

Abstract
This article describes the effect of re-interrogation of electron-transfer dissociation (ETD) data with newly developed analytical tools. MS/MS-based characterization of O-linked glycopeptides is discussed using data acquired from a complex mixture of O-linked glycopeptides, featuring mucin core 1-type carbohydrates with and without sialic acid, as well as after partial deglycosylation to leave only the core GalNAc units (Darula and Medzihradszky in Mol Cell Proteomics 8:2515, 2009). Information content of collision-induced dissociation spectra generated in collision cell (in QqTOF instruments) and in ion traps is compared. Interpretation of the corresponding ETD data using Protein Prospector is also presented. Search results using scoring based on the frequency of different fragment ions occurring in ETD spectra of tryptic peptides are compared with results obtained after ion weightings were adjusted to accommodate differential ion frequencies in spectra of differing charge states or cleavage specificities. We show that the improved scoring is more than doubled the glycopeptide assignments under very strict acceptance criteria. This study illustrates that “old” proteomic data may yield significant new information when re-interrogated with new, improved tools.
Electronic supplementary material
The online version of this article (doi:10.1007/s00726-010-0692-2) contains supplementary material, which is available to authorized users.
Keywords: Mass spectrometry, Electron-transfer dissociation (ETD), Collision-induced dissociation (CID), O-linked glycopeptides, O-glycosylation, Database search
Introduction
Extracellular O-glycosylation is a somewhat neglected research area considering the widespread occurrence of the modification on the extracellular domains of membrane proteins or on secreted proteins. This can be partly explained by the fact that characterization of O-glycosylation represents a formidable analytical challenge: there are no consensus sequences for predicting sites of modification; extracellular O-glycosylation features a series of different core structures (GalNAc, Man, Fuc, Xyl, and Glc), with complex and diverse elongations; there are no efficient enrichment strategies for any of them, and the modifications are difficult to study using mass spectrometry (Varki et al. 2009; Peter-Katalinic 2005).
Mass spectrometry has been successfully used for the characterization of a wide variety of post-translational modifications (PTMs) for the last two decades, even in a high throughput manner. Collision-induced dissociation (CID) of O-linked glycopeptides has produced quite a few success stories (Harris et al. 1993; Agarwala et al. 1994; Medzihradszky et al. 1996; Alving et al. 1999; Balog et al. 2010). However, these studies have also highlighted the problems associated with such molecules. Glycosidic bonds are weaker than peptide bonds, and thus, glycopeptide CID spectra are dominated by carbohydrate fragmentation. In addition, O-linked glycopeptides undergo gas-phase deglycosylation that eliminates the carbohydrate completely, leaving the unmodified, and thus, “unlabeled” Ser or Thr residue(s) behind (Medzihradszky et al. 1996; Peter-Katalinic 2005). Hence, from the CID spectra of O-linked glycopeptides the size, the building blocks and the order of the carbohydrate units can be determined as well as the molecular weight of the peptide (minus the modification). In most instances, prior knowledge of the protein identity is necessary because the peptide fragmentation observed is usually not sufficient for peptide sequence identification on the basis of observed peptide fragment ions. In addition, the bigger the size of the carbohydrate substituent, the more fragment ions from the sugar structure are observed and the less from the modified peptide sequence, making peptide identification increasingly more difficult. CID data from ion traps that permit only a single activation step contain almost exclusively carbohydrate fragments and practically no information about the peptide sequence. Data obtained from instruments equipped with collision cells that permit multiple collisions/activation steps (e.g. quadrupole collision cells) usually provide more comprehensive fragmentation information than ion trap data (Peter-Katalinic 2005).
With the advent of electron-capture (Zubarev et al. 2000) and electron-transfer (Syka et al. 2004) dissociation (ECD and ETD, respectively), the identification and site assignment of fragmentation-prone side-chain modifications have become more easily achievable goals. ECD has become popular for intact protein characterization, making use of the high resolution and mass accuracy of the FT-ICR instruments that typically perform this type of fragmentation. ETD has been extensively used for the identification of unmodified peptides and PTMs in large scale, high throughput studies due to its high sensitivity in quadrupole ion trap instruments. However, since these electron-based MS/MS techniques yield very different data from CID spectra, which has almost exclusively been acquired in earlier proteomic studies, search engines have to be modified and optimized to permit efficient analysis of ETD fragmentation data.
Large scale, high throughput proteomic experiments are performed mostly on tryptic digests. However, the application of other proteolytic cleavage methods has been promoted for ETD data acquisition due to peptides with higher charge densities being advantageous for ETD analysis. Thus, optimizing search engines for analysis of peptides produced by different cleavage specificity is of increased importance for ETD data. Thus, we recently performed a statistical analysis of the ETD fragments detected in digests produced by different cleavage specificities (Chalkley et al. 2010). A new version of Protein Prospector was then created (version 5.4) that applies an altered scoring system based on the results of this statistical analysis (Baker et al. 2010). It was recognized that doubly charged precursor ions show dramatically different fragmentation to precursor ions of higher charge states. It was also apparent that the presence of a basic residue at either terminus also influences the weighing of the different ion types (Chalkley et al. 2010). Hence, the new scoring system in Protein Prospector allows for different fragment ion weighting depending on precursor charge and also the presence of basic residues at either terminus of the peptide. This improved scoring has been shown to be particularly beneficial for cleavage products that do not display basic residues at their C-terminus, such as AspN and LysN digests, where the improvement can be dramatic: 40% more peptides identified at the same false discovery rate threshold (Baker et al. 2010). Since a significant portion of serum-derived glycopeptides are not fully tryptic peptides due to the presence of active proteases in serum, we decided to test the “new” search engine and re-interrogate the data acquired for our published glycopeptide study.
We previously reported that ETD analysis using an earlier version of Protein Prospector successfully identified 49 glycopeptides bearing mucin core 1-type structures (GalNAcGal) with or without sialic acid (Darula and Medzihradszky 2009). However, in order to find these glycopeptides, a species-specific search was conducted with the SwissProt database, and manual validation was essential for each glycopeptide match with an expectation value of lower than 0.5. In that study, we also analyzed partially deglycosylated glycopeptide mixtures, featuring only the core GalNAc residues.
In this paper, we present a brief overview of the options available for large-scale glycopeptide data interpretation, compare the performance of Protein Prospector tuned for tryptic peptides to the newer scoring optimized for alternative cleavage methods, and illustrate the improved performance of the search engine by the identification of novel glycopeptides and glycosylation sites. Our results demonstrate that “old” proteomic data may yield significant valuable information when re-interrogated with new, improved tools.
Experimental
Datasets
O-linked (mucin core-1 type) glycopeptides were isolated from bovine serum utilizing Jacalin-affinity chromatography. The isolation protocol, partial deglycosylation, and mass spectrometric analysis of glycopeptides have been described previously (Darula and Medzihradszky 2009). LC/MS/MS data discussed here were acquired using an LTQ-Orbitrap hybrid mass spectrometer. Precursor masses were measured in the Orbitrap; CID and ETD data were acquired in the linear trap.
Data interpretation
For data interpretation, raw mass spectrometric data were converted into peaklists using Bioworks 3.3.1 SP1 or in-house software, PAVA (Lynn et al. 2008). The same peaklists were used by both search engine versions (Protein Prospector v.5.3 and v5.4). Database searching was performed against the UniProt database (downloaded on Dec 15, 2009) supplemented with a random sequence for each entry, and species specified as Bos taurus (31074/21095996 entries searched). Trypsin was specified as the enzyme; 1 missed cleavage and non-specific cleavage at one of the peptide termini were permitted. Mass accuracy was set to 15 ppm for precursor ions and 0.6 Da for fragment ions. Carbamidomethylation of Cys residues was set as fixed modification, and the following were allowed as variable modifications: acetylation of protein N-termini; Met oxidation; cyclization of N-terminal Gln residues; and HexHexNAc or SAHexHexNAc (for intact glycopeptides) and HexNAc (for partially deglycosylated glycopeptides) modification on Thr and Ser residues. A maximum of three modifications per peptide were considered. Acceptance criteria for the reported results were as follows: minimal scores for proteins and peptides were 22 and 15, respectively, and maximum expectation values for proteins and peptides were 0.05 and 0.1, respectively. Results from different fractions of affinity enrichment were merged, and only the best scoring identifications are reported. From proteins that share sequences only the top hit is listed.
The new scoring is implemented on the Protein Prospector public website at: http://prospector.ucsf.edu.
Results
As published earlier (Darula and Medzihradszky 2009), mucin core 1-type glycopeptides were enriched from bovine serum and analyzed by mass spectrometry. LC/MS/MS analysis of such a mixture using a QqTOF instrument (QTOF Premier, Waters) indicated the presence of more than 100 glycopeptides based on the presence of diagnostic carbohydrate fragment ions, but only a handful of these yielded sufficiently informative CID spectra to allow identification by automated database searching or even manual sequencing (e.g. Fig. 1). When the same mixtures were subjected to LC/MS/MS analysis in an LTQ-Orbitrap, CID spectra almost exclusively contained only carbohydrate fragments (Fig. 2). From the intact glycopeptide datasets discussed here only four glycopeptides produced sufficient peptide fragmentation to be identified using CID data for database searching. The rest of the CID data, although generally not good enough for peptide identification, were nevertheless useful for manual confirmation of the glycopeptide identification. For example, when the modified peptide contains a proline residue, which is frequently the case with O-glycosylation, then usually both halves are detected as illustrated in Fig. 2, with the glycosylated part frequently present at different degrees of deglycosylation. (Similar CID fragmentation is presented in Supplemental Figures 5A, 5B, and 10 in online resource, Supplementary material 1). The corresponding ETD spectra contained a lot more information; however, the necessary data interpretation tools were still being developed when we performed the previous analysis. In order to be able to identify modified peptides, database searching had to be restricted to bovine proteins within the Swiss Prot database, and very liberal acceptance criteria were applied, followed by manual analysis of all candidate modified spectra. We also experimented with permitting non-specific cleavages at both termini and multiple missed cleavages. Altogether, we reported the identification of 49 glycopeptides (Darula and Medzihradszky 2009).
Fig. 1.
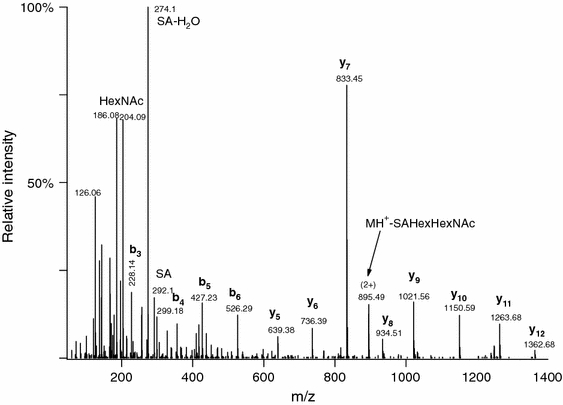
Q-TOF CID spectrum of m/z 815.73 (3+). The peptide sequence can be deduced as AVGAQVLESTPPPHVMR; site of the SAGalGalNAc modification cannot be determined. The identity of the sugar units also cannot be determined from the CID data, but only from the lectin specificity
Fig. 2.
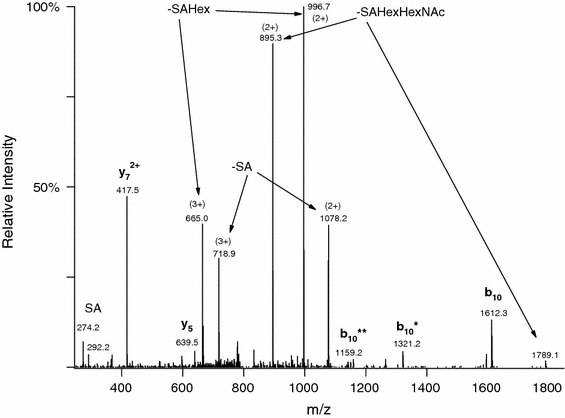
Linear ion trap CID spectrum of the same glycopeptide AVGAQVLESTPPPHVMR modified with SAGalGalNAc as in Fig. 1. Precursor ion was at m/z 815.7307(3+). The abundant peptide fragments in the spectrum were formed by cleavage N-terminal to the first Pro residue. The glycosylated half of the structure (b 10) can be observed with the sugar attached as well as partially or completely deglycosylated—the number of asterisks indicates the number of carbohydrate units lost
Since the publication of this study the performance of the search engine Protein Prospector for analysis of ETD data has been significantly improved (Baker et al. 2010). Thus, we decided to re-visit the data to see if the improved software performance would allow new components to be identified using the larger UniProt database, which contains roughly three times as many bovine protein entries, but is going to return less confident expectation value estimates as a result. Thus, in the present study, the glycopeptide dataset was interrogated against the UniProt database supplemented by randomized sequences, and the performance of two different versions of the search engine Protein Prospector was compared. ETD scoring in version 5.3 was developed based on fragmentation observed in ETD spectra from tryptic digests. ETD scoring in version 5.4 was optimized based on our statistical analysis of ETD fragmentation of peptides representing different cleavage specificities and also compensates for fragmentation differences between different charge-state precursor ions (Chalkley et al. 2010; Baker et al. 2010). Table 1 compares the performance of the two generations of the search engine at different acceptance criteria. The newly identified glycosylated sequences are listed in Tables 2 and 3. New glycosylation sites are presented in Table 4. (Supporting ETD spectra can be found in online resource, Supplementary material 1). Supplemental Tables 1 (online resource, Supplementary material 2) and 2 (online resource, Supplementary material 3) show the full search results.
Table 1.
Comparison of the performance of different versions of Protein Prospector Batch-Tag
| Version 5.3.3 | Version 5.4.2 | Improvement | |
|---|---|---|---|
| Intact glycopeptides | |||
| E < 0.10 | 21 | 61 | 2.9 |
| E < 0.05 | 16 | 42 | 2.63 |
| E < 0.01 | 10 | 28 | 2.8 |
| Partially deglycosylated peptides | |||
| E < 0.10 | 23 | 35 | 1.52 |
| E < 0.05 | 21 | 30 | 1.42 |
| E < 0.01 | 17 | 23 | 1.35 |
Table 2.
New—previously not reported—intact glycopeptides identified in this study (protein score > 22, protein E < 0.05, peptide score > 15 and E < 0.1)
| Uniprot ID | Protein name | Sequence | Score | Expected |
|---|---|---|---|---|
| A5D7R6 | ITIH2 protein | (Y)HGSKVS(GalNAcGalSA)PNSVPSWVNPSPAPVLPMPAVGAQVLES(GalNAcGalSA)TPPPHVMR | 19.7 | 0.09 |
| A5D7R6 | ITIH2 protein | (M)PAVGAQVLEST(GalNAcGalSA)PPPHVM(Oxidation)R | 21 | 0.062 |
| A5D7R6 | ITIH2 protein | (M)PAVGAQVLEST(GalNAcGalSA)PPPHVMR | 25 | 0.062 |
| A5D7R6 | ITIH2 protein | (P)AVGAQVLES(GalNAcGal)TPPPHVMR | 23.1 | 0.09 |
| A5D7R6 | ITIH2 protein | (P)AVGAQVLEST(GalNAcGalSA)PPPHVM(Oxidation)R | 40.7 | 0.0022 |
| A5D7R6 | ITIH2 protein | (P)AVGAQVLEST(GalNAcGalSA)PPPHVMR | 24.8 | 0.047 |
| P01044 | Kininogen-1 | (K)C(Carbamidomethyl)PSRPWKPVNGVNPT(GalNAcGalSA)VEM(Oxidation)K | 27.7 | 0.084 |
| P01044 | Kininogen-1 | (Y)EC(Carbamidomethyl)LGC(Carbamidomethyl)VHPISTKS(GalNAcGalSA)PDLEPVLR | 46.7 | 2.20E−04 |
| P01044 | Kininogen-1 | (Y)EC(Carbamidomethyl)LGC(Carbamidomethyl)VHPIST(GalNAcGal)KSPDLEPVLR | 46.6 | 6.60E−05 |
| B8QGI3 | Insulin-like growth factor 2 | (R)EAKS(GalNAcGalSA)HRPLIALPT(GalNAcGalSA)QDPATHGGASSK | 38 | 6.90E−04 |
| A4IFA5 | VASN protein | (R)VRPGPRPS(GalNAcGalSA)PAPAT(GalNAcGalSA)PRPLPLGIEPASPTSLR | 30.7 | 0.015 |
| Q3T052 | ITIH4 | (R)IKGTTPT(GalNAcGalSA)ALPFAPVQAPSVILPLPGQSVDR | 18.8 | 0.059 |
| Q3T052 | ITIH4 | (R)LVLPELMS(GalNAcGalSA)PLAPASAPS(GalNAcGalSA)PTSGPGGASHDTDFR | 29.8 | 0.025 |
| Q3T052 | ITIH4 | (R)LVLPELM(Oxidation)SPLAPASAPSPT(GalNAcGalSA)S(GalNAcGalSA)GPGGASHDTDFR | 32.9 | 0.0042 |
| Q58D62 | Fetuin-B | (N)Q(Gln→pyro-Glu)RPANPSKTEELQQQNT(GalNAcGalSA)APTNSPTK | 29.4 | 0.011 |
| Q0VCM5 | ITIH1 | (Q)ASQPAPT(GalNAcGalSA)HSSLDIK | 36.2 | 0.028 |
Table 3.
New—previously not reported—partially deglycosylated glycopeptides identified in this study (protein score > 22, protein E < 0.05, peptide score > 15 and E < 0.1)
| UniProt ID | Protein name | Peptide | Score | Expected |
|---|---|---|---|---|
| Q3T052 | ITIH4 | (R)LVLPELMS(GalNAc)PLAPASAPSPTSGPGGAS(GalNAc)HDTDFRIK | 24.2 | 0.02 |
| A5D7R6 | ITIH2 protein | (Y)HGSKVSPNSVPSWVNPS(GalNAc)PAPVLPM(Oxidation)PAVGAQVLEST(GalNAc)PPPHVMR | 22.3 | 0.025 |
| A5D7R6 | ITIH2 protein | (Y)HGSKVSPNSVPSWVNPS(GalNAc)PAPVLPMPAVGAQVLES(GalNAc)TPPPHVM(Oxidation)R | 28.7 | 0.0017 |
| A5D7R6 | ITIH2 protein | (Y)HGSKVSPNSVPSWVNPS(GalNAc)PAPVLPMPAVGAQVLEST(GalNAc)PPPHVMR | 30.6 | 6.90E−04 |
| A5D7R6 | ITIH2 protein | (K)VSPNSVPSWVNPS(GalNAc)PAPVLPMPAVGAQVLEST(GalNAc)PPPHVMR | 31.4 | 0.0023 |
| A5D7R6 | ITIH2 protein | (P)AVGAQVLEST(GalNAc)PPPHVM(Oxidation)R | 20.1 | 0.067 |
| A5D7R6 | ITIH2 protein | (A)QVLEST(GalNAc)PPPHVMR | 29.3 | 0.0074 |
| A5D7R6 | ITIH2 protein | (Q)VLEST(GalNAc)PPPHVM(Oxidation)R | 27.6 | 0.013 |
| A5D7R6 | ITIH2 protein | (Q)VLEST(GalNAc)PPPHVMR | 22.4 | 0.094 |
| P07456 | Insulin-like growth factor 2 | (K)SHRPLIALPT(GalNAc)QDPATHGGASSK | 45.5 | 5.90E−06 |
| P07456 | Insulin-like growth factor 2 | (L)IALPT(GalNAc)QDPATHGGAS(GalNAc)SKASSD | 25.1 | 0.016 |
Table 4.
New glycosylation sites identified in this study
| UniProt ID | Protein name | Site spectrum |
|---|---|---|
| A5D7R6 | ITIH2 proteina | Ser-673 (Supplemental Figure 2) |
| Ser-690 or Thr-691 (Supplemental Figure 2) | ||
| Thr-691 (Supplemental Figure 3) | ||
| A4IFA5 | VASN protein | Ser-455 (Supplemental Figure 4) |
| Thr-460 (Supplemental Figure 4) | ||
| P01044 | Kininogen-1 | Ser-149 (Supplemental Figures 6, 7) |
| Thr-150 or Ser-152 (Supplemental Figures 6, 7) | ||
| Thr-605 (Supplemental Figure 8) | ||
| Q2KIU3 | Putative uncharacterized protein | Thr-72 (Supplemental Figure 9) |
| B8QGI3 | Insulin-like growth factor 2 | Ser-173 (Fig. 3) |
From this comparison, it is clear that modifying the search engine made the identification of digestion products dramatically more sensitive. Obviously some of the novel glycoproteins, such as ITI H2 and VASN, were overlooked earlier because the smaller Swiss Prot database was used to try to get higher confidence matches, and these proteins are not present in the Swiss Prot database (as described earlier during the software development phase, search space for database searches with ETD data had to be more limited in order to identify intact glycopeptides that frequently do not undergo very efficient fragmentation because of their relatively low charge density). However, there are clear examples where the glycoprotein was identified in the original study, but certain glycopeptides were assigned only by the improved search engine. For example, new glycosylation sites have been revealed in insulin-like growth factor and in kininogen (Table 4; Fig. 3; Supplemental Figures 6–8 in online resource, Supplementary material 1).
Fig. 3.
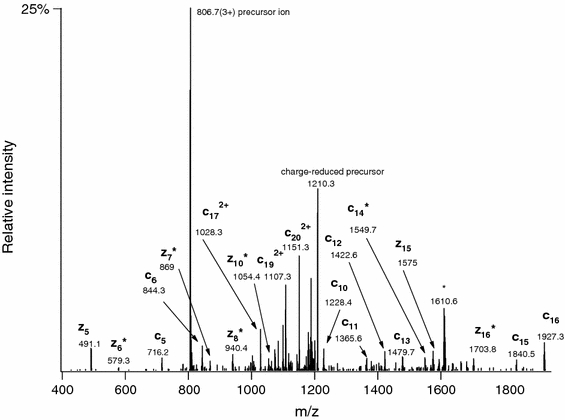
Linear ion trap ETD spectrum of m/z 806.3813 (3+) acquired during the analysis of the partially deglycosylated mixture. The corresponding structure was identified as 159IALPT(GalNAc)QDPATHGGAS(GalNAc)SKASSD179 of insulin-like growth factor 2. Asterisks indicate c − 1• and z + 1 ions. The ion at m/z 1610 is the charge-reduced ion for a 2+ ion within the precursor selection window
The improvement in glycopeptide assignment is more striking with the intact glycopeptide spectra. However, it should be pointed out that this analysis involved a much bigger dataset, namely 16 LC/MS experiments, while the partially deglycosylated data are from a single LC/MS file. Hence, there was more “room for improvement” with the intact glycopeptide dataset. Nevertheless, the results still illustrate some of the advantages of partial deglycosylation. This is more formally illustrated by the comparison of ETD spectra of the doubly glycosylated ITI H2 peptide HGSKVSPNSVPSWVNPSPAPVLPMPAVGAQVLESTPPPHVMR with intact trisaccharides (Supplemental Figure 1 in online resource, Supplementary material 1) and bearing only the core GalNAc units (Supplemental Figure 2 in online resource, Supplementary material 1), where the partially deglycosylated peptide produced a more informative spectrum. In addition, while the search engine displayed a much improved performance, some assignments are still questionable despite the acceptable score and expectation values (Supplemental Figures 11 and 12 in online resource, Supplementary material 1).
Discussion
Data analysis tools are continually being developed and optimized. In particular, when a new technique first becomes available, the software for interpretation of the results may be sub-optimal. When a significant improvement is made in software tools, the re-analysis of previously generated datasets is a potentially rich resource for extracting new biological insight. Indeed, the presence and rapid population of public proteomic resources acknowledges the potential benefit of allowing re-analysis of data with new tools.
In this study, we compared the performance of a search engine using an older scoring system based on the frequency of occurence of different ion types in ETD spectra of tryptic peptides (Protein Prospector v5.3) to a new version that was developed to better accommodate the products of different cleavage specificities (v5.4) for the analysis of glycopeptides spectra from bovine serum (Darula and Medzihradszky 2009). The newer version of Protein Prospector has been optimized based on a statistical analysis of ETD data of digests of different enzyme specificities. Most importantly, an improved scoring system has been introduced that applies ion type weighting depending on the charge state and the localization of the basic residue within the precursor peptide. In addition, ETD spectra are filtered to remove the most common neutral loss fragment ions of charge-reduced precursor ions and multiply charged fragment ions are also considered for precursor ions of charge state of 3+ or higher. Finally, a correction in the calculation of the expectation values was established that results in a more reliable statistical analysis of PTMs. The sum of all these changes led to significant improvements in the number of peptides identified in all datasets tested, and by comparing the results to those reported by other search engines it indicated that the older version of Protein Prospector (version 5.3) appeared to already outperform alternative tools, and the new version further increased this edge (Baker et al. 2010).
In this study, the new version of Protein Prospector identified 190% more intact glycopeptides (using a peptide expectation value threshold at of <0.1). This improvement corresponded to 42 unique glycopeptides, while there were two identifications unique to the former version 5.3. In the case of partially deglycosylated peptides, the new scoring identified 50% more glycopeptides corresponding to 16 unique hits. There were four glycopeptides that only the former 5.3 version identified. Identifications unique to the earlier version sometimes corresponded to weak spectra and thus results of borderline reliability that did not meet the acceptance criteria with the new version of the software. For example, glycopeptides VVVGPS(HexNAcHex)VVAVPLPLHR and TPIVGQPS(HexNAc)IPGGPVR belong to this category (score = 17.7; E = 0.034 vs. score = 17.3; E = 0.13 and score = 16.4; E = 0.042 vs. score = 17.8; E = 0.17, respectively). In addition, sometimes the same sequences but with different modification sites indicated are included in the list. This reflects the general problem of PTM site assignments. The software in different searches may position the modification to different sites when there is insufficient information for unambiguous site determination and neither assignment contradicts the data. For example, this phenomenon was seen for the spectra of glycopeptides KTFMLQAS(HexNAcHexSA)QPAPTHSSLDIK, QVLES(HexNAc)TPPPHVMR and KIQEVPPAVT(HexNAc)TAPPGSR.
Site assignment issues
Although careful manual validation of ETD spectra confirmed the peptide identification for the majority of the novel glycopeptides, site assignment adds another layer of complexity to the characterization of PTMs, and is far less reliable, even when using ETD for fragmentation analysis. In our tables some sequences are listed with different modification sites indicated. These assignments may reflect real structural differences, for example, HVGKT(HexNAc)PIVGQPSIPGGPVR and HVGKTPIVGQPS(HexNAc)IPGGPVR in fetuin, or just indicate the ambiguity of the site assignment, when the same precursor ion was selected for MS/MS analysis multiple times and produced slightly different results, like the three different versions of the trisaccharide-modified (140–160) kininogen peptide. This problem is a recurrent issue in PTM analysis, and it is acknowledged that peptide identification is far more reliable compared to modification site assignment. There are several factors that contribute to this phenomenon. First of all, search engines always designate a site of modification even if there is not any decisive information for a particular site assignment. Sometimes the peaks that may suggest the site of modification are too close to the noise level to be reliable, or some fragments are mis-assigned (e.g. supplemental activation results in neutral losses of sugar moieties from charge-reduced precursor ions, which are not considered by search engines for ETD spectra. These fragment ions can then be mis-assigned leading to incorrect site determination.). Therefore, manual evaluation of search results on modified peptides is still an inevitable job. Nonetheless, improvements in the Protein Prospector search engine do result in more reliable identifications that render manual inspection much faster and less tedious.
Due to rampant proteolytic activity in serum, the majority of the newly identified peptides refer to the same glycosylation sites that were reported in our previous study (Darula and Medzihradszky 2009). However, seven novel glycosylation sites were found. Two of these sites have been reported to be glycosylated in the human homolog; glycosylation of the other five sites has not been reported according to UniProt. In two further cases, the site of modification could not be assigned unambiguously. The novel glycosylation sites partly belonged to proteins that we earlier found glycosylated at other sites (kininogen 1 and insulin-like growth factor 2; Darula and Medzihradszky 2009). Identification of these new glycosylation sites clearly illustrates the enhanced performance of the improved scoring in Prospector version 5.4. On the other hand, novel glycoproteins have also been identified. These are the ITIH2 protein (A5D7R6), and VASN protein (A4IFA5). These two proteins are not included in the SwissProt database, and our original search needed to be restrained to the bovine entries of the Swissprot database because of the shortcomings in sensitivity of early database searches with ETD data. As described above, the developmental version of Protein Prospector used in our original study was accommodating ETD fragmentation, but its scoring had not been optimized. Thus, even some valid glycopeptide hits displayed expectation values as high as 0.4 (Darula and Medzihradszky 2009). A further novel glycosylation site, Thr-72 of a collagen-domain containing protein (Q2KIU3, putative uncharacterized protein), has been identified by both versions of Protein Prospector.
Conclusions
Our results illustrate the common knowledge that proteomic datasets contain more information than is typically interpreted from a first analysis. Different search engines may extract additional information and identify new sequences as well as unexpected modifications. This is especially true when the first results were published at the beginning of the application of a novel analytical tool. In such studies, data interpretation is also at the beginner’s level; fragmentation rules are not fully characterized, and scoring has not been optimized. Hence, important information can potentially be missed. We demonstrate that re-interrogation of “old” datasets with new tools may yield valuable results.
Optimization of ETD scoring in the search engine led to a dramatic improvement in processing ETD spectra and identifying glycopeptides. However, despite these advances manual evaluation of the spectra is still essential for reliable site assignments. We also highlight how CID data provide confirmatory information for the glycopeptide assignment in support of the ETD data.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by NIH grant NCRR P41RR001614 (to the UCSF Mass Spectrometry Facility, director: A.L. Burlingame) and a Hungarian Science Foundation Grant, OTKA T60283 (to KFM).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Agarwala KL, Kawabata S, Takao T, Murata H, Shimonishi Y, Nishimura H, Iwanaga S. Activation peptide of human factor IX has oligosaccharides O-glycosidically linked to threonine residues at 159 and 169. Biochemistry. 1994;33:5167–5171. doi: 10.1021/bi00183a021. [DOI] [PubMed] [Google Scholar]
- Alving K, Paulsen H, Peter-Katalinic J. Characterization of O-glycosylation sites in MUC2 glycopeptides by nanoelectrospray QTOF mass spectrometry. J Mass Spectrom. 1999;34:395–407. doi: 10.1002/(SICI)1096-9888(199904)34:4<395::AID-JMS771>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Baker PR, Medzihradszky KF, Chalkley RJ (2010) Improving software performance for peptide ETD data analysis by implementation of charge-state and sequence-dependent scoring. Mol Cell Proteomics (in press) [DOI] [PMC free article] [PubMed]
- Balog CI, Mayboroda OM, Wuhrer M, Hokke CH, Deelder AM, Hensbergen PJ (2010) Mass spectrometric identification of aberrantly glycosylated human apolipoprotein C-III peptides in urine from Schistosoma mansoni-infected individuals. Mol Cell Proteomics 9(4):667–681 [DOI] [PMC free article] [PubMed]
- Chalkley RJ, Medzihradszky KF, Lynn AJ, Baker PR, Burlingame AL. Statistical analysis of peptide electron transfer dissociation fragmentation mass spectrometry. Anal Chem. 2010;82:579–584. doi: 10.1021/ac9018582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darula Z, Medzihradszky KF. Affinity enrichment and characterization of mucin core-1 type glycopeptides from bovine serum. Mol Cell Proteomics. 2009;8:2515–2526. doi: 10.1074/mcp.M900211-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flahaut C, Capon C, Balduyck M, Ricart G, Sautiere P, Mizon J. Glycosylation pattern of human inter-alpha-inhibitor heavy chains. Biochem J. 1998;333:749–756. doi: 10.1042/bj3330749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RJ, van Halbeek H, Glushka J, Basa LJ, Ling VT, Smith KJ, Spellman MW. Identification and structural analysis of the tetrasaccharide NeuAcα(2-6)Galβ(1-4)GlcNAcβ (1-3)Fucα1-O-linked to serine 61 of human factor IX. Biochemistry. 1993;32:6539–6547. doi: 10.1021/bi00077a007. [DOI] [PubMed] [Google Scholar]
- Lynn A, Chalkley RJ, Baker PR, Medzihradszky KF, Guan S, Burlingame AL(2008) The effect of peaklist generation software on database search results. In: 56th ASMS conference on mass spectrometry, Denver, CO
- Medzihradszky KF, Gillece-Castro BL, Townsend RR, Burlingame AL, Hardy MR. Structural elucidation of O-linked glycopeptides by high energy collision-induced dissociation. J Am Soc Mass Spectrom. 1996;7:319–328. doi: 10.1016/1044-0305(95)00682-6. [DOI] [PubMed] [Google Scholar]
- Olsen EH, Rahbek-Nielsen H, Thogersen IB, Roepstorff P, Enghild JJ. Posttranslational modifications of human inter-alpha-inhibitor: identification of glycans and disulfide bridges in heavy chains 1 and 2. Biochemistry. 1998;37:408–416. doi: 10.1021/bi971137d. [DOI] [PubMed] [Google Scholar]
- Peter-Katalinic J. O-glycosylation of proteins. Methods Enzymol. 2005;405:139–171. doi: 10.1016/S0076-6879(05)05007-X. [DOI] [PubMed] [Google Scholar]
- Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of glycobiology. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.