

Abstract
Orotate phosphoribosyltransferases (OPRTs) form and break the N-ribosidic bond to pyrimidines by way of ribocation-like transition states (TSs) and therefore exhibit large α-secondary [1′-3H] kcat/Km kinetic isotope effects (KIEs). Substrate binding isotope effects (BIEs) with OPRTs report on the degree of ground state destabilization for these complexes and permit resolution of binding and transition state effects from the kcat/Km KIEs. The BIEs for [1′-3H]orotidine 5′-monophosphate (OMP) interactions with the catalytic sites of Plasmodium falciparum and human OPRTs are 1.104 and 1.108, respectively. These large BIEs establish altered sp3 bond hybridization of C1′ toward the sp2 geometry of the transition states upon OMP binding. Thus, the complexes of these OPRTs distort OMP part of the way toward the transition state. As the [1′-3H]OMP kcat/Km KIEs are approximately 1.20, half of the intrinsic kcat/Km KIEs originate from BIEs. Orotidine, a slow substrate for these enzymes, binds to the catalytic site with no significant [1′ 3H]orotidine BIEs. Thus, OPRTs are unable to initiate ground-state destabilization of orotidine by altered C1′ hybridization because of the missing 5′-phosphate. However the kcat/Km KIEs for [1′-3H]orotidine are also approximately 1.20. The C1′-distortion for OMP happens in two steps, half upon binding and half on going from the Michaelis complex to the TS. With orotidine as substrate, there is no ground-state destabilization in the Michaelis complexes, but the C1′-distortion at the TS is equal to that of OMP. The large single barrier for TS formation with orotidine slows the rate of barrier crossing.
Intrinsic kinetic isotope effects (KIEs) are commonly used to provide bond structural information for enzymatic transition states (TSs). Competing isotopic label methods provide KIEs on the kinetic parameter of kcat/Km (1, 2). But the kcat/Km KIEs include all kinetically important steps between reactants free in solution and the first irreversible step of the reaction (Figure 1) (3). By measuring equilibrium binding isotope effects (BIEs), bond vibrational changes upon formation of the Michaelis complex are obtained and KIEs from the chemical step can be resolved.(4). Reactions involving N-ribosidic bond-loss commonly form ribocation transition states where the anomeric carbon undergoes rehybridization from sp3 in the reactant to sp2 at the transition state. α-Secondary [1′-3H] BIEs are a sensitive way to measure bonding changes in reactants upon binding to catalytic sites and therefore report on the degree of ground state destabilization for the enzyme-substrate complexes. Enzyme-based ground state destabilization is established for several enzymes and been summarized by Anderson (5), although earlier reports have questioned the ability of enzymes to distort substrates (6).
Figure 1.

Reactions catalyzed by PfOPRT and HsOPRT and the relationship between BIE, kcat/Km KIE and kcat KIE. The substrate pair of orotidine–pyrophosphate (PPi) is shown. A similar transition state is formed when PPi is replaced with phosphonoacetic acid. Transition state features are shown in brackets.
Recent BIE studies with purine nucleoside phosphorylase (PNP) reported a [5′-3H] BIE remote from the site of chemical bond breaking and gave a BIE of 1.5%. For TS analogues of PNP, normal [5′-3H] BIEs of 13 to 29% were reported for human PNP, indicating that TS analogue interactions differ considerably from those in Michaelis complexes or at the TS (7). BIEs with PNP were explained by local C5′-H5′ bond distortion and are not necessarily linked to ground-state destabilization. Ground-state destabilization can be established by BIEs of atoms involved in the chemical reaction by showing that the distortion leads toward the transition state.
Orotate phosphoribosyltransferases from Plasmodium falciparum (PfOPRT) and human sources (HsOPRT) catalyze the reversible pyrophosphorolysis of orotidine and orotidine 5′-monophosphate (OMP). Transition state analyses from isotope effects and quantum chemical calculations have established ribocation TS structures with fully dissociated dianionic orotates (Figure 1) (8, 9). Large normal α-secondary [1′-3H] kcat/Km intrinsic KIEs of 19 to 26% were measured for reactions catalyzed by PfOPRT and HsOPRT. These isotope effects are primarily the result of out-of-plane bending mode changes as C1′ geometry is distorted from sp3 in the reactant to sp2 at the SN1 TS. Secondary [1′-3H] kcat/Km KIEs from in vacuo calculations provide a two-state model for the conversion of unbound reactants to the transition state. In reality, kcat/Km KIEs also include changes to the [1′-3H] bond vibrational changes in the Michaelis complex. Here we use BIE to resolve these effects.
The X-ray crystal structure of HsOPRT in complex with OMP (PDB code: 2WNS) proposes multiple catalytic site contacts to OMP. Structural models predict an extended hydrogen-bond network surrounding the pyrimidine group to enforce leaving group activation. Leaving group forces have been confirmed in OPRT by isotope-edited Fourier transform infrared (FTIR) spectroscopy of the pyrimidine group (10). Enzyme-bound OMP differs in geometry from the in vacuo energy minima, most obviously for the N-ribosyl torsion angle between the orotate base and ribose ring. The [1′-3H] bond vibrations are sensitive to the N-ribosyl torsion angle (11). When the enzyme stabilizes a conformation away from the energetic minimum of unbound substrate, the degree of sp3 hybridization at C1′ may be altered to cause a BIE (12).
Here, [1′-3H] BIEs of OMP and orotidine were measured in binary and ternary complexes of PfOPRT and HsOPRT. Large normal BIEs with [1′-3H]OMP (alone or with sulfate, a competitive inhibitor with pyrophosphate) demonstrate ground state destabilization toward the TS. Lack of [1′-3H]orotidine BIEs indicate that without the 5′-phosphate, substrate undergoes no significant distortion in the Michaelis complexes. This is the first work to report a [1′-3H] BIE for any N-ribosyltransferase. In combination with experimental KIEs, the [1′-3H] BIEs reveal the 5′-phosphate to assist is a two-step C1′-distortion for OPRT-catalyzed OMP pyrophosphorolysis.
Materials and Methods
Reagents.
D-[1-3H]Ribose and D-[6-14C]glucose were purchased from American Radiolabeled Chemicals Inc. Hexokinase (HK), glucose 6-phosphate dehydrogenase (G6PDH), phosphogluconic acid dehydrogenase (PGDH), L-glutamic acid dehydrogenase (GDH), phosphoriboisomerase (PRI), adenylate kinase (AK) and pyruvate kinase (PK) were purchased from Sigma-Aldrich. Phospho-D-ribosyl-α-1-pyrophosphate synthase (PRPPase) and ribokinase (RK) were prepared as described before (13, 14). Alkaline phosphatase (AP) was purchased from Roche Applied Science. PfOPRT and HsOPRT were expressed in Escherichia coli cells and purified as described previously (8). All other reagents were purchased from readily available commercial sources and used without further purification.
Synthesis of Isotopically Labeled OMPs and Orotidines.
Isotopically labeled OMPs were synthesized enzymatically as previously described (8). Orotidines were prepared from OMPs based on a previous report (9).
Quantum Chemical Calculations.
The OMP structures were calculated in vacuo using hybrid density functional theory implemented in Gaussian 03 (15). The initial model of OMP bound to HsOPRT was from the crystal structure of HsOPRT in complex with OMP (PDB code: 2WNS). At the B3LYP/6-31G (d,p) level, geometry optimizations were performed for HsOPRT-bound OMP with dihedral angles fixed to those in the crystal structure and free OMPs with restricted O4′-C1′-N1-C2 dihedral angles. Bond frequencies of the optimized OMPs were calculated at the same level of theory and basis set. BIEs were calculated from the computed frequencies of free OMPs and HsOPRT-bound OMP at 298 K using ISOEFF98 program (16).
Determination of Binding Isotope Effects.
Competitive binding of [1′-3H]OMP and [5′-14C]OMP and of [1′-3H]orotidine and [5′-14C]orotidine were measured with and without sulfate, using the ultrafiltration method (12, 17). Sulfate binds competitively to pyrophosphate (PPi) binding sites of PfOPRT and HsOPRT and is chemically inert. With OMP as substrate the Ki of sulfate is 3.1 ± 0.3 mM for PfOPRT and 17.6 ± 2.8 mM for HsOPRT. With orotidine as substrate, sulfate has a Ki of 2.7 ± 0.3 mM for PfOPRT and 10.8 ± 1.6 mM for HsOPRT. Measurements of [1′-3H] BIEs in binary complexes were made in 50 mM Tris–HCl (pH 8.0), 20–500 μM OMP or orotidine (dpm of 3H/14C = 4:1), 10–300 μM PfOPRT/HsOPRT in a final volume of 100 μL. Measurements of [1′-3H] BIEs in ternary complexes included, in addition, 30–200 mM sulfate (11 × Ki) and one molar equivalent of magnesium in a volume of 100 μL. Samples were loaded into the upper well of the ultrafiltration apparatus and 30 psi argon pressure was applied until approximately half of the solution had passed through the dialysis membrane (12–14 kDa molecular weight cutoff) into the lower well (60–90 min). Samples (30 μL) from upper and lower wells were mixed with 10 mL of scintillation fluid (Ultima Gold) and counted for at least 6 cycles (10 min per cycle). Spectral deconvolution of 3H and 14C was performed using a 14C standard in a matrix identical to that of the BIE samples. The BIE was calculated from eq 1,
| (1) |
where 14CT and 14CB are the 14C counts in the top and bottom wells, respectively, and 3HT and 3HB are the 3H counts in the top and bottom wells, respectively (7).
Results and Discussion
[1′-3H] BIEs of OMP and Orotidine.
On formation of the binary complexes, [1′-3H]OMP generates a BIE of 1.104 for PfOPRT and of 1.108 for HsOPRT (Table 1). The large normal BIEs indicate that C1′–H1′ bonds become less constrained in the Michaelis complexes relative to those of free OMPs in solution. α-Secondary [1′-3H] BIEs are dominated by out-of-plane bending modes, and these isotope effects establish sp3 distortion at C1′, toward the sp2 geometry of the transition state. The result is an unequivocal demonstration of reactant ground-state destabilization by enzymatic interactions. Comparison of [1′-3H]OMP BIEs and KIEs reveal that C1′ distortion for OMP occurs in two steps, half upon binding and half on going from the Michaelis complex to the TS.
Table 1.
[1′-3H] BIEs and kcat/Km KIEs for PfOPRT and HsOPRT.
| OMP [1′-3H] vs [5′-14C] |
|||
|---|---|---|---|
| binary complex OPRT•OMP |
ternary complex OPRT•OMP•SO42− |
||
| BIEsa | PfOPRT | 1.104 ± 0.002 | 1.091 ± 0.003 |
| HsOPRT | 1.108 ± 0.003 | 1.096 ± 0.004 | |
|
Kd (μM) |
PfOPRT | 3.1 ± 1.6 | 5.6 ± 1.8 |
| HsOPRT | 2.5 ± 1.2 | 4.1 ± 2.5 | |
|
|
|||
| expt | calcd | ||
| KIEsb | PfOPRT | 1.261 ± 0.014 | 1.335 |
| HsOPRT | 1.199 ± 0.015 | 1.330 | |
|
Kmb (μM) |
PfOPRT | 3.7 ± 1.4 | |
| HsOPRT | 1.6 ± 0.6 | ||
| Orotidine [1′-3H] vs [5′-14C] |
|||
|---|---|---|---|
| binary complex OPRT•orotidine |
ternary complex OPRT•orotidine•SO42− |
||
| BIEsa | PfOPRT | 0.997 ± 0.003 | 0.991 ± 0.006 |
| HsOPRT | 0.999 ± 0.003 | 0.991 ± 0.005 | |
|
Kd (μM) |
PfOPRT | 130 ± 34 | 160 ± 30 |
| HsOPRT | 140 ± 27 | 150 ± 22 | |
|
|
|||
| expt | calcd | ||
| KIEsb | PfOPRT | 1.189 ± 0.006 | 1.405 |
| HsOPRT | 1.209 ± 0.004 | 1.411 | |
|
Kmb (μM) |
PfOPRT | 96 ± 40 | |
| HsOPRT | 91 ± 37 | ||
In contrast to OMP, [1′-3H]orotidine in the binary complexes show BIEs of 0.997 and 0.999 for PfOPRT and HsOPRT, respectively (Table 1). The near-unity [1′-3H] BIEs for orotidine reflect no significant C1′ distortion upon binding to the OPRT active sites. Although the 5′-phosphate of OMP is remote from the reaction center, it is essential in anchoring the orotidine group to permit ground-state destabilization at the anomeric carbon. Without the 5′-phosphate group, orotidine is docked into the active site without significant C1′ ground-state destabilization, but the C1′ distortion at the TS is equal to that of OMP since kcat/Km KIE values are similar. Relative to OMP, orotidine has a 25-fold higher Km and 240-fold lower kcat/Km for both OPRT enzymes. The single large barrier for TS formation with orotidine as substrate slows the rate of barrier crossing. The [1′-3H]orotidine BIEs demonstrate the importance of the 5′-phosphate group for ground-state destabilization in the OPRT complexes. Altered kinetic properties have been reported for other enzymes with phosphate groups remote from the site of chemistry (18-21). Here, BIEs demonstrate that the remote phosphate binding effect is required for anomeric carbon distortion toward the TS.
In the presence of sulfate the ternary complexes give [1′-3H]OMP BIEs of 1.091 for PfOPRT and 1.096 for HsOPRT. In contrast, [1′-3H]orotidine BIEs are 0.991 for both enzymes (Table 1). Compared to BIEs determined for the binary complexes, [1′-3H] BIEs from the ternary complexes decrease about 1%. Decreased [1′-3H] BIEs correspond to small conformational constraints in the C1′-H1′ bonds to demonstrate that bound sulfate causes a small constraint to the H1′ out-of-plane modes.
Dissociation Constants.
The ultrafiltration method used for BIE experiments also permits measurement of dissociation constants of OMP and orotidine in the binary and ternary complexes. For PfOPRT, the Kd of OMP with and without sulfate is 5.6 and 3.1 μM, respectively, similar to its Km value of 3.7 μM (Table 1). Orotidine has a Kd of 160 and 130 μM for PfOPRT with and without sulfate respectively, similar to its Km of 96 μM with pyrophosphate. In the HsOPRT ternary complexes, Kds of OMP and orotidine are 4.1 and 150 μM, respectively. For the binary complexes, the Kd values are 2.5 and 140 μM for OMP and orotidine, similar to their corresponding Km values of 1.6 and 91 μM with pyrophosphpate. The similarity of dissociation constants with sulfate compared to the Km values for OMP and orotidine with pyrophosphate support similar interactions at the catalytic sites. These values are consistent with the transition state structures of the OPRTs where only weak van der Waals interactions are present to the attacking nucleophile until the TS has been reached (8, 9).
Origin of the [1′-3H] BIE.
Large normal [1′-3H] BIEs reflect bonding changes at the anomeric carbon upon formation of the Michaelis complexes. As the geometry of the O4′–C1′–N1–C2 dihedral angle changes from the energetic minimum of 51° for free OMP in vacuo to the specific value of 167° for OMP bound to HsOPRT, (PDB code: 2WNS), the change in C1′-hybridization results in a predicted [1′-3H] isotope effect (Figure 2). On changing the torsion angle from 51° to 167°, isotope effects of approximately 1.20 are predicted in vacuo and in water (Figures. 2, 3). Although [1′-3H] isotope effects are notorious in their difficulty to match computationally (22), the calculations predict BIEs in the correct direction and toward the TS and demonstrate relatively small changes in C1′–H1′ bond length (1.092 Å and 1.094 Å). However, the C2′–C1′–H1′ bond angles reveal significant sp3 distortion, changing from 108.7° and 113.6°, a change in C1′ hybridization from sp2.7 to sp2.47 (NBO analysis). These stereoelectronic changes establish OMP ground state destabilization toward the sp2 TSs of the OPRTs.
Figure 2.
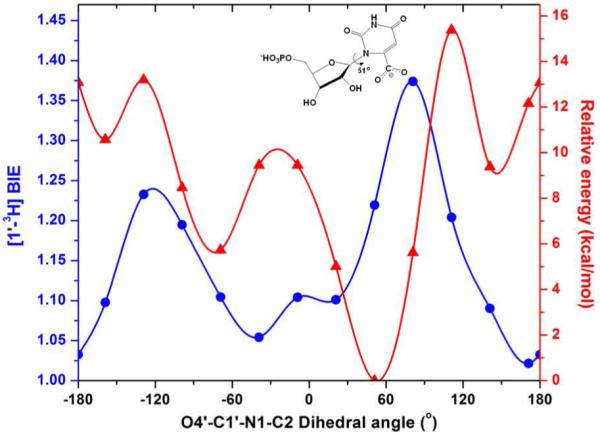
Calculated effects (in vacuo) of rotating the O4′–C1′–N1–C2 dihedral angle of free OMP on its relative energy ( ). The calculated distortional [1′-3H]OMP BIE (
). The calculated distortional [1′-3H]OMP BIE ( ) compares different OMP solution state geometries to the OPRT crystallographic torsion angle for bound OMP of 167°. The distortional BIEs were calculated using ISOEFF98 (16). The energy-minimized OMP with an O4′–C1′–N1–C2 dihedral angle of 51° is shown. Note the BIE is at a minimum at 167° (free and bound substrate at 167°). For a solution structure of 51°, distorted to 167°, the geometry found on the enzyme, the predicted distortional BIE is 1.22.
) compares different OMP solution state geometries to the OPRT crystallographic torsion angle for bound OMP of 167°. The distortional BIEs were calculated using ISOEFF98 (16). The energy-minimized OMP with an O4′–C1′–N1–C2 dihedral angle of 51° is shown. Note the BIE is at a minimum at 167° (free and bound substrate at 167°). For a solution structure of 51°, distorted to 167°, the geometry found on the enzyme, the predicted distortional BIE is 1.22.
Figure 3.

Calculated effects (in water) of rotating the O4′–C1′–N1–C2 dihedral angle of free OMP on its relative energy ( ). The calculated distortional [1′-3H]OMP BIE (
). The calculated distortional [1′-3H]OMP BIE ( ) compares different OMP solution state geometries to the OPRT crystallographic torsion angle for bound OMP of 167°. Using free OMPs with varied O4′–C1′–N1–C2 dihedral angles and HsOPRT-bound OMP at 167°, theoretical BIEs were calculated using ISOEFF98. Calculations in water were performed using the Onsager model implemented in Gaussian 03. The recommended radii for calculations in water were determined through volume calculations.
) compares different OMP solution state geometries to the OPRT crystallographic torsion angle for bound OMP of 167°. Using free OMPs with varied O4′–C1′–N1–C2 dihedral angles and HsOPRT-bound OMP at 167°, theoretical BIEs were calculated using ISOEFF98. Calculations in water were performed using the Onsager model implemented in Gaussian 03. The recommended radii for calculations in water were determined through volume calculations.
The [1′-3H]OMP BIEs in Figures 2 and 3 are calculated purely on the N-ribosyl torsion angle and associated structural changes and overestimate the experimental BIEs in vacuo and in water. Clearly, the enzymes contribute additional changes to the C1′-3H1′ bond, changes that are known to influence the C1′-3H1′ bond (23-27). Upon formation of the Michaelis complex, distorted OMP displays a C4′–O4′–C1′–N1 dihedral angle of −118°, compared with −151° for that calculated for free OMP.
The strained OMP causes orbital overlap between a lone pair of O4′ and the antibonding orbital (σ*) of C1′–N1, promoting N-glycosidic bond cleavage. Ground state distortion of OMP at the OPRT binding site is assisted by this effect. The antibonding orbital (σ*) of C1′-H1′ is antiperiplanar to a lone pair of O4′. The orbital interactions combined with an anomeric effect collectively contribute to the [1′-3H] BIEs .
Computational Analysis of [1′-3H]OMP BIE.
X-ray crystal structures of OPRT homologues reveal large conformation changes involving catalytic site loop-motions upon binding of substrates or products (28-31). These motions form contacts to catalytic site reactants and are components of the reaction coordinate (32-36). One mechanism to generate [1′-3H]OMP BIE effects is to immobilize the pyrimidine and ribosyl groups in different positions on the enzyme relative to those free in solution. Comparison of free and enzyme-bound dihedral angles gives a calculated [1′-3H]OMP BIE of 1.219, greater than the experimental [1′-3H]OMP BIE of 1.108, and indicating that forces other than the N-ribosidic torsion angle contribute to the BIE. As required by this analysis, the calculated BIE values are unity at 167°, comparing like states on and off the enzyme.
Differences in calculated and experimental BIEs could be due to the anionic 6-carboxyl group of OMP (Figure 2). In vacuo calculations can overestimate intramolecular interactions between the 6-carboxylate anion and the 2′-hydroxyl group.
Calculation of the reactant state of OMP in water also overestimates the [1′-3H] BIE to be 1.225 (Figure 3). Calculated dihedral torsion angle energy barriers in water are decreased substantially but still give large [1′-3H] BIE values (Figure 4).
Figure 4.
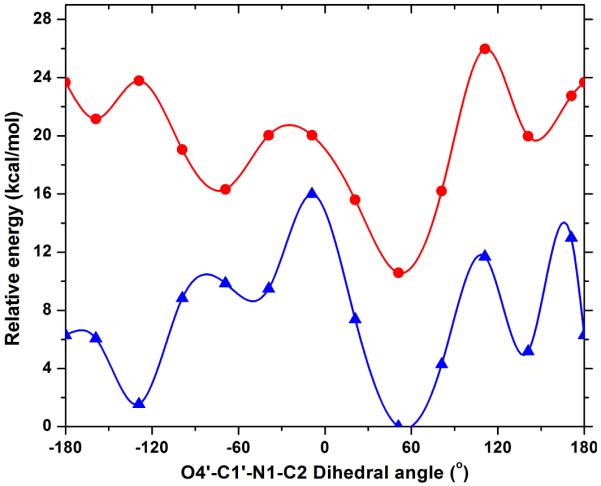
Effects of rotating O4′–C1′–N1–C2 dihedral angle in OMP on the relative energy for free OMP calculated in vacuo ( ) and in water (
) and in water ( ). Calculations in water were performed using the Onsager model implemented in Gaussian 03. The recommended radii for calculations in water were determined through volume calculations.
). Calculations in water were performed using the Onsager model implemented in Gaussian 03. The recommended radii for calculations in water were determined through volume calculations.
The calculations described above used the energetic minimum as a fixed value for calculation, and free-energy-weighted averages of [1′-3H]OMP BIEs were calculated to be 1.216 in vacuo and 1.223 in water, close to those values determined from free OMP geometries as global minima.
[1′-3H]OMP BIEs and KIEs for HsOPRT.
Resolution of [1′-3H]OMP kcat/Km KIE into binding and transition state effects shows that half of the kcat/Km KIE originates from BIE and establishes two-step distortion of OMP at the OPRT active sites, half upon binding and half on going from the Michaelis complex to the TS. In comparison, the near-unity [1′-3H]orotidine BIE for HsOPRT indicates that the experimental kcat/Km KIE of 1.209 has no contribution from binding step and there is no significant ground-state destabilization in the Michaelis complexes.
Conclusion
These are the first reports of [1′-3H] BIEs in any N-ribosyltransferases. The large normal [1′-3H]OMP BIEs reveal significant ground-state destabilization upon formation of the Michaelis complexes of PfOPRT and HsOPRT. The result is an unequivocal demonstration of substrate distortion toward the transition state. Half of the [1′-3H]OMP KIE found at the sp2 TS for these enzymes originates on formation of the enzyme-substrate complexes. The ability of the OPRT enzymes to distort the sp3 geometry of C1’ requires substrate to be anchored by the 5′-phosphate. Orotidine passes through the same sp2 TS but without ground-state destabilization and a 240-fold penalty in catalytic efficiency. Distortion of the geometry of C1′ is also known to occur on binding of a transition state analogue to human PNP (37). Here we demonstrate that similar changes can occur on substrate binding. [1′-3H] BIEs for OMP and orotidine with and without sulfate provide unique and valuable insights into interactions within the enzyme-substrate complexes.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (AI049512 and GM041916).
Abbreviations and Textual Footnotes
- BIEs
binding isotope effects
- OMP
orotidine 5′-monophosphate
- PfOPRT
Plasmodium falciparum orotate phosphoribosyltransferase
- HsOPRT
human orotate phosphoribosyltransferase
- KIEs
kinetic isotope effects
- TS
transition state
- TP
thymidine phosphorylase
- PNP
purine nucleoside phosphorylase
- PPi
pyrophosphate
- FTIR
Fourier transform infrared
Footnotes
Supporting Information Available: Complete calculation results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cleland WW. The use of isotope effects to determine transition-state structure for enzymic reactions. Methods Enzymol. 1982;87:625–641. doi: 10.1016/s0076-6879(82)87033-x. [DOI] [PubMed] [Google Scholar]
- 2.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Annu. Rev. Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 3.Schramm VL. Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch. Biochem. Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 4.Schramm VL. Binding isotope effects: boon and bane. Curr. Opin. Chem. Biol. 2007;11:529–536. doi: 10.1016/j.cbpa.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson VE. Quantifying energetic contributions to ground state destabilization. Arch. Biochem. Biophys. 2005;433:27–33. doi: 10.1016/j.abb.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 6.Fersht A. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W. H. Freeman; 1999. p. 373. [Google Scholar]
- 7.Murkin AS, Tyler PC, Schramm VL. Transition-state interactions revealed in purine nucleoside phosphorylase by binding isotope effects. J. Am. Chem. Soc. 2008;130:2166–2167. doi: 10.1021/ja7104398. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Luo M, Schramm VL. Transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J. Am. Chem. Soc. 2009;131:4685–4694. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Schramm VL. Pyrophosphate interactions at the transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J. Am. Chem. Soc. 2010;132:8787–8794. doi: 10.1021/ja102849w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Deng H, Schramm VL. Leaving group activation and pyrophosphate ionic state at the catalytic site of Plasmodium falciparum orotate phosphoribosyltransferase. J. Am. Chem. Soc. 2010;132:17023–17031. doi: 10.1021/ja107806j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocinero EJ, Carcabal P, Vaden TD, Simons JP, Davis BG. Sensing the anomeric effect in a solvent-free environment. Nature. 2011;469:76–79. doi: 10.1038/nature09693. [DOI] [PubMed] [Google Scholar]
- 12.Lewis BE, Schramm VL. Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J. Am. Chem. Soc. 2003;125:4785–4798. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 13.Parkin DW, Leung HB, Schramm VL. Synthesis of nucleotides with specific radiolabels in ribose. Primary 14C and secondary 3H kinetic isotope effects on acid-catalyzed glycosidic bond hydrolysis of AMP, dAMP, and inosine. J. Biol. Chem. 1984;259:9411–9417. [PubMed] [Google Scholar]
- 14.Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry. 2005;44:11647–11659. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 15.Frisch MJ, et al. Gaussian 03, Revision E.01. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 16.Anisimov V, Paneth P. ISOEFF98. A program for studies of isotope effects using Hessian modifications. J. Math. Chem. 1999;26:75–86. [Google Scholar]
- 17.Schramm VL. Comparison of initial velocity and binding data for allosteric adenosine monophosphate nucleosidase. J. Biol. Chem. 1976;251:3417–3424. [PubMed] [Google Scholar]
- 18.Sievers A, Wolfenden R. The effective molarity of the substrate phosphoryl group in the transition state for yeast OMP decarboxylase. Bioorg. Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Morrow JR, Amyes TL, Richard JP. Phosphate binding energy and catalysis by small and large molecules. Acc. Chem. Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsang WY, Amyes TL, Richard JP. A substrate in pieces: allosteric activation of glycerol 3-phosphate dehydrogenase (NAD+) by phosphite dianion. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Go MK, Amyes TL, Richard JP. Hydron transfer catalyzed by triosephosphate isomerase. Products of the direct and phosphite-activated isomerization of [1-(13)C]-glycolaldehyde in D(2)O. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westaway KC, Fang YR, MacMillar S, Mattsson O, Poirier RA, Islam SM. Determining the transition-state structure for different SN2 reactions using experimental nucleophile carbon and secondary alpha-deuterium kinetic isotope effects and theory. J. Phys. Chem. A. 2008;112:10264–10273. doi: 10.1021/jp804237g. [DOI] [PubMed] [Google Scholar]
- 23.Thatcher Gregory RJ. The Anomeric Effect and Associated Stereoelectronic Effects. American Chemical Society; 1993. Anomeric and Associated Stereoelectronic Effects; pp. 6–25. [Google Scholar]
- 24.Carr CA, Ellison SLR, Robinson MJT. Diverse origins of conformational equilibrium isotope effects for hydrogen in 1,3-dioxans. Tetrahedron Lett. 1989;30:4585–4588. [Google Scholar]
- 25.Forsyth DA, Hanley JA. Conformationally dependent intrinsic and equilibrium isotope effects in N-methylpiperidine. J. Am. Chem. Soc. 1987;109:7930–7932. [Google Scholar]
- 26.Anet FAL, Kopelevich M. Deuterium isotope and anomeric effects in the conformational equilibria of molecules containing CHD-O groups. J. Am. Chem. Soc. 1986;108:2109–2110. [Google Scholar]
- 27.Kirby AJ. Mechanism and stereoelectronic effects in the lysozyme reaction. CRC Crit. Rev. Biochem. 1987;22:283–315. doi: 10.3109/10409238709086959. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Segura L, Witte JF, McClard RW, Hurley TD. Ternary complex formation and induced asymmetry in orotate phosphoribosyltransferase. Biochemistry. 2007;46:14075–14086. doi: 10.1021/bi701023z. [DOI] [PubMed] [Google Scholar]
- 29.Henriksen A, Aghajari N, Jensen KF, Gajhede M. A flexible loop at the dimer interface is a part of the active site of the adjacent monomer of Escherichia coli orotate phosphoribosyltransferase. Biochemistry. 1996;35:3803–3809. doi: 10.1021/bi952226y. [DOI] [PubMed] [Google Scholar]
- 30.Scapin G, Grubmeyer C, Sacchettini JC. Crystal structure of orotate phosphoribosyltransferase. Biochemistry. 1994;33:1287–1294. doi: 10.1021/bi00172a001. [DOI] [PubMed] [Google Scholar]
- 31.Scapin G, Ozturk DH, Grubmeyer C, Sacchettini JC. The crystal structure of the orotate phosphoribosyltransferase complexed with orotate and alpha-D-5-phosphoribosyl-1-pyrophosphate. Biochemistry. 1995;34:10744–10754. doi: 10.1021/bi00034a006. [DOI] [PubMed] [Google Scholar]
- 32.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem. Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 33.Gao J, Ma S, Major DT, Nam K, Pu J, Truhlar DG. Mechanisms and free energies of enzymatic reactions. Chem. Rev. 2006;106:3188–3209. doi: 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 35.Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Atomic detail of chemical transformation at the transition state of an enzymatic reaction. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16543–16548. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz SD, Schramm VL. Enzymatic transition states and dynamic motion in barrier crossing. Nat Chem Biol. 2009;5:551–558. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauve AA, Cahill SM, Zech SG, Basso LA, Lewandowicz A, Santos DS, Grubmeyer C, Evans GB, Furneaux RH, Tyler PC, McDermott A, Girvin ME, Schramm VL. Ionic states of substrates and transition state analogues at the catalytic sites of N-ribosyltransferases. Biochemistry. 2003;42:5694–5705. doi: 10.1021/bi034003a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.