivo binding affinities, these data suggest a potential regulatory role for the acetylation of Lys120 by controlling the ratio between specific to nonspecific binding of p53 to DNA and hence its activity as a transcription factor.
Structural Consequence of Acetylation.
AcK120DBD had the same crystal structure as p53DBD, with the acetylated side chain too mobile to be located. We have been unable to crystallize complexes of AcK120DBD with DNA, but obtained useful data from mass spectrometry of complexes formed between a RE and a mixture of excess p53DBD and AcK120DBD. According to the spectra in Fig. 4, the homocomplexes DNA(p53DBD)4 or DNA(AcK120DBD)4 were preferentially formed relative to DNA(p53DBD)2(AcK120DBD)2 (Fig. 5). Lys120 is at the DNA–protein interface of DNA(p53)4 complexes and not the protein–protein interfaces (13, 14). Accordingly, DNA(p53DBD)4 and DNA(AcK120DBD)4 must have different protein–protein interfaces, induced by geometrical changes at the protein–DNA interfaces. These results are entirely consistent with recent electron microscopy observations that p53 binds well to nonspecific DNA at 211 mM I, but in different, relaxed, quaternary states from p53 bound to specific sequences (34).
Implications for Searching for Target Response Elements.
p53 binds tightly to nonspecific DNA. p53 has to search for its target sequences among the nonspecific DNA, which is in vast excess so that, even if it binds less tightly, would sequester p53. Electron microscopy shows that the binding to nonspecific sequences occurs from p53 adopting different conformational states, leading to speculation that accessory proteins could force p53 tetramers to adopt the “specific binding conformation” (34). Acetylation of Lys120 is now seen to increase the specificity of p53 and change its conformational preferences. If the data in vitro extend to the situation in vivo, then p53 prior to acetylation of Lys120 would not be in a search process for its targets, but would bind to any exposed nonspecific sequences, possibly sliding along them (35). Acetylation would then start the search process by weakening the binding to nonspecific sequences.
Materials and Methods
Protein Expression and Purification.
Nonacetylated versions of full-length p53 and DBD (residues 94–293) were expressed in BL21 cells as previously described (36). Briefly, cells induced with 1 mM IPTG and incubated at 22 °C overnight in 2×TY medium supplemented with 0.1 mM ZnCl2. Cells were lysed in 50 mM phosphate buffer pH 8, 300 mM NaCl, protease inhibitors (Roche), and 10 mM β-mercaptoethanol, and clear lysate was loaded onto a Ni column. Protein was eluted (50 mM phosphate buffer pH 8, 300 mM NaCl, 10 mM β-mercaptoethanol, and 250 mM Imidazole) and dialyzed overnight (25 mM Tris buffer pH 7.5, 300 mM NaCl, 10% glycerol, 5 mM DTT) with tobacco etch virus (TEV) protease (p53) or thrombin (p53DBD). Dialyzed solution was diluted (1∶10, ice-cold 25 mM Tris buffer pH 7.5, 10% glycerol, 5 mM DTT), and cleaved p53 was separated from the His-tagged lipoyl domain using a heparin column. Protein was eluted from the heparin column with a gradient over 20 column volumes of elution buffer (25 mM Tris buffer pH 7.5, 1 M NaCl, 10% glycerol, and 5 mM DTT). Combined fractions were concentrated using Centricon concentrators (Millipore) and further purified by gel filtration. DBD was purified with a HiLoad 26/60 Superdex 75 column (GE Healthcare) using citrate buffer (20 mM citrate buffer pH 6.1, 150 mM NaCl, and 10 mM DTT). Full-length p53 was purified with a HiLoad 26/60 Superdex 200 column (GE Healthcare) using phosphate buffer (25 mM phosphate buffer pH 7.2, 300 mM NaCl, 5 mM DTT, and 10% glycerol). All purification steps were carried out at 4 °C.
Acetylated proteins were expressed in a similar manner, except for the following modifications. A DNA fragment of K120TAG mutant p53 was cloned as N-terminal fusion of His6, lipoyl domain, and TEV protease cleavage site (p53) or thrombin cleavage site (p53DBD) between XhoI and NcoI sites on pCDF-Duet vector encoding the pyrrolysine tRNA gene from M. barkeri with an lpp promoter and rrnC terminator (16). BL21 cells were cotransformed with the above plasmid and a pAcKRS-3 plasmid (16) (coding for M. barkeri pyrrolysine tRNA synthetase with the mutations L266M, L270I, Y271F, L274A, and C313F) and grown in 2×TY medium supplied with 10 mM acetylated lysine. Before induction with 1 mM IPTG, cells were supplemented with 20 mM Nicotinamide. Purification of acetylated proteins was carried out as described above for nonacetylated proteins.
DNA-Binding Affinity and Specificity.
DNA-binding affinities to specific REs were measured by fluorescence anisotropy, using a Cary Eclipse (Varian) spectrometer equipped with a Microlab M dispenser (Hamilton). Excitation and emission wavelength were 480 nm and 530 nm, respectively. All measurements were done at 20 °C. Buffers of varying salt concentrations were used (90 or 150 or 225 mM NaCl plus 25 mM phosphate buffer pH 7.2, 10% (vol/vol) glycerol, 5 mM DTT, 0.2 mg/mL bovine serum albumin). The 150 mM KCl buffer was identical to the NaCl buffer, except that potassium salts were used instead of sodium salts. Measurements in the presence of Mg2+ ions were made in 145 mM NaCl, 2 mM MgCl2 plus 25 mM phosphate buffer pH 7.2, 10% (vol/vol) glycerol, 5 mM DTT, 0.2 mg/mL bovine serum albumin. In a typical experiment, 250 μL of 1–5 μM protein solution were titrated into 900 μL of 20 nM labeled DNA solution. Data were corrected for the resulting dilution effect. Fluorescence intensities were measured 60 s after each titration step to allow equilibration. All data analysis was done using laboratory software (results are described in Table 1). All sequences were 5′ fluorescein-labeled, except for 20-bp RE, which was 5′ Alexa488-labeled.
General DNA binding was measured by fluorescence anisotropy in 96-well plates using a Pherastar plate reader (BMG Labtech) equipped with a Bravo 96-channel pipetting robot (Velocity 11) as previously described (21). In a typical titration experiment, 120 nM (final monomer concentration) of full-length p53 was allowed to form a complex with 20 nM of the 20-base-pair reporter sequence GGACATGTCCGGACATGTCC, labeled at the 5′ end with Alexa488 fluorophore, and the complex was then titrated with unlabeled DNA (50 μM) at 22 °C. The reporter sequence consists of two identical copies of the half-site GGACATGTCC, known to be one of the tightest-binding sequences for human p53. First, binding affinity was measured between AcK120p53 and a reference DNA—a nonlabeled version of the reporter DNA. This binding affinity was then compared to that of the other 31 competitor DNA sequences, in which a single mutation was introduced systematically at each position within one half-site of the palindromic reference sequence. Buffer conditions for all experiments were 25 mM sodium phosphate pH 7.2, 225 mM NaCl (286 mM total I), 10% vol/vol glycerol, 5 mM DTT and 0.2 mg/mL bovine serum albumin. Data were analyzed according to cooperative binding and competition models using laboratory-developed software (21).
Crystallization and Structure Solution.
Crystals of acetylated DBD were grown using the sitting drop vapor diffusion technique with 26% (wt/vol) PEG 3350, 43 mM sodium acetate, and 100 mM Hepes, pH 7.5 as the crystallization solution. Drops composed of 200 nL of protein at 200 μM (20 mM citrate buffer pH 6.1, 150 mM NaCl, and 10 mM DTT) and 200 nL of crystallization solution were equilibrated for a minimum of 3 d at 17 °C above a reservoir solution of 100 μL. Crystals were transferred to mother liquor containing 20% glycerol and flash-frozen in liquid nitrogen. The protein crystals belonged to space group P21 with a = 68.926 Å, b = 69.581 Å, c = 83.494 Å, and β = 90.12°. An X-ray dataset was collected on beamline ID14-2 at the European Synchrotron Radiation Facility. Data were indexed and integrated using MOSFLM (37) and were further processed using the CCP4 package (38) (Table 2). The structure was solved by molecular replacement using PHASER (39) with the chain A of PDB ID code 1TSR (40) as a search model. Model building and structure refinement were performed using COOT (41) and PHENIX (42). Data collection and refinement statistics are summarized in Table 2.
Mass Spectrometry.
Acetylated and nonacetylated p53 DBDs were dialyzed against 500 mM ammonium acetate pH 6.9 at 4 °C. Acetylated DBD was mixed with the nonacetylated variant at increased ratios up to a final ratio of 1∶1. DNA was added to each of the mixtures, and the mass of the formed complex was immediately measured using nanoflow ESI-MS. Data were recorded on a Synapt HDMS system (Waters Corp.) optimized for the transmission of noncovalent complexes (43). In a typical experiment, 3 μL of the mixture were introduced by electrospray from gold-coated borosilicate capillaries prepared in-house, as described (44). The following experimental parameters were applied: capillary voltage = 1.3–1.5 kV, sample cone = 100 V, trap and transfer collision energy = 100 V; backing pressure = 5 mbar, source pressure = 0.06–0.07 mbar; trap pressure = 0.05 mbar; IMS pressure = 0.5 mbar; time-of-flight analyzer pressure = 1.2 × 10-6 mbar. The mass spectrometer was calibrated with cesium iodide solution (100 mg/mL). Data were processed with MassLynx 4.0 software (Waters/Micromass) and are shown with minimal smoothing and without background subtraction.
Supplementary Material
Acknowledgments.
This work was partly supported by European Molecular Biology Grant ALTF-650-2006 (to E.A.) and MRC Programme Grant G0901534 (to A.R.F.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors of the Lys120-acetylated DBD have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2YBG).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1105028108/-/DCSupplemental.
References
- 1.Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 3.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 4.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 5.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toledo F, Wahl GM. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 7.Sykes SM, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 9.Sykes SM, Stanek TJ, Frank A, Murphy ME, McMahon SB. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J Biol Chem. 2009;284:20197–20205. doi: 10.1074/jbc.M109.026096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA. 2005;102:10188–10193. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Dey R, Chen L. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure. 2010;18:246–256. doi: 10.1016/j.str.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitayner M, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Zupnick A, Prives C. Mutational analysis of the p53 core domain L1 loop. J Biol Chem. 2006;281:20464–20473. doi: 10.1074/jbc.M603387200. [DOI] [PubMed] [Google Scholar]
- 16.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding Nϵ-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 17.Nikolova PV, Henckel J, Lane DP, Fersht AR. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc Natl Acad Sci USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joerger AC, Allen MD, Fersht AR. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J Biol Chem. 2004;279:1291–1296. doi: 10.1074/jbc.M309732200. [DOI] [PubMed] [Google Scholar]
- 19.Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol. 2005;348:589–596. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J Mol Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 21.Veprintsev DB, Fersht AR. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36:1589–1598. doi: 10.1093/nar/gkm1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy LA, Murphy E, Raju B, London RE. Measurement of cytosolic free magnesium ion concentration by 19F NMR. Biochemistry. 1988;27:4041–4048. doi: 10.1021/bi00411a021. [DOI] [PubMed] [Google Scholar]
- 23.Mouat MF, Manchester KL. The intracellular ionic strength of red cells and the influence of complex formation. Comp Hematol Int. 1998;8:58–60. [Google Scholar]
- 24.Rotevatn S, et al. Cytosolic free magnesium concentration in cultured chick heart cells. Am J Physiol. 1989;257:C141–C146. doi: 10.1152/ajpcell.1989.257.1.C141. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Rosengarth A, Luecke H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr D Biol Crystallogr. 2007;63:276–281. doi: 10.1107/S0907444906048499. [DOI] [PubMed] [Google Scholar]
- 26.Joerger AC, Ang HC, Veprintsev DB, Blair CM, Fersht AR. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J Biol Chem. 2005;280:16030–16037. doi: 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- 27.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 28.Sharon M, Robinson CV. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- 29.Wendt S, et al. Quantitative evaluation of noncovalent chorismate mutase-inhibitor binding by ESI-MS. J Am Soc Mass Spectrom. 2003;14:1470–1476. doi: 10.1016/j.jasms.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Freund J, Kalbitzer HR. Physiological buffers for NMR spectroscopy. J Biomol NMR. 1995;5:321–322. doi: 10.1007/BF00211760. [DOI] [PubMed] [Google Scholar]
- 31.Record MT, Jr, deHaseth PL, Lohman TM. Interpretation of monovalent and divalent cation effects on the lac repressor-operator interaction. Biochemistry. 1977;16:4791–4796. doi: 10.1021/bi00641a005. [DOI] [PubMed] [Google Scholar]
- 32.Kao-Huang Y, et al. Nonspecific DNA binding of genome-regulating proteins as a biological control mechanism: Measurement of DNA-bound Escherichia coli lac repressor in vivo. Proc Natl Acad Sci USA. 1977;74:4228–4232. doi: 10.1073/pnas.74.10.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Hippel PH, Revzin A, Gross CA, Wang AC. Non-specific DNA binding of genome regulating proteins as a biological control mechanism: 1. The lac operon: Equilibrium aspects. Proc Natl Acad Sci USA. 1974;71:4808–4812. doi: 10.1073/pnas.71.12.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melero R, et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc Natl Acad Sci USA. 2011;108:557–562. doi: 10.1073/pnas.1015520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci USA. 2011;108:563–568. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bullock AN, et al. Thermodynamic stability of wild-type and mutant p53 core domain. Proc Natl Acad Sci USA. 1997;94:14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 38.CCP4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 43.Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 44.Nettleton EJ, et al. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: Evidence from electrospray mass spectrometry. J Mol Biol. 1998;281:553–564. doi: 10.1006/jmbi.1998.1937. [DOI] [PubMed] [Google Scholar]
- 45.Brandt T, Petrovich M, Joerger AC, Veprintsev DB. Conservation of DNA-binding specificity and oligomerisation properties within the p53 family. BMC Genomics. 2009;10:628. doi: 10.1186/1471-2164-10-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajagopalan S, Huang F, Fersht AR. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2010;39:2294–2303. doi: 10.1093/nar/gkq800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider TD, Stephens RM. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.