

Abstract
The motor symptoms of Parkinson's disease (PD) are due primarily to the degeneration of the dopaminergic neurons in the nigrostriatal pathway. However, several other brain areas and neurotransmitters other than dopamine such as noradrenaline, 5-hydroxytryptamine and acetylcholine are affected in the disease. Moreover, adenosine because of the extensive interaction of its receptors with the dopaminergic system has been implicated in the in the pathophysiology of the disease. Based on the involvement of these nondopaminergic neurotransmitters in PD and the sometimes severe adverse effects that limit the mainstay use of dopamine-based antiparkinsonian treatments, recent assessments have called for a broadening of therapeutic options beyond the traditional dopaminergic drug arsenal.
In this review we describe the interactions between dopamine and adenosine receptors that underpin the preclinical and clinical rationale for pursuing adenosine A2A receptor antagonists as symptomatic and potentially neuroprotective treatment of PD. The review will pay particular attention to recent results regarding specific A2A receptor-receptor interactions and recent findings identifying urate, the end product of purine metabolism, as a novel prognostic biomarker and candidate neuroprotectant in PD.
1) Localization of adenosine receptors and functional interactions with dopamine receptors
Extensive interactions between adenosine A1 and A2A receptors and the various dopamine receptors are present in brain at several levels, whereas the interactions between adenosine A2A and dopamine D2 receptors are restricted within the basal ganglia where they are of particular relevance to the characteristic motor dysfunction of PD.
High densities of adenosine A2A receptors are present in both the ventral and dorsal striatum of rodents and primates, including humans. These receptors colocalize in the striatum with the dopamine D2 receptor in the dendritic spines of enkephalin-rich striatopallidal GABA neurons and on glutamatergic terminals (Schiffmann et al., 1991; Rosin et al., 1998). This anatomical framework provides an important structural basis to our understanding of previously discovered A2A/D2 functional interactions.
In addition, A2A receptors are highly expressed in the globus pallidus (GP), mainly in the neuropil, where their stimulation enhances striatopallidal GABA outflow, and their blockade reduces it (Rosin et al., 1998; Ochi et al., 2000; Shindou et al., 2003). In 6-hydroxydopamine (6-OHDA)-lesioned rats, intrapallidal infusion of A2A receptor antagonists, while not eliciting any motor response per-se, does potentiate motor activity induced by l-DOPA or dopaminergic agonists. This suggests that blockade of pallidal A2A receptors, by reducing extracellular GABA, may stabilize GP activity and in turn subthalamic nucleus (STN) activity (Simola et al., 2006). Therefore, both structures may contribute to the therapeutic action of A2A receptor antagonists.
Adenosine A2A receptors exert an excitatory influence on striatopallidal neurons, in part through their antagonistic effect on dopamine D2 receptor activation (Fig. 1). The basis of this antagonistic action of adenosine A2A receptors is their ability to decrease the binding affinity of D2 receptors for dopamine as demonstrated in rat striatal membrane, in human striatal tissue and in different cell lines (Ferré et al., 1991; Diaz-Cabiale et al., 2001; Hillion et al., 2002; Canals et al., 2003). In agreement with these studies, stimulation of adenosine A2A receptors counteracts the D2 receptor-mediated inhibition of cAMP formation and D2 receptor-induced intracellular Ca2+ responses (Kull et al., 1999; Olah et al., 2000; Salim et al., 2000). Of great importance, A2A receptors exert a strong influence on DARPP-32, a dopamine and cAMP-regulated phosphoprotein, which is expressed at high levels in the GABAergic efferent neurons and is deeply involved in dopamine-mediated signalling (Lindskog et al., 2002) (Fig. 1).
Fig 1.
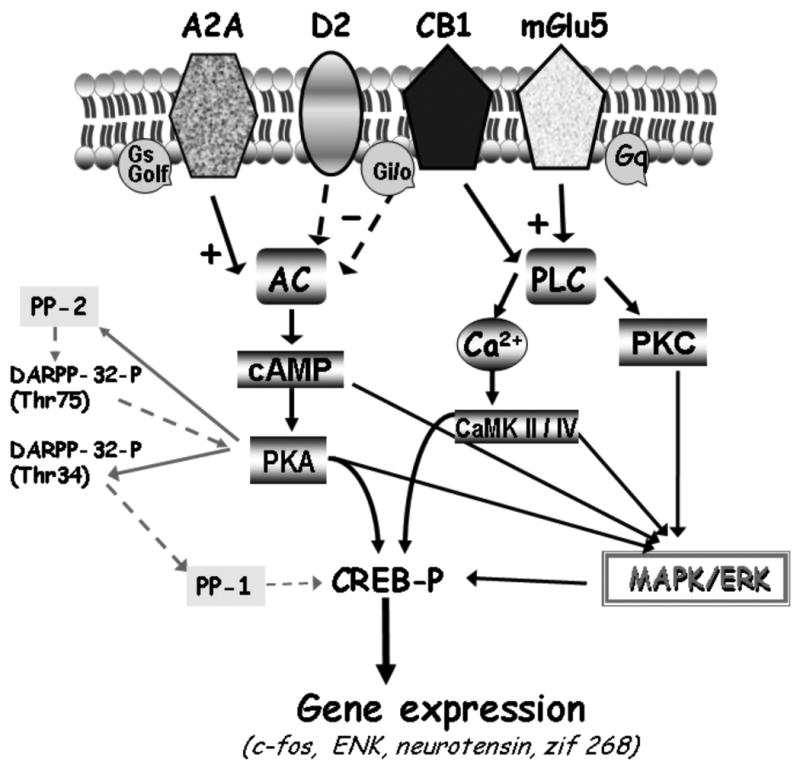
Functional interactions between dopamine D2, adenosine A2A, cannabinoid CB1 and glutamate mGlu5 receptors in striatopallidal neurons. Adenosine A2A receptors interact antagonistically with D2 and CB1 receptors at the intramembrane level and at the adenylyl cyclase level; Metabotropic glutamate mGlu5 and adenosine A2A receptors act synergistically to counteract the D2 dopamine receptor signalling in striatopallidal neurons. Synergistic interactions exist between A2A and mGlu5 receptors at the level of c-fos expression, MAP kinases and phosphorylation of DARPP-32 protein; for further explanation see text. broken arrows – inhibitory effect; ‘+’- stimulation; ‘- ’ – inhibition; AC – adenylyl cyclase; Ca2+ - calcium ions; CaMK II/IV calcium/calmodulin –dependent protein kinase type II/IV; cAMP – cyclic AMP; CREB – cAMP response element-binding protein; K+ - potassium channel; DARPP-32 -dopamine and cAMP-regulated phosphoprotein; DARPP-32-P (Thr75) and DARPP-32-P (Thr34) – DARPP-32-phopshorylated at threonine residues 75 and 34, respectively; Gi, Go – inhibitory G-proteins, Gq, Gs, Golf – stimulatory G-proteins; MAPK – mitogen-activated protein kinase; PKA - protein kinase A; PKC - protein kinase C; PLC – phospholipase C; PP-1 – protein phosphatase-1 ; PP-2 - protein phosphatase-2.
The regulation of dopaminergic signal transduction by A2A receptors is also illustrated by the regulation of CREB activity by A2A receptor stimulation, which increases cAMP formation and in turn phosphorylation of CREB. Selective D2 receptor agonists dose-dependently counteracted these effects (Kull et al., 1999). Furthermore, a variety of in vivo studies support the reciprocal antagonistic influence of A2A and D2 receptors in induction of immediate early-gene expression (e.g., c-fos, zif/268, NGFI-B and jun-B) (Morelli et al., 1995; Svenningsson et al., 1999; Tronci et al., 2006). Interestingly, the gene expression of striatal A2A receptors and the A2A/D2 receptor interaction are increased in the dopamine-denervated striatum (Ferré and Fuxe 1992) (Fig. 1). Despite these mentioned findings, it has been shown that stimulation, as well as blockade, of adenosine A2A receptors induces behavioral and biochemical responses in mice lacking dopamine D2 receptors, suggesting that adenosine A2A receptor actions can occur independently of dopamine signaling (Aoyama et al., 2000).
2) Receptor-receptor interaction: heterodimeric complexes as basis for new antiparkinsonian therapies
An important finding, with respect to A2A receptors, is the formation of functional receptor complexes (receptor mosaics) with other G-protein-coupled receptors (GPCRs). A2A receptors, like many other GPCRs, form both homo and heterodimers (Agnati et al., 2003). This discovery has further increased our understanding of the biology of the A2A receptor with particular emphasis on molecular interactions with receptors for other neurotransmitters such as dopamine D2, D3, cannabinoid CB1 and metabotropic glutamate mGlu5 receptors (Fig. 1). Heterodimerization may have functional and pharmacological consequences; therefore, the presence of heterodimeric complexes has constituted a considerable step forward in the neurobiology of adenosine, suggesting new ways of modulating neuronal activity by targeting the A2A receptor (Ferré et al., 2009; Fuxe et al., 2003).
Coimmunoprecipitation studies with FRET and BRET analyses have demonstrated the existence of constitutive A2A/D2 heteromers as well as A2A homodimers within the plasma membrane, indicating less than 10 nm separating the receptors (Hillion et al., 2002; Canals et al., 2003).
Functional implications of the A2A/D2 intramembrane receptor/receptor interaction through heteromerization, include a decrease in receptor affinity for dopamine agonists acting on D2 receptors, as well as a reduction of D2 receptor G protein coupling and signaling. Thus, the essence of this A2A/D2 receptor heteromerization may be to convert the D2 receptor into a state of strongly reduced functional activity. The discovery of heteromeric A2A/D2 complexes has added to the substantial evidence for antagonistic molecular, cellular, electrophysiological and behavioural interactions between A2A and D2 receptors. These form the basis for antiparkinsonian strategies that simultaneously block adenosine A2A and stimulate dopamine D2 receptors (Morelli et al., 2007, 2009).
Moreover, evidence for functional A2A/D3 heteromers has recently been obtained in cotransfected A2A/D3 cells using FRET. A2A activation reduces the affinity of D3 agonist binding sites as well as D3 signaling (Torvinen et al., 2005). It should be noted, however, that while this interaction could provide interesting clues for mediation of ventral striatal (limbic) functions, it might not be relevant for dorsal striatum functions where the D3 receptor, which is expressed only after dopamine denervation, and the A2A receptor are segregated in different neuronal populations (of striatonigral and striatopallidal neurons respectively) (Bordet et al., 2000).
Although functional properties of multiple receptor heteromers remain to be determined, the interaction of A2A receptors with receptors other than those for dopamine, is also relevant (Fig. 1). Coimmunoprecipitation evidence shows that A2A and mGlu5 receptors form heteromeric complexes and combined stimulation of both of these receptor types, synergistically reduced the affinity of the D2 receptor agonist binding sites in striatal membranes (Fuxe et al., 2003). These observations were supported by the high degree of A2A and mGlu5 colocalization in primary cultures of striatal neurons and in striatal glutamatergic nerve terminals (Rodrigues et al., 2005). Coactivation of A2A and mGlu5 receptors causes a synergistic interaction at the level of c-fos expression and on ERK as well DARPP-32 phosphorylation, indicating a possible role of this heteromeric complex in striatal plasticity (Ferré et al., 2002; Nishi et al., 2003) (Fig. 1). Combined A2A and mGlu5 receptor activation may also produce synergistic cellular effects on striatal output neurons in vivo, as demonstrated by a greater than additive increase in GABA release from ventral striatopallidal neurons after local perfusion with both A2A and mGlu5 agonists (Diaz-Cabiale et al., 2002). Similarly, the discovery of functional A2A/mGlu5 receptor interactions and heteromeric A2A/mGlu5 complexes, led to recent findings of a synergistic antiparkinsonian potential of combining A2A and mGlu5 antagonists (Coccurello et al., 2004; Kachroo et al., 2005).
Another interesting interaction that may have important implications for the design of new drugs useful in the treatment of PD, is that of adenosine or dopamine receptors with cannabinoid CB1 receptors whose presence has been described in basal ganglia, most specifically in GABAergic striatal neurons (Egertova and Elphick, 2000) (Fig. 1). CB1 receptors colocalize with D2 and A2A receptors predominantly in the soma and dendrites of the striatopallidal GABA neurons and in corticostriatal glutamate terminals. CB1-D2 heteromers as well as CB1-A2A heteromeric complexes have been described in HEK-293 cell lines (Marcellino et al., 2008; Ferré et al., 2009). Interestingly post-synaptic CB1 receptor signaling was found to be dependent on A2A receptor activation. Accordingly, blockade of A2A receptors counteracted the motor depressant effects and extracellular field potentials, in corticostriatal neurons, produced by cannabinoid CB1 agonist (Carriba et al., 2007; Tebano et al., 2009). At the same time, CB1 receptors mediate psychomotor activation by A2A receptors antagonists (Lerner et al., 2010).
Antagonistic CB1/D2 interactions have been described at the behavioural level as well. The CB1 receptor agonist CP 55,940 at a dose that did not change basal locomotion is able to block quinpirole-induced increases in locomotor activity. In addition, not only the CB1 receptor antagonist rimonabant but also the specific A2A antagonist MSX-3 blocked the inhibitory effect of CB1 receptor agonists on D2-like receptor agonist-induced hyperlocomotion (Marcellino et al., 2008). These results find support in the existence of antagonistic CB1/D2 receptor-receptor interactions within CB1/D2 heteromers in which A2A receptors might also participate (Marcellino et al., 2008) (Fig. 1). Based on this biochemical evidence showing how CB1 receptors interact with both A2A and D2 receptors, it has been proposed that these receptors are putative targets for PD.
3) Role of adenosine receptors in neuroprotection
Arresting disease progression at the level of its underlying neuronal degeneration, remains a critical unmet goal of neurotherapeutics for PD. More than a decade of research has suggested that manipulating adenosine neurotransmission might offer a valuable strategy to achieve neuroprotection in PD.
The first studies investigating adenosine and neuroprotection were conducted in models of ischemic and excitotoxic brain injury (reviewed in Fredholm et al., 2005). Under these conditions, increased extracellular adenosine in response to brain injury has been shown to act as a neuroprotectant (Evans et al., 1987; Dux et al., 1990). However, a pro-neurotoxic role of adenosine has also been demonstrated, suggesting that blockade of adenosine receptors may confer neuroprotection across a range of neurodegenerative disorders (Jones et al., 1998; Chen et al., 1999; de Mendonca et al., 2000; Popoli et al., 2002; Melani et al., 2003; Pinna et al., 2010). This apparent paradox reflects the complexity of adenosine transmission, with several receptor subtypes selectively localized in brain areas and uniquely coupled to G proteins and signaling pathways, as described above in this chapter. Moreover, the important role played by adenosine in the immune response in the CNS should be considered as a component of degenerative and protective processes. Hence, the same adenosine receptor subtype expressed in different cell types such as neurons and glia, may mediate opposing effects in response to different neurotoxic insults (Fig. 2).
Fig. 2.
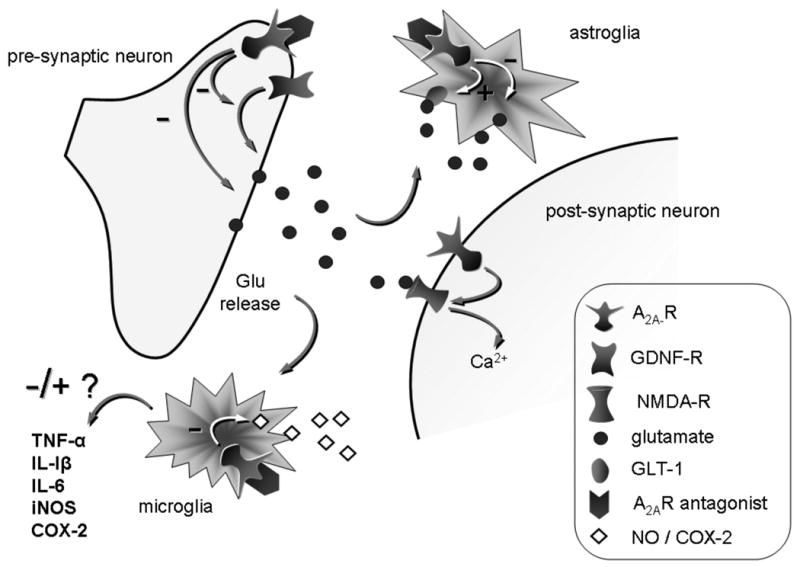
Schematic representation of possible cellular mechanisms affected by A2A receptor blockade in a neuroprotective model of PD. In presynaptic neurons, A2A antagonism inhibits glutamate efflux either directly or indirectly through an inhibition of GDNF receptors (see text for more details on this mechanism). A decrease in glutamate release results in reduced glial response and as a consequence release of toxic factors. In microglia, direct A2A receptor blockade inhibits NO and COX-2 production. In astroglia, direct A2A receptor blockade inhibits glutamate release directly or indirectly through an inhibition of GLT-1.
In PD, the initial suggestion that manipulating adenosine neurotransmission might be beneficial in terms of affecting disease onset or progression came from the epidemiological evidence that consumption of caffeine, a non-specific A1/A2A receptor antagonist, is associated with a reduced the risk of developing PD (Ascherio et al., 2001; Schwarzschild et al., 2003b). Convergent laboratory studies investigating the effect of caffeine and more specific adenosine receptor antagonists suggested that blockade of the A2A receptor subtype prevents nigrostriatal degeneration in several models of PD (Schwarzschild et al., 2006). Interestingly, epidemiological studies have identified a second purine as another robust inverse risk factor for PD. Blood levels of urate (the end product of adenosine metabolism in humans) is also strongly linked to a reduced risk of PD and, more recently, of its progression, as described below.
Neuroprotection and A1 adenosine receptors
Preclinical evidence for a role of A1 receptors in neuroprotection in PD is sparse; therefore this section will focus mostly on a general role of A1 receptors on glutamate release and toxic cytokine control.
A1 adenosine receptors are widely distributed throughout the CNS, being expressed both in neuronal and glial cells (Svenningsson et al., 1997; Ochiishi et al., 1999; Daré et al., 2007). Neuronal A1 adenosine receptors exist in presynaptic terminals as well as in postsynaptic membranes. However the most striking effect of their stimulation is the inhibition of neurotransmitter release, mediated by a reduction of presynaptic calcium influx (Brown et al., 1990; Ochiishi et al., 1999; Masino et al., 2002; Borycz et al., 2007). Although similar effects have been reported for different neurotransmitters, including glutamate, dopamine, acetylcholine and GABA, the inhibition of glutamate release holds the main interest for neurodegeneration. Based on this mechanism, A1 receptor agonists have been mainly pursued for their potential to protect under conditions characterized by a massive release of glutamate, as occurs in ischemic stroke. Few studies have investigated the effects of A1 receptor agonism in models of PD. Lau and Mouradian (1993) have shown that an adenosine A1 agonist was able to prevent the decrease in striatal dopamine levels induced by a single injection of the neurotoxin MPTP. Accordingly, the A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxathine (CPX) may have slightly exacerbated striatal MPTP toxicity (Chen et al., 2001). Although the mechanism has not been fully clarified, an inhibition of excessive release of glutamate, which may contribute to MPTP toxicity, might be involved in the protective effect displayed by A1 receptor agonists in the striatum. In line with this interpretation, blockade of NMDA glutamate receptors achieved a similar protective effect against an acute MPTP insult (Chan et al., 1993). Other clues to A1-mediated neuroprotection in PD models came from evidence that caffeine pre-treatment protected against methamphetamine-induced nigrostriatal toxicity via an upregulation of A1 receptors (Delle Donne and Sonsalla, 1994). Accordingly, Alfinito and colleagues have shown that administration of an A1 antagonist exacerbated dopamine neuron degeneration induced by the mitochondrial inhibitor malonate, although nigral but not striatal A1 receptors were selectively involved in the effect (Alfinito et al., 2003).
Glial A1 receptors (Haskó et al., 2008) may play an important role in dopamine neuron demise in PD. The involvement of this receptor in neuroinflammatory responses has been well documented, although direct evidence toward its involvement in neuroinflammation associated with nigrostriatal damage is lacking. In the presence of ischemia and brain injury, extracellular adenosine stimulates glial A1 receptors, resulting in inhibition of astrocyte proliferation and excessive reactive astrogliosis, as well as increased production of trophic factors, such as nerve growth factor (NGF), S100beta protein and transforming growth factor beta, which in turn may help to protect neurons from injury. Interestingly, the anti-inflammatory cytokine IL-6 enhances the expression of A1 receptors in astrocytes, suggesting that a self-modulating loop involving A1 receptors is triggered in presence of a neuronal damage (Schubert et al., 1997; Ciccarelli et al., 2001; van Calker and Biber 2005). Although the topic is still poorly investigated, based on this evidence it is tempting to speculate that a modulation of the astroglial response through the A1 receptor might be beneficial in PD nigrostriatal degeneration.
Neuroprotection and adenosine A2A receptors
In contrast to the A1 receptor, the A2A receptor has been consistently implicated as a mediator or modulator of dopaminergic neuron degeneration across a range of laboratory models of PD.
An early suggestion of the neuroprotective potential of A2A receptor blockade in PD came with the demonstration that caffeine can attenuate the loss of striatal dopamine induced by acute MPTP administration in mice (Chen et al., 2001). Caffeine has also been shown to be neuroprotective in other models of PD such as the unilaterally 6-OHDA-lesioned rat (Joghataie et al., 2004) and more recently in a dual pesticide, chronic exposure model (Kachroo et al., 2010), in which repeated systemic administration of paraquat plus maneb leads to degeneration of nigral dopaminergic neurons. In addition, two major demethylation metabolites of caffeine, namely theophylline and paraxanthine, both adenosine receptor antagonists themselves, have also been shown to protect against MPTP-induced neurotoxicity in mice (Xu et al., 2010). These consistent findings of neuroprotection by caffeine and its metabolites in multiple models of PD have strengthened the hypothesis that a true protective effect of caffeine is the basis for its inverse association with PD risk in epidemiological studies (see below).
A similar protective effect was observed upon administration of the selective A2A receptor antagonist KW-6002, but not with the A1 receptor antagonist CPX (Chen et al., 2001; Pierri et al., 2005; Schwarzschild et al., 2006). In line with these results, genetic deletion of A2A receptors prevented the loss of striatal dopamine induced by acute MPTP (Chen et al., 2001). Thereafter, a protective effect by A2A antagonists was reported in a different toxin model of PD, in which 6-OHDA is infused within the rat striatum (Ikeda et al., 2002). In these studies, neuroprotection was assessed as attenuation of the striatal dopamine depletion or of the loss of TH-positive cells in the substantia nigra pars compacta (Table 1). Recently, the A2A receptor antagonists SCH-58261 and ANR 94, were shown to prevent the death of nigral dopaminergic neurons induced by subchronic MPTP administration in mice (Carta et al., 2009; Pinna et al., 2010). Therefore, blockade of the A2A receptor seems to confer a functional protection of striatal dopamine transmission, as well as to prevent the loss of nigral dopaminergic neurons induced by neurotoxin exposure.
Table 1. Neuroprotective outcome of A2AR manipulation in different rodent PD models.
Summary of recent data highlighting the neuroprotective outcome observed in both the striatum and the substantia nigra by pharmacologically and genetically targeting the A2A receptor in different PD models.
| PD model | A2AR manipulation | Functional Protection in Str | SNc neurons survival | Attenuation of glial response | Refs |
|---|---|---|---|---|---|
| 6-OHDA in Str | A2A antag. | + | + | NA | Ikeda et al, 2002 |
|
| |||||
| MPTP acute | A2A antag. | + | + | + |
Chen et al, 2001 Ikeda et al, 2002 Pierri et al, 2005 Yu et al, 2008 |
| totA2AKO | + | NA | NA | ||
| fbnA2AKO | - | NA | NA | ||
|
| |||||
| MPTP subchronic | A2A antag. | + | + | + | Carta et al, 2009 |
| fbnA2AKO | + | + | + | ||
Despite considerable evidence suggesting the neuroprotective potential of A2A receptor blockade in PD, the underlying mechanism is still a matter of debate. In neurons, A2A adenosine receptors have been identified both pre- and post-synaptically, where they control neurotransmitter release and neuronal stimulation, respectively (Schiffman et al., 1991; Rosin et al., 1998; Svenningsson et al., 1999; Rebola et al., 2005) (Fig. 2). Moreover, cells involved in the neuroinflammatory response such as astroglia, microglia and bone marrow-derived cells all express the A2A receptor (Fiebich et al., 1996; Saura et al., 2005). Since many factors may contribute to neuronal demise in PD, including neuronal pathological processes and chronic neuroinflammation, several mechanisms have been investigated to explain the neuroprotective effect of A2A receptor blockade. Interestingly, recent reports have highlighted that multiple mechanisms, involving either neuronal or glial receptors, may be recruited in different PD models to achieve neuroprotection by A2A receptor blockade (Yu et al., 2008; Carta et al., 2009).
Neuronal A2A receptors and neuroprotection
An alternative strategy employed to complement pharmacological approaches in demonstrating the neuroprotective efficacy of A2A receptor blockade has been offered by targeted gene mutations producing mice lacking this receptor. In general, A2A receptor knockout mice display attenuated brain damage in models of ischemia or excitotoxin-induced brain injury as had been previously observed using traditional pharmacological antagonists (Phillis, 1995; Monopoli et al., 1998; Chen et al., 1999; Pedata et al., 2005). More recently, studies' using a conditional knockout (Cre/loxP) system to generate mice with a selective postnatal depletion of forebrain neuronal A2A receptors has contributed to the understanding of cellular mechanisms of A2A receptor-mediated neuroprotection in PD. In a subchronic MPTP model of PD, loss of these A2A receptors fully prevented neurotoxin-induced degeneration of nigral dopaminergic neurons, endorsing a primary role of neuronal A2A receptors in the neuroprotective effects of A2A antagonists in this model (Carta et al., 2009). However, neuronal A2A receptors seemed to play a minor role in striatal dopamine loss induced by a more acute MPTP intoxication, since Yu et al. (2008) reported that neuronal A2A receptors inactivation did not protect striatal terminals from a one-day, high-dose MPTP exposure (Yu et al., 2008). Interestingly, in this study mice globally lacking A2A receptors were partially protected against striatal MPTP-toxicity. This suggests that blockade solely of neuronal A2A receptors might not be sufficient to prevent neurotoxicity induced by acute MPTP in the striatum, or alternatively that cells other than forebrain neurons, likely glial cells, may play a role in A2A-mediated protection in the striatum (Yu et al., 2008) (Fig 2). Studies of toxin-induced striatal neuron death modeling the core neurodegenerative features of Huntington disease rather than PD have also given opposing results regarding the role of A2A receptors. In corticostriatal slices A2A receptor antagonism reduced the irreversible functional alterations caused by rotenone in striatal neurons, supporting a beneficial role of neuronal A2A receptor blockade against neurotoxic insults (Belcastro et al., 2009). By contrast, Huang et al reported that in a model of acute striatal neuron damage produced by local infusion of the mitochondrial toxin 3-nitropropionic acid (3-NP), selective deletion of A2A receptors on forebrain neurons was unable to protect against neurotoxicity (Huang et al., 2006). In this model global A2A receptor inactivation actually exacerbated 3-NP-induced neurotoxicity. Moreover, the cell-type-specific inactivation of A2A receptors located in bone marrow-derived cells also exacerbated striatal damage (Huang et al., 2006). All together these data, while supporting a neuroprotective outcome of neuronal A2A receptor blockade in PD, highlight the complexities of the roles played by A2A receptors, pointing to distinct actions of cell-type specific receptors in different neurodegenerative conditions.
In neurons, A2A receptors are enriched in the striatum, but have been also described in other basal ganglia nuclei, such as GP, and at lower levels in the substantia nigra (Schiffmann et al., 1991; Cunha et al., 1994; Johansson et al., 1997; Rosin et al., 1998; Brooks et al., 2008).
At the presynaptic level striatal A2A receptors are mostly located on glutamatergic terminals where they modulate the efflux of glutamate (Marchi et al., 2002; Popoli et al., 2002; Melani et al., 2003; Tebano et al., 2004) (Fig 2). It has been extensively demonstrated that antagonism of the A2A receptor protects against ischemic damage and toxin-induced excitotoxicity in the hippocampus or striatum (Jones et al., 1998; Popoli et al., 2002; Melani et al., 2006). Excessive excitotoxic glutamate efflux from the STN to substantia nigral neurons is a component of PD neuropathology, likely contributing to nigral neuron death. Therefore a reduction of glutamate efflux in the substantia nigra, and perhaps in the striatum, might provide a mechanism for protection against dopaminergic degeneration in PD models (Wallace et al., 2007).
It is important to point out that the effect of A2A receptor antagonists on striatal glutamate levels is dependent on the dose and experimental conditions (Tebano et al., 2004). In intact striatum, the A2A antagonist SCH-58261 decreased evoked glutamate efflux at low doses only, whereas it lost this effect at higher doses (Pintor et al., 2001). Moreover local infusion of an A2A antagonist directly into the 6-OHDA-lesioned striatum increased glutamate outflow (Corsi et al., 2003). Tebano and colleagues have shown that in corticostriatal slices A2A antagonists inhibit glutamate outflow, whereas in striatal neurons they amplify excitotoxic mechanisms due to direct NMDA receptor stimulation (Tebano et al., 2004). Therefore, the potential neuroprotective effect mediated by pre-synaptic A2A receptor blockade might emerge specifically at the low-dose range, whereas other mechanisms, likely mediated by post-synaptic A2A receptors, might mask this effect at higher doses. The higher binding affinity displayed by pre- versus post-synaptic A2A receptors supports this concept (Cunha et al., 1996).
An interesting mechanism of adenosine-glutamate interaction that has emerged recently involves cross-talk between adenosine and GDNF receptors, co-localized on striatal glutamatergic nerve endings (Gomes et al., 2009). In rat striatal synaptosomes, GDNF enhanced glutamate release by 13% from corticostriatal terminals, an effect potentiated by A2A agonist CGS21268 and prevented by the A2A receptor antagonist SCH58261 (Gomes et al., 2009). Therefore, it is suggested that A2A receptor blockade would impair GDNF-stimulated increase of corticostriatal glutamate release, providing a beneficial effect on neurodegeneration (Gomes et al., 2009).
Glial A2A receptors and neuroprotection
Neuroinflammation, characterized by reactive astrocytes and activated microglia, is a hallmark of PD and may play a pathogenic role in dopamine neuron degeneration. Classical studies have described activated microglia in the substantia nigra of PD patients, whereas most recent studies have reported a more widespread distribution of activated microglia in the brain, both in early and late stages of the disease, involving pons, basal ganglia, striatum, and frontal and temporal cortex (Gerhard et al., 2006; Mc Geer and McGeer, 2008). Persistent microglial activation leads to elevated levels of glial-derived cytokines and chronic neuroinflammation, which exert neurotoxic effects on highly vulnerable dopaminergic neurons. On the basis of this evidence it appears of great importance that, in addition to neurons, adenosine A2A receptors are located on both microglia (Fiebich et al., 1996; Saura et al., 2005) and astrocytes (Lee et al., 2003) (Fig. 2). Recent reports in vivo have shown that A2A receptor antagonists were able to prevent the astroglial and microglial activation induced by acute or subchronic administration of MPTP in mice (Pierri et al., 2005; Carta et al., 2009). These results are in line with that obtained in an in vivo model of ischemia, where the specific A2A receptor antagonist SCH58261 inhibited phosphorylation of P38-MAPK (mitogen activated protein kinase), supporting an inhibitory effect of A2A receptor antagonism on the response in microglial cells (Melani et al., 2006). Therefore, an effect on glial cells has been considered as a potential mechanism of neuroprotection by A2A receptor antagonists. However, the effect on glial response observed in different models of neurodegeneration might either result from a direct effect through stimulation of A2A receptors on these cells, or might be secondary to an effect mediated by A2A receptors on neurons.
The molecular effects of glial A2A receptor stimulation have been investigated by a number of in vitro studies, leading to somewhat contradictory results and suggesting that receptors located on microglia and astroglial cells might play opposing roles in neurodegeneration. Supporting a beneficial effect of glial A2A receptor blockade, A2A receptor agonist CGS21680 potentiated lipopolysaccharide-induced NO release and NO synthase-II expression by microglial cells in a concentration-dependent manner, whereas an A2A antagonist suppressed this effect (Saura et al, 2005). Accordingly, A2A receptors mediated the induction of cyclooxygenase-2 and nitric oxide synthase in microglia (Fiebich et al., 1996, 1998).
However, A2A receptor agonists inhibited cytokines production by activated microglia (van der Putten et al, 2009). In cultured astroglia, both adenosine kinase inhibitors and the A2A receptor agonist CGS21280 counteracted LPS-induced production of NO (Brodie et al., 1998; Lee et al., 2005). In line with a positive role of A2A receptor blockade, the A2A receptor agonist CGS21680 increased the astrocytic release of glutamate via an A2A receptor/PKA signaling pathway and via inhibition of glutamate transporter GLT-1, resulting in increased synaptic concentrations of this neurotransmitter, and likely in deleterious effects on neurons survival. Most importantly, this effect was inhibited by the A2A receptor antagonist DMPX (Nishizaki, 2004). Furthermore, adenosine and the A2A receptor agonist CPCA increased astrocyte proliferation after brain injury in rat cortex, whereas adenosine antagonists where shown to counteract astrocytic proliferation in both in vitro and in vivo models of brain injury (Hindley et al., 1994; Brambilla et al., 2003; Pugliese et al., 2009). Therefore, whereas substantial evidence suggests that manipulating glial A2A receptors might be beneficial in neurodegenerative conditions, their selective stimulation or blockade might be beneficial depending on neurodegenerative conditions and time of intervention (Fig. 2). Moreover, reports on glial A2A receptors manipulation in in vivo PD models are currently lacking.
Lately, a new alternative mechanism of neuroprotection has been suggested, involving A2A receptors located on oligodendrocytes. In a stroke model A2A receptor antagonist SCH58261 reduced striatal Olig 2 transcription factor induced by ischemia (Melani et al., 2009).
4) Epidemiology and clinical trials of adenosine antagonists
Caffeine Epidemiology
Case control studies: coffee
Considerable epidemiological and laboratory data have suggested that A2A receptor blockade by caffeine, a nonselective adenosine receptor antagonist, may protect against the underlying neurodegeneration of PD. Drinking caffeinated beverages (coffee and to a lesser extent tea) has emerged as the dietary factor most consistently linked to an altered risk of PD, with greater consumption associated with a reduced risk (Hellenbrand et al., 1996; Fall et al., 1999; Benedetti et al., 2000; Ross et al., 2000; Ascherio et al., 2001; Checkoway et al., 2002; Ragonese et al., 2003; Tan et al., 2003; Hu et al., 2007). In the early 90's case-control studies suggested a reduced risk of developing PD associated with drinking coffee but the reduction either was not statistically significant (Jimenez-Jimenez et al., 1992; Morano et al., 1994), or was significant but partially attributable to the confounding association of smoking in coffee drinkers (Grandinetti et al., 1994) and thus difficult to interpret. Following on from this, larger, case-control studies, using better-matched cohorts demonstrated that even after adjusting for tobacco smoking and other potential confounding factors, the significant inverse relationship between prior coffee drinking exposure and PD remained (Hellenbrand et al., 1996; Fall et al., 1999; Benedetti et al., 2000). These latter studies also specifically investigated dose-response relationships, with increasing coffee consumption (measured in cups per day) associated with decreasing likelihood of having developed PD. In these retrospective analyses PD patients were 4 to 8 times less likely than control subjects to have reported being heavy coffee drinkers in the past. While case control studies have some advantages, weaknesses of these designs for investigating dietary etiology of chronic diseases are well known (Willett, 1998) and include for example difficulty selecting appropriate control subjects and recall bias. The introduction of large cohort prospective investigations (see below) has overcome many of these limitations through follow-up evaluations to determine disease incidence in subjects from a single population in which the exposure in question (i.e., coffee or caffeine consumption) had been reported years earlier.
Case-control studies: tea
Relatively few studies have considered the effect of tea drinking and its association with PD risk. This could be attributed to its low consumption in North America and Europe (Ascherio et al., 2001; Chan et al., 1998). These studies usually (Ho et al., 1989; Hellenbrand et al., 1996; Ayuso-Peralta et al., 1997; Chan et al., 1998; Checkoway et al., 200; Tan et al., 2003, 2007), although not always (Preux et al., 2000), indicated a reduced risk of developing PD amongst frequent tea drinkers. Interestingly, epidemiological studies from China have observed that the prevalence of PD is much lower than in the Caucasian population (Li et al., 1985; Zhang et al., 1993). Barranco et al., 2009 reviewed observational studies that evaluated tea consumption and the risk of PD (11 case-control and 1 cohort) between 1981 and 2003. The studies represented documented cases from North America, Europe and Asia. Amongst the case-control studies, the pooled OR was 0.8 (95% CI 0.71 to 0.90) suggesting that tea consumption is inversely associated with the risk of PD. It is unclear whether the active ingredient(s) mediating this observed protective effect is caffeine or some biologically active substance(s) present in tea but not coffee.
Cohort studies
Caffeinated beverages
More convincing epidemiological evidence that caffeine as well as coffee consumption are linked to a reduced risk of developing PD has been obtained from the study of prospectively followed, large populations (Ross et al., 2000; Ascherio et al., 2001). Three decades after ∼8000 Japanese-American men were enrolled in the Honolulu Heart Program (and provided details of their dietary caffeine consumption), over 100 had gone on to develop PD. Higher initial coffee intake was dose-dependently associated with a reduced incidence of PD, with a 5-fold lower risk amongst those who drank over 24 oz per day (Ross et al., 2000). Confirmation of these findings was provided by two prospective studies of larger, multiethnic populations, namely the Health Professionals Follow-up Study (HPFS) of 50,000 men followed for a period of 10 years, and the Nurses Health Study (NHS) of 90,000 women followed over 16 years (Ascherio et al., 2001). Amongst the men, increased coffee, tea and non-coffee caffeine consumption, as well as total caffeine consumption were all significantly, dose-dependently and negatively correlated with the incidence of subsequent PD. These associations were independent of smoking and other potential confounding factors. By contrast, the rates of consuming decaffeinated coffee were unrelated to the risk of PD, implicating caffeine as the component in coffee that is inversely associated with PD risk. Similar findings were observed in 2 separate cohort studies of Finnish men and women free from PD at baseline (Hu et al., 2007; Saaksjarvi et al., 2008). Heavier coffee consumption was associated with a reduced risk of PD even after adjustment for confounding factors. The Finnish population is of particular interest since it exhibits one of the world's highest rates of coffee consumption (Fredholm et al., 1999).
Gender differences (estrogen interactions)
Interestingly, a stark gender difference in how caffeine relates to PD risk emerged from comparison of female and male cohorts within a large protective study of health care workers (Ascherio et al., 2001). Initially, analysis of women in the NHS study revealed no clear relationship between PD and caffeine or coffee intake. This gender difference was consistent with the observations of PD incidence rates in Olmstead County, MN, which were strongly inversely related to prior coffee drinking in men (with a ∼17-fold reduction in risk amongst drinkers vs. non drinkers; p < 0.01) but did not vary with coffee exposure in women (with a relative risk of 1.0; Benedetti et al., 2000). Ascherio and colleagues gained insight upon stratification of the women by estrogen exposure history. In two separate prospective studies (Ascherio et al., 2003, 2004) they showed that amongst women who did not use postmenopausal estrogens, caffeine was in fact associated with a reduction in the risk of subsequent PD (just as in men). Conversely, for women who had used estrogen replacement caffeine use did not carry a lower risk of PD, suggesting a hormonal basis for the gender difference in caffeine's association with PD.
Interestingly, in contrast to the result from studies by Ascherio and colleagues, the two prospective cohort studies of Finnish populations (Hu et al., 2007; Saaksjarvi et al., 2008) reported no gender differences in the (inverse) relationship between coffee consumption and PD risk. However as Saaksjarvi and colleagues note in their study, the effect of postmenopausal hormone use could not be examined due to small number of users, which was reported to be ∼5% of women and markedly less than in the US cohorts. For example in the NHS, a third of the women were currently taking postmenopausal estrogens and 54% reported ever taking them. Thus, the variability in an overall association between caffeine and PD risk among women between cohorts may be explained by wide variation in their use of postmenopausal estrogen, which appears to complicate the relationship between caffeine and PD.
Recent laboratory experiments have explored the biology that may underlie gender differences in the caffeine-PD link observed in populations with higher rates of estrogen use. Xu et al., 2006 found that the ability of caffeine to attenuate MPTP toxicity in a mouse model of PD was greater in male versus female mice, and in ovariectomized versus sham-operated female mice. They also demonstrated that chronic estrogen replacement undermined caffeine's protective effect both in male and in ovariectomized female mice, providing direct evidence of a hormonal influence on caffeine's neuroprotective properties in lab animals. The study also implicated a biological basis for the gender difference in the association between caffeine consumption and PD risk. In general, these and other laboratory studies (as reviewed above) support – but do not prove -- the hypothesis that the consistent epidemiological association between caffeine and a reduced risk of PD is causal.
PD progression
Convergent epidemiological and laboratory data support the possibility that dietary caffeine reduces the risk of developing PD. Less well known is the relationship between caffeine intake and the rate of progression of the disease. A preliminary investigation (Schwarzschild et al, 2003a) reported a secondary analysis of the ‘Comparison of the agonist pramipexole versus levodopa on the motor complications of PD’ (CALM-PD) trial database, which included data over 5 years from 268 participating PD patients (87% of the original CALM-PD cohort). No association was found between caffeine intake (from coffee, tea and soda sources) and rate of PD progression even after adjustment for treatment group, gender, tobacco and alcohol use. Outcome measures assessed progression by change in the Unified Parkinson Disease Rating Scale (UPDRS), or by alteration in the striatal uptake of iodine-123-labeled 2-β-carboxymethoxy-3-β-(4-iodophenyl) tropane ([123I] β-CIT) in the subset of patients that underwent neuroimaging. The study also allowed for assessment of caffeine consumption and dyskinesia development. A trend albeit non-significant toward lower risk of dyskinesias with increasing caffeine intake was observed. The lack of an association between caffeine consumption and rate of subsequent progression was also observed in a NET-PD trial cohort (Simon et al., 2008) monitoring changes in the UPDRS score and likelihood of disease progression to the point of requiring symptomatic therapy as measurable outcomes. These studies do not lend support to a protective benefit of caffeine by PD patients. However, they leave open the possibility that disease progression among individuals who develop PD despite ingesting caffeine at higher levels may be less influenced by any true protective effect it may have.
5) Clinical trials of A2A antagonists for Parkinson's disease
Trials for symptomatic anti-parkinsonian indications
Non-selective adenosine antagonists: caffeine and theophylline
Although caffeine is a non-specific adenosine receptor antagonist that blocks with similar potency at the A1, A2A and A2B subtypes, (Fredholm et al., 1999) it appears to have its main psychomotor stimulant actions primarily through blockade of CNS A2A receptors (Xu et al., 2004; Yu et al., 2008). Thus the first trials of A2A antagonism in PD may have been a pair of double-blind, placebo-controlled studies of caffeine effects on parkinsonian symptoms of patients taking levodopa or a dopamine agonist in the 1970s (Shoulson et al., 1975; Kartzinel et al., 1976). Both studies found no benefit of caffeine. However, their small size (<10 subjects), clinical heterogeneity (some subjects did not have PD) and extremely high dosing 1-1.5 grams of caffeine daily precludes meaningful conclusions about the symptomatic utility of caffeine for PD from these studies. A more rigorously designed randomized, placebo-controlled trial of caffeine at more pharmacologically informative doses (100 or 200 mg bid) is currently underway and projects enrollment of some 50 subjects with idiopathic PD and excessive daytime somnolence (McGill University Health Center; clinical trials.gov identifier # NCT00459420). Although its primary outcome focuses on daytime sleepiness, a secondary analysis of parkinsonian motor symptoms is planned using the standard Unified Parkinson's Disease Rating Scale.
Theophylline, a demethylation metabolite of caffeine as well as an anti-asthmatic agent in common use, is also a non-specific adenosine antagonist. Although small open-label trials of theophylline in PD seemed to suggest antiparkinsonian benefit without exacerbation of dykinesias (Mally & Stone, 1994; Kostic et al., 1999), a subsequent double-blind, placebo-controlled trial of theophylline in PD did not clearly demonstrate relief from symptoms (Kulisevsky et al., 2002). However, the power and design of all these trials like the earlier caffeine studies in PD were not sufficient to rule out clinically useful symptomatic effects, as have been suggested by long-standing preclinical studies of motoric enhancement in PD models (Stromberg & Waldeck, 1973).
Selective adenosine A2A antagonists
A clear indication of the promise of adenosine A2A receptor antagonism for PD is the depth of industry investment in programs to develop a variety of xanthine and non-xanthine structure based adenosine receptor antagonists as antiparkinsonian agents. Amongst at least ten publicly announced programs are some five that have entered human studies, with four being now actively pursued through phase II and III clinical trials: istradefylline (aka KW-6002) from Kyowa-Kirin (Kyowa Hakko Kirin Co., 01.15.2009 announcement). Preladenant (aka SCH 420814) from Schering Plough [now Merck] (Schering Plough Co., 11.24.2008 announcement). BIIB014 (aka V2006) from BiogenIdec [licensed from Vernalis] (Papapetropoulos et al., 2010) and Syn-115 from Synosia [licensed from Roche] (Black et al., 2010 a and b).
The early frontrunner amongst these clinical development programs was istradefylline, which had progressed through encouraging phase II trial results (Bara-Jimenez et al., 2003; Hauser et al., 2003; Kase et al., 2003; Hauser et al., 2008; Lewitt et al., 2008; Stacy et al., 2008; Fernandez et al., 2010) to as yet unpublished, reportedly mixed results of phase III studies (Kyowa Hakko Kirin Co., 06.03.2007 announcement) before submission of a New Drug Application (NDA) to US Food and Drug Administration (FDA). However, the FDA in response issued a ‘Non-Approvable letter’ in 2008 based on concerns of the adequacy of overall efficacy (Kyowa Hakko Kirin Co., 02.08.2008 announcement). Since then clinical development of istradefylline has continued, but the company has pulled back from an international program to focus on trials and a potential indication in Japan.
Based on available data for the published phase II trials, istradefylline employed (at once-daily doses from 20-80 mg) as adjunctive therapy in relatively advanced subjects, produced a modest but significant reduction in “off” time (i.e., in motor dysfunction). Of note, these ‘positive’ outcome measures were based on reports of the patients themselves, whereas no significant treatment-induced improvement was observed based on the clinician-scored UPDRS. Although preclinical evidence supporting the original study designs had suggested adjunctive A2A antagonism might assuage PD symptoms without exacerbating dyskinesias (reviewed in Xu et al., 2004), istradefylline treatment in this relatively advanced PD population actually increased dyskinesias in most studies. However when dyskinesias were stratified into “troublesome” and “non-troublesome”, only the latter were significantly increased on the drug.
Recently, istradefylline was also tested as monotherapy for its potential symptomatic benefit in early PD (Fernandez et al., 2010). Despite a trend toward improvement on istradefylline, again the difference between placebo and A2A antagonist-treated groups did not reach significance for the change in UPDRS score, the primary outcome in this study. Retrospective assessment of why this early A2A antagonist development program fell short in its initial efforts to gain an indication for PD treatment have focused on trial design elements, including dose selection (for both istradefylline and concomitant levodopa) and disease stage of the targeted PD subpopulation (i.e., possibly too advanced in adjunctive trials, though possibly too early in the monotherapy trial).
In addition, none of the trials reports addressed the potential confound of concomitant A2A antagonism by caffeine use, which apparently was neither excluded nor monitored. At doses relevant to typical human consumption, caffeine and more specific A2A antagonists (including istradefylline) bind to striatal A2A receptors in vivo to a similar extent (El Yacoubi et al., 2001; Moresco et al., 2005). Given also the well-known psychomotor stimulant properties of caffeine and its continued consumption in PD with a mean intake of approximately 200 mg/day among early in the disease (Schwarzschild et al., 2003a; Simon et al., 2008), its use is important to consider in A2A antagonist trials. As caffeine use tends to decrease over the course of the disease, controlling for its use in the analysis of early PD/monotherapy trials may be particularly informative. If specific adenosine A2A receptor antagonists were found to be more effective amongst those consuming less caffeine (i.e., less of a general adenosine antagonist), it would support the distinct possibility of a shared antiparkinsonian effect through a common mechanism. It would then remain to be determined whether specific A2A receptor antagonism offers an advantage of greater efficacy or tolerability (given possibly adverse effects of blocking other adenosine receptors) to offset the lower cost and greater availability of caffeine.
Trials for disease-modification in PD
Convergent epidemiological and laboratory studies have suggested that adenosine A2A antagonism may confer disease modifying benefits beyond the antiparkinsonian motor effects now being actively pursued in clinical trials as above (Schwarzschild et al., 2006; see Figure 3). Although caffeine itself was listed amongst the most attractive candidate neuroprotectants under consideration for clinical investigation (Ravina et al., 2003), if a specific A2A antagonist were to receive an indication for symptomatic relief it may be more likely than caffeine to first undergo testing in a ‘neuroprotection’ trial given the high costs of conducting the necessarily large and long-term trials required.
Fig 3.
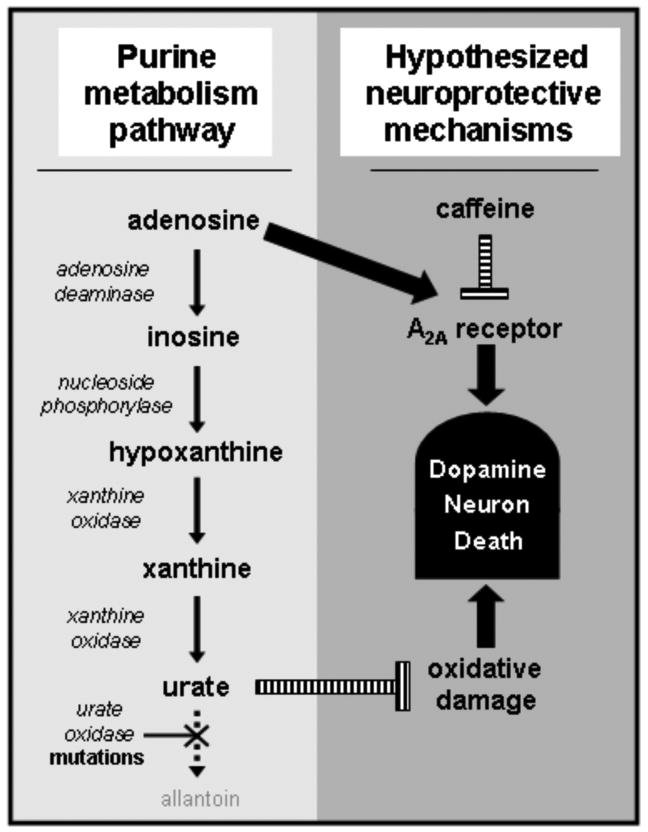
Therapeutic targets along the purine metabolic pathway. Adenosine A2A antagonists (including caffeine) and urate have emerged as realistic candidate neuroprotectants. In humans the enzymatic metabolism of purines such as adenosine ends with urate due to multiple mutations within the urate oxidase gene during primate evolution (see text). The schematic suggests a possible homeostatic mechanism linking an adenosinergic neurodegenerative influence with an offsetting neuroprotective influence of urate.
The prospects of favorable disease modification by A2A receptor blockade may extend beyond its neuroprotective potential as A2A receptors have also been implicated in the maladaptive neuroplasticity that underlies the development of levodopa-induced dyskinesia (LID) in PD (Morelli et al., 2009). Accordingly, early treatment with an A2A antagonist versus placebo as an adjunct to newly initiated levodopa may also be considered as a novel trial design to assess for both an early synergistic benefit (possibly with greater sensitivity than a monotherapy design), as well as for a prophylactic effect on LID during a second long-term phase of observation with subjects maintained in their blinded treatment arms.
6) Urate as a novel target for neuroprotection
Biology
Evolutionary significance
During primate evolution a series of mutations occurred in the urate oxidase gene (UOx) likely accounting for the relatively high levels of urate (the physiologically dissociated form of uric acid) in apes and humans compared to other mammals (Oda et al., 2002; see Figure 3). Urate is not only the end product of the metabolism of purines like adenosine. It also possesses antioxidant properties comparable to those of ascorbate (Ames et al., 1981) and accounts for most of the antioxidant capacity in human plasma (Yeum et al., 2004), supporting the hypothesis that our ancestors gained antioxidant benefits from UOx mutations and a resulting elevation of urate concentrations (Proctor, 1970). While higher levels of urate may have conferred an evolutionary advantage through bolstered defenses against oxidative damage (Ames et al., 1981) today they are also the core molecular culprit in gout and uric acid kidney stones.
Neuroprotective effects in cellular models of PD
In addition to its antioxidant actions, urate has also been shown to possess other potentially protective properties in vitro. It has been shown to scavenge peroxynitrite as well as oxygen free radicals in vitro (Whiteman et al., 2002; Franzoni et al., 2006) and displays potent iron-chelating activity independent of its direct antioxidant action (Davies et al., 1986). Direct evidence for a neuroprotective effect of urate has come initially from cellular and animal models of multiple sclerosis, (Hooper et al., 1997, 1998; Scott et al., 2002) stroke (Yu et al., 1998; Romanos et al., 2007) and spinal cord injury (Scott et al., 2002; Du et al., 2007).
Urate also confers protection in cellular models of PD. In PC12 cells, urate blocked apoptosis and oxidant production induced by dopamine (Jones et al., 2000) or the pesticide rotenone in combination with homocysteine (Duan et al., 2002) and reduced cell death induced by MPP+ or Fe2+ (Haberman et al., 2007). Urate also attenuated toxin-induced loss of primary neurons in culture. A recent study of dopaminergic neurons in primary midbrain culture of rat ventral mesencephalon found that their physiological function and survival were significantly enhanced by urate, (Guerriero et al., 2009) at concentrations (≥ 30-50 µM) corresponding to those in human CSF associated with a reduced of clinical decline in PD (Ascherio et al., 2009).
Epidemiology
When first considered in case-control studies, lower urate levels were found in serum (Larumbe et al., 2001; Annanmaki et al., 2007; Bogdanov et al., 2008; Johansen et al., 2009; Andreadou et al., 2009), possibly in cerebrospinal fluid (Tohgi et al, 1993), and in post-mortem nigrostriatal tissue samples (Church & Ward, 1994) of PD patients compared to those of controls. These studies suggested that low CNS as well as peripheral levels of urate are associated with PD.
A series of epidemiological investigations of prospectively followed cohorts has more incisively linked higher blood urate with a reduced risk of developing PD (Davis et al., 1996; De Lau et al., 2005; Weisskopf et al., 2007; Chen et al., 2009). For example, in the largest of these cohorts, 18,000 men were followed for more than 8 years in the HPFS. Weisskopf and colleagues found that those in the top quartile of plasma urate concentration had a 2- to 3-fold lower risk of PD than subjects in the bottom quartile (p<0.02 for trend across all quartiles). Amongst the subset of cases for whom blood was collected at least four years before the diagnosis of PD, an even greater reduction of PD risk was observed – with those in the highest urate quartile having a 5-fold lower risk of PD compared to the lowest quartile (p<0.01 for trend). This further analysis suggests that the low uricemia among individuals with PD precedes the onset of neurological symptoms and is thus unlikely to be a consequence of changes in diet, behavior, or medical treatment early in the course of the disease. This inverse association was independent of age, smoking, caffeine consumption and other aspects of lifestyle that have been related to PD or uricemia.
Similarly, urate-elevating diet was also associated with a lower risk of PD (Gao et al., 2008). In the HPFS cohort, the authors found that a higher dietary uricemic index (reflecting dietary patterns linked to higher plasma urate) predicted a reduced risk of developing PD (p<0.001 for trend). The association between the index and PD risk remained strong and significant in models further adjusted for age, smoking and caffeine intake. Their findings suggest that dietary interventions that raise blood urate concentrations might reduce the risk of PD.
In a related set of epidemiological studies of large prospectively followed cohorts, a diagnosis of gout (a form of arthritis due to urate crystallization in joints and associated with hyperuricemia) was linked to a lower risk of later being diagnosed with PD (Alonso et al., 2007; De Vera et al., 2008). Together these epidemiological data establish urate exposure – assessed by laboratory, dietary or pathological indicators – as a robust inverse risk factor for PD.
Clinical studies of urate and PD progression
The emergence of robust epidemiological data linking higher urate levels amongst healthy populations to a reduced risk of developing PD, prompted a corollary hypothesis: Amongst people already diagnosed with PD, do higher urate levels predict a slower rate of clinical decline? To test the hypothesis, incidentally measured urate levels in two large completed ‘neuroprotection’ trials were related to rates of clinical decline over years. Although neither the PRECEPT (Parkinson Study Group, 2007) nor the DATATOP (Parkinson Study Group, 1993) trial had demonstrated efficacy of candidate neuroprotectants, each had collected data for routine safety lab tests -- including serum urate -- at enrollment of some 800 recently diagnosed ‘de novo’ PD patients, who were then followed closely for two years. For both trials the primary outcome was time to disability warranting the initiation of levodopa or dopaminergic agonist therapy, with secondary outcomes including rate of UPDRS change and, in the case of the PRECEPT trial, rate of loss of dopamine transporter (DAT) ligand uptake in the striatum.
In the PRECEPT cohort, subjects with higher (but still normal) levels of serum urate at baseline were significantly less likely to develop disability warranting dopaminergic therapy and also retained significantly more striatal DAT binding capacity during the study (Schwarzschild et al., 2008). For example, subjects in the top quintile of serum urate (∼7-8 mg/dL with normal value reference ranges typically 3-8 mg/dL) reached the end point at only half the rate of subjects in the lowest quintile (hazard ratio, 0.51; 95% confidence interval, 0.37-0.72; p for trend <0.001). In the DATATOP cohort higher baseline urate concentrations in CSF as well as in serum were similarly associated with a slower rate of reaching the primary disability endpoint (Ascherio et al., 2009). In both cohorts higher urate levels were also predictive of a favorable rate of clinical decline measured by the change in UPDRS score. Although several descriptive clinical features of PD have been identified as probable predictors of the rate of clinical decline in PD (Post et al., 2007), urate may be the first molecular factor clearly linked to clinical progression of idiopathic PD.
In addition to its emerging utility as a prognostic biomarker of PD in research studies, the demonstrated antioxidant and neuroprotective properties of urate raise the possibility of its potential for direct therapeutic benefit. Convergence of these biological, epidemiological and clinical findings has prompted rapid translation to clinical application (Parkinson Study Group; clinical trials.gov identifier # NCT00833690). A multi-center, randomized, placebo-controlled trial of inosine (a precursor of urate as well as the deamination product of adenosine in purine metabolism; see Figure 3) in early PD is currently underway to assess its safety and ability to elevate serum and CSF urate levels, and its potential for further development as a novel strategy to impede progression of the disease.
Acknowledgments
Supported by NIH grants K24NS060991 and R01NS054978, DoD W81XWH-04-1-0881 and the RJG Foundation
List of abbreviations
- BRET
Bioluminescence Resonance Energy Transfer
- cAMP
cyclic adenosine monophosphate
- CNS
central nervous system
- CPX
8-cyclopentyl-1,3-dipropylxathine
- CREB
cAMP response element-binding
- DARPP-32
dopamine and cAMP regulated phosphoprotein-32
- DAT
dopamine transporter
- ERK
Extracellular signal-regulated kinases
- FDA
US Food and Drug Administration
- FRET
fluorescence resonance energy transfer
- GABA
gamma-aminobutyric acid
- GDNF
glial derived neurotrophic factor
- GLT-1
glutamate transporter-1
- HPFS
Health Professionals Follow-up Study
- L-DOPA
L-3,4-dihydroxyphenylalanine
- LID
L-DOPA induced dyskinesia
- LPS
lipopolysaccharide
- MAPK
mitogene-activated protein kinase
- MPTP
1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine
- NDA
New Drug Application
- NGF
nerve growth factor
- NHS
Nurses Health Study
- NO
nitric oxide
- 3-NP
3-nitropropionic acid
- 6-OHDA
6-hydroxydopamine
- PD
Parkinson's disease
- STN
subthalamic nucleus
- TH
tyrosine hydroxylase
- UPDRS
Unified Parkinson's Disease Rating Scale
Contributor Information
Micaela Morelli, Email: morelli@unica.it, Department of Toxicology, University of Cagliari, via Ospedale 72, 09124 Cagliari, Italy. Tel 0039-3804335298.
Anna R Carta, Email: acarta@unica.it, Department of Toxicology, University of Cagliari, via Ospedale 72, 09124 Cagliari, Italy. Tel 0039-3804335298.
Anil Kachroo, Email: AKACHROO@PARTNERS.ORG, Mass General Institute for Neurodegenerative Disease, Massachusetts General Hospital, Boston, MA 02129, USA. Tel 617-724-9611.
Michael A. Schwarzschild, Email: MichaelS@helix.mgh.harvard.edu, Mass General Institute for Neurodegenerative Disease, Massachusetts General Hospital, Boston, MA 02129, USA. Tel 617-724-9611.
References
- Agnati LF, Ferré S, Lluis C, Franco R, Fuxe K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev. 2003;55(no. 3):509–560. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK. Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci. 2003;23(no. 4):10982–10987. doi: 10.1523/JNEUROSCI.23-34-10982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Rodríguez LA, Logroscino G, Hernán MA. Gout and risk of Parkinson disease: a prospective study. Neurology. 2007;69:1696–1700. doi: 10.1212/01.wnl.0000279518.10072.df. [DOI] [PubMed] [Google Scholar]
- Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78:6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadou E, Nikolaou C, Gournaras F, Rentzos M, Boufidou F, Tsoutsou A, Zournas C, Zissimopoulos V, Vassilopoulos D. Serum uric acid levels in patients with Parkinson's disease: their relationship to treatment and disease duration. Clin Neurol Neurosurg. 2009;111(no. 9):724–728. doi: 10.1016/j.clineuro.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Annanmaki T, Muuronen A, Murros K. Low plasma uric acid level in PD. Mov Disord. 2007;22:1133–1137. doi: 10.1002/mds.21502. [DOI] [PubMed] [Google Scholar]
- Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci. 2000;20(no. 15):5848–5852. doi: 10.1523/JNEUROSCI.20-15-05848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascherio A, Zhang SM, Hernán MA, Kawachi I, Colditz GA, Speizer FE, Willett WC. Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol. 2001;50(no. 1):56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Schwarschild MA, Zhang SM, Colditz GA, Speizer FE. Caffeine, postmenopausal estrogen, and risk of Parkinson's disease. Neurology. 2003;60:790–795. doi: 10.1212/01.wnl.0000046523.05125.87. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, O'Reilly EJ, McCullough ML, Calle EE, Rodriguez C, Thun MJ. Coffee consumption, gender, and Parkinson's disease mortality in the cancer prevention study II cohort : the modifying effects of estrogen. Am J Epidemiol. 2004;160(no. 1):977–984. doi: 10.1093/aje/kwh312. [DOI] [PubMed] [Google Scholar]
- Ascherio A, LeWitt PA, Xu K, Eberly S, Watts A, Matson WR, Marras C, Kieburtz K, Rudolph A, Bogdanov MB, Schwid SR, Tennis M, Tanner CM, Beal MF, Lang AE, Oakes D, Fahn S, Shoulson I, Schwarzschild MA. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch Neurol. 2009;66:1460–1468. doi: 10.1001/archneurol.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso-Peralta L, Jiménez-Jiménez FJ, Cabrera-Valdivia F, Molina JA, Javier MR, Almazán, Pedro-Cuesta JD, Tabernero C, Giménez-Roldan S. Premorbid Dietetic Habits and Risk for Parkinson's Disease. Parkinsonism & Related Disorders. 1997;3:55–61. doi: 10.1016/s1353-8020(96)00047-8. [DOI] [PubMed] [Google Scholar]
- Bara-Jimenez W, Sherzai A, Dimitrova T, Favit A, Bibbiani F, Gillespie M, Morris MJ, Mouradian MM, Chase TN. Adenosine A(2A) receptor antagonist treatment of Parkinson's disease. Neurology. 2003;61(no. 3):293–296. doi: 10.1212/01.wnl.0000073136.00548.d4. [DOI] [PubMed] [Google Scholar]
- Barranco Quintana JL, Allam MF, Del Castillo AS, Navajas RF. Parkinson's disease and Tea: A quantitative review. J Am Coll Nutr. 2009;28(no. 1):1–6. doi: 10.1080/07315724.2009.10719754. [DOI] [PubMed] [Google Scholar]
- Belcastro V, Tozzi A, Cantucci M, Costa C, Di Filippo M, Autori A, Picconi B, Siliquini S, Luchetti E, Borsini F, Calabresi P. A2A adenosine receptor antagonists protect the striatum against rotenone-induced neurotoxicity. Exp Neurol. 2009;217(no. 1):231–234. doi: 10.1016/j.expneurol.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Benedetti MD, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA. Smoking, alcohol, and coffee consumption preceding Parkinson's disease: a case-control study. Neurology. 2000;55:1350–1358. doi: 10.1212/wnl.55.9.1350. [DOI] [PubMed] [Google Scholar]
- Black KJ, Campbell MC, Dickerson W, Creech ML, Koller JM, St Chung C, Bandak SI. A Randomized Double-Blind, Placebo-Controlled Cross-Over Trial of the Adenosine A2A antagonist SYN115 in Parkinson's disease. Abstract [PD4.001]: 62nd AAN annual meeting; Toronto. 2010a. [Google Scholar]
- Black KJ, Koller MJ, Campbell MC, Bandak SI. Perfusion MRI in Parkinson Disease Reveals Pharmacodynamics of the Adenosine A2A Antagonist SYN115. Abstract [S33.005]: 62nd AAN annual meeting; Toronto. 2010b. [Google Scholar]
- Bogdanov M, Matson WR, Wang L, Matson T, Saunders-Pullman R, Bressman SS, Flint Beal M. Metabolomic profiling to develop blood biomarkers for Parkinson's disease. Brain. 2008;131:389–396. doi: 10.1093/brain/awm304. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Schwartz JC, Sokoloff P. Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur J Neurosci. 2000;12(no. 6):2117–2123. doi: 10.1046/j.1460-9568.2000.00089.x. [DOI] [PubMed] [Google Scholar]
- Borycz J, Pereira MF, Melani A, Rodrigues RJ, Köfalvi A, Panlilio L, Pedata F, Goldberg SR, Cunha RA, Ferré S. Differential glutamate-dependent and glutamate-independent adenosine A1 receptor-mediated modulation of dopamine release in different striatal compartments. J Neurochem. 2007;101(no. 2):355–363. doi: 10.1111/j.1471-4159.2006.04386.x. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Cottini L, Fumagalli M, Ceruti S, Abbracchio MP. Blockade of A2A adenosine receptors prevents basic fibroblast growth factor-induced reactive astrogliosis in rat striatal primary astrocytes. Glia. 2003;43(no. 2):190–194. doi: 10.1002/glia.10243. [DOI] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM, Jacobson KA. Activation of the A2A adenosine receptor inhibits nitric oxide production in glial cells. FEBS Lett. 1998;429(no. 2):139–142. doi: 10.1016/s0014-5793(98)00556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DJ, Doder M, Osman S, Luthra SK, Hirani E, Hume S, Kase H, Kilborn J, Martindill S, Mori A. Positron emission tomography analysis of [11C]KW-6002 binding to human and rat adenosine A2A receptors in the brain. Synapse. 2008;62(no. 9):671–681. doi: 10.1002/syn.20539. [DOI] [PubMed] [Google Scholar]
- Brown SJ, James S, Reddington M, Richardson PJ. Both A1 and A2a purine receptors regulate striatal acetylcholine release. J Neurochem. 1990;55(no. 1):31–38. doi: 10.1111/j.1471-4159.1990.tb08817.x. [DOI] [PubMed] [Google Scholar]
- Canals M, Marcellino D, Fanelli F, Ciruela F, De Benedetti P, Goldberg SR, Neve K, Fuxe K, Agnati LF, Woods AS, Ferré S, Lluis C, Bouvier M, Franco R. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer 2003. J Biol Chem. 2003;278(no. 47):46741–46749. doi: 10.1074/jbc.M306451200. [DOI] [PubMed] [Google Scholar]
- Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, Müller C, Woods AS, Hope BT, Ciruela F, Casadó V, Canela EI, Lluis C, Goldberg SR, Moratalla R, Franco R, Ferré S. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32(no. 11):2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- Carta AR, Kachroo A, Schintu N, Xu K, Schwarzschild MA, Wardas J, Morelli M. Inactivation of neuronal forebrain A receptors protects dopaminergic neurons in a mouse model of Parkinson's disease. J Neurochem. 2009;111(no. 6):1478–1489. doi: 10.1111/j.1471-4159.2009.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Langston JW, Di Monte DA. MK-801 temporarily prevents MPTP-induced acute dopamine depletion and MPP+ elimination in the mouse striatum. J Pharmacol Exp Ther. 1993;267(no. 3):1515–1520. [PubMed] [Google Scholar]
- Chan DK, Woo J, Ho SC, Pang CP, Law LK, Ng PW, Hung WT, Kwok T, Hui E, Orr K, Leung MF, Kay R. Genetic and environmental risk factors for Parkinson's disease in a Chinese population. J Neurol Neurosurg Psychiatry. 1998;65:781–784. doi: 10.1136/jnnp.65.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth WT, Jr, Swanson PD. Parkinson's disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J of Epidemiol. 2002;155(no. 8):732–738. doi: 10.1093/aje/155.8.732. [DOI] [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19(no.21):9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci. 2001;21(no. RC143):1–6. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Mosley TH, Alonso A, Huang X. Plasma urate and Parkinson's disease in the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol. 2009;169:1064–1069. doi: 10.1093/aje/kwp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church WH, Ward VL. Uric acid is reduced in the substantia nigra in Parkinson's disease: effect on dopamine oxidation. Brain Res Bull. 1994;33(no. 4):419–425. doi: 10.1016/0361-9230(94)90285-2. [DOI] [PubMed] [Google Scholar]
- Ciccarelli R, Ballerini P, Sabatino G, Rathbone MP, D'Onofrio M, Caciagli F, Di Iorio P. Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci. 2001;19(no. 4):395–414. doi: 10.1016/s0736-5748(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Coccurello R, Breysse N, Amalric M. Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology. 2004;29:1451–1461. doi: 10.1038/sj.npp.1300444. [DOI] [PubMed] [Google Scholar]
- Corsi C, Pinna A, Gianfriddo M, Melani A, Morelli M, Pedata F. Adenosine A2A receptor antagonism increases striatal glutamate outflow in dopamine-denervated rats. Eur J Pharmacol. 2003;464(no. 1):33–38. doi: 10.1016/s0014-2999(03)01352-9. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Johansson B, van der Ploeg I, Sebastião AM, Ribeiro JA, Fredholm BB. Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res. 1994;27(no. 649 (1-2)):208–216. doi: 10.1016/0006-8993(94)91066-9. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Johansson B, Constantino MD, Sebastião AM, Fredholm BB. Evidence for high-affinity binding sites for the adenosine A2A receptor agonist [3H] CGS 21680 in the rat hippocampus and cerebral cortex that are different from striatal A2A receptors. Naunyn Schmiedebergs Arch Pharmacol. 1996;353(no. 3):261–271. doi: 10.1007/BF00168627. [DOI] [PubMed] [Google Scholar]
- Daré E, Schulte G, Karovic O, Hammarberg C, Fredholm BB. Modulation of glial cell functions by adenosine receptors. Physiol Behav. 2007;10(no. 92 (1-2)):15–20. doi: 10.1016/j.physbeh.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Davies KJ, Sevanian A, Muakkassah-Kelly SF, Hochstein P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem J. 1986;235:747–754. doi: 10.1042/bj2350747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM. Observations on serum uric acid levels and the risk of idiopathic Parkinson's disease. Am J Epidemiol. 1996;144(no. 5):480–484. doi: 10.1093/oxfordjournals.aje.a008954. [DOI] [PubMed] [Google Scholar]
- De Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Ann Neurol. 2005;58(no. 5):797–800. doi: 10.1002/ana.20663. [DOI] [PubMed] [Google Scholar]
- Delle Donne KT, Sonsalla PK. Protection against methamphetamine-induced neurotoxicity to neostriatal dopaminergic neurons by adenosine receptor activation. J Pharmacol Exp Ther. 1994;271(no. 3):1320–1326. [PubMed] [Google Scholar]
- de Mendonça A, Sebastião AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Rev. 2000;33(no. 2-3):258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- De Vera M, Rahman MM, Rankin J, Kopec J, Gao X, Choi H. Gout and the risk of Parkinson's disease: a cohort study. Arthritis Rheum. 2008;59:1549–1554. doi: 10.1002/art.24193. [DOI] [PubMed] [Google Scholar]
- Diaz-Cabiale Z, Hurd Y, Guidolin D, Finnman UB, Zoli M, Agnati LF, Vanderhaeghen JJ, Fuxe K, Ferré S. Adenosine A2A agonist CGS 21680 decreases the affinity of dopamine D2 receptors for dopamine in human striatum. Neuroreport. 2001;12(no. 9):1831–1834. doi: 10.1097/00001756-200107030-00014. [DOI] [PubMed] [Google Scholar]
- Diaz-Cabiale Z, Vivo M, Del Arco A, O'Connor WT, Harte MK, Muller CE, Martinez E, Popoli P, Fuxe K, Ferré S. Metabotropic glutamate mGlu5 receptor-mediated modulation of the ventral striopallidal GABA pathway in rats. Interactions with adenosine A(2A) and dopamine D(2) receptors. Neurosci Lett. 2002;324(no. 2):154–158. doi: 10.1016/s0304-3940(02)00179-9. [DOI] [PubMed] [Google Scholar]
- Du Y, Chen CP, Tseng CY, Eisenberg Y, Firestein BL. Astrogliamediated effects of uric acid to protect spinal cord neurons from glutamate toxicity. Glia. 2007;55(no. 5):463–472. doi: 10.1002/glia.20472. [DOI] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homoocysteine levels endanger dopaminergic neurons in models of Parkinson's disease. J Neurochem. 2002;80(1):101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Dux E, Fastbom J, Ungerstedt U, Rudolphi K, Fredholm BB. Protective effect of adenosine and a novel xanthine derivative propentofylline on the cell damage after bilateral carotid occlusion in the gerbil hippocampus. Brain Res. 1990;21(no. 516):248–256. doi: 10.1016/0006-8993(90)90925-2. [DOI] [PubMed] [Google Scholar]
- Egertová M, Elphick MR. Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal tail of CB. J Comp Neurol. 2000;26(no. 422):159–171. doi: 10.1002/(sici)1096-9861(20000626)422:2<159::aid-cne1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Ongini E, Costentin J, Vaugeois JM. In vivo labelling of the adenosine A2A receptor in mouse brain using the selective antagonist [3H]SCH 58261. Eur J Neurosci. 2001;14:1567–1570. doi: 10.1046/j.0953-816x.2001.01771.x. [DOI] [PubMed] [Google Scholar]
- Evans MC, Swan JH, Meldrum BS. An adenosine analogue, 2-chloroadenosine, protects against long term development of ischaemic cell loss in the rat hippocampus. Neurosci Lett. 1987;29(no. 83):287–292. doi: 10.1016/0304-3940(87)90101-7. [DOI] [PubMed] [Google Scholar]
- Fall PA, Fredrikson M, Axelson O, Granerus AK. Nutritional and occupational factors influencing the risk of Parkinson's disease: a case-control study in southeastern Sweden. Mov Disord. 1999;14:28–37. doi: 10.1002/1531-8257(199901)14:1<28::aid-mds1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Fernandez HH, Greeley DR, Zweig RM, Wojcieszek J, Mori A, Sussman NM. for the 6002-US-051 Study Group. Istradefylline as monotherapy for Parkinson disease: Results of the 6002-US-051 trial. Parkinsonism Relat Disord. 2010;16(no. 1):16–20. doi: 10.1016/j.parkreldis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Ferré S, von Euler G, Johansson B, Fredholm BB, Fuxe K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci USA. 1991;88(no. 16):7238–7241. doi: 10.1073/pnas.88.16.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Fuxe K. Dopamine denervation leads to an increase in the intramembrane interaction between adenosine A2 and dopamine D2 receptors in the neostriatum. Brain Res. 1992;594(no. 1):124–130. doi: 10.1016/0006-8993(92)91036-e. [DOI] [PubMed] [Google Scholar]
- Ferré S, Karcz-Kubicha M, Hope BT, Popoli P, Burgueno J, Gutierrez MA, Casado V, Fuxe K, Goldberg SR, Lluis C, Franco R, Ciruela F. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc Natl Acad Sci USA. 2002;99(no. 18):11940–011945. doi: 10.1073/pnas.172393799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Goldberg SR, Lluis C, Franco R. Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology. 2009;56 1:226–234. doi: 10.1016/j.neuropharm.2008.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2areceptors. Glia. 1996;18:152–60. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Butcher RD, Gebicke-Haerter PJ. Protein kinase C-mediated regulation of inducible nitric oxide synthase expression in cultured microglial cells. J Neuroimmunol. 1998;1(no. 92):170–178. doi: 10.1016/s0165-5728(98)00201-x. [DOI] [PubMed] [Google Scholar]
- Franzoni F, Colognato R, Galetta F, Laurenza I, Barsotti M, Di Stefano R, Bocchetti R, Regoli F, Carpi A, Balbarini A, Migliore L, Santoro G. An in vitro study on the free radical scavenging capacity of ergothioneine: comparison with reduced glutathione, and trolox. Biomed Pharmacother. 2006;60(no. 8):453–457. doi: 10.1016/j.biopha.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacological reviews. 1999;51(no. 1):83–133. [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Agnati LF, Jacobsen K, Hillion J, Canals M, Torvinen M, Tinner-Staines B, Staines W, Rosin D, Terasmaa A, Popoli P, Leo G, Vergoni V, Lluis C, Ciruela F, Franco R, Ferré S. Receptor heteromerization in adenosine A2A receptor signaling: relevance for striatal function and Parkinson's disease. Neurology. 2003;9(no. 61) 6:S19–23. doi: 10.1212/01.wnl.0000095206.44418.5c. [DOI] [PubMed] [Google Scholar]
- Gao X, Chen H, Choi HK, Curhan G, Schwarzschild MA, Ascherio A. Diet, Urate and PD risk in men. Am Journal of epidemiology. 2008;167:831–838. doi: 10.1093/aje/kwm385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21(no. 2):404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Gomes CA, Simões PF, Canas PM, Quiroz C, Sebastião AM, Ferré S, Cunha RA, Ribeiro JA. GDNF control of the glutamatergic cortico-striatal pathway requires tonic activation of adenosine A2A receptors. J Neurochem. 2009;108:1208–1219. doi: 10.1111/j.1471-4159.2009.05876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandinetti A, Morens DM, Reed D, MacEachern D. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson's disease. Am J Epidemiol. 1994;139(no. 12):1129–1138. doi: 10.1093/oxfordjournals.aje.a116960. [DOI] [PubMed] [Google Scholar]
- Guerreiro S, Ponceau A, Toulorge D, Martin E, Alvarez-Fischer D, Hirsch EC, Michel PP. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: potentiation by low-level depolarization. J Neurochem. 2009;109:1118–1128. doi: 10.1111/j.1471-4159.2009.06040.x. [DOI] [PubMed] [Google Scholar]
- Haberman F, Tang SC, Arumugam TV, Hyun DH, Yu QS, Cutler RG, Guo Z, Holloway HW, Greig NH, Mattson MP. Soluble neuroprotective antioxidant uric acid analogs ameliorate ischemic brain injury in mice. Neuromolecular Med. 2007;9(no. 4):315–323. doi: 10.1007/s12017-007-8010-1. [DOI] [PubMed] [Google Scholar]
- Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(no. 9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser RA, Hubble JP, Truong DD. Istradefylline US-001 Study Group. Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology. 2003;61(no. 3):297–303. doi: 10.1212/01.wnl.0000081227.84197.0b. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Shulman LM, Trugman JM, Roberts JW, Mori A, Ballerini R, Sussman NM. Istradefylline 6002-US-013 Study Group. Study of istradefylline in patients with Parkinson's disease on levodopa with motor fluctuations. Mov Disord. 2008;23(no. 1):2177–2185. doi: 10.1002/mds.22095. [DOI] [PubMed] [Google Scholar]
- Hellenbrand W, Seidler A, Boeing H, Robra HP, Vieregge P, Nischan P, Joerg J, Oertel WH, Schneider E, Ulm G. Diet and Parkinson's disease. II Diet and Parkinson's disease. I: A possible role for the past intake of specific foods and food groups. Results from a self administered food-frequency questionnaire in a case-control study. Neurology. 1996;47:636–643. doi: 10.1212/wnl.47.3.636. [DOI] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferré S, Fuxe K. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277(no. 20):18091–18097. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- Hindley S, Herman MA, Rathbone MP. Stimulation of reactive astrogliosis in vivo by extracellular adenosine diphosphate or an adenosine A2 receptor agonist. J Neurosci Res. 1994;38(no. 4):399–406. doi: 10.1002/jnr.490380405. [DOI] [PubMed] [Google Scholar]
- Ho SC, Woo J, Lee CM. Epidemiologic study of Parkinson's disease in Hong Kong. Neurology. 1989;39:1314–1318. doi: 10.1212/wnl.39.10.1314. [DOI] [PubMed] [Google Scholar]
- Hooper CD, Bagasra O, Marini JC, Zborek A, Ohnishi ST, Kean R, Champion JM, Sarker AB, Bobroski L, Farber JL, Akaike T, Maeda H, Koprowski H. Prevention of experimental allergic encephalomyelitis by targeting nitric oxide and peroxynitrite: implications for the treatment of multiple sclerosis. Proc Natl Acad Sci. 1997;94:2528–2533. doi: 10.1073/pnas.94.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chudhry I, Koprowski H. Uric acid, a natural scavenger of peroxynitrite in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea consumption and the risk of Parkinson's disease. Mov Disord. 2007;22(15):2242–2248. doi: 10.1002/mds.21706. [DOI] [PubMed] [Google Scholar]
- Huang QY, Wei C, Yu L, Coelho JE, Shen HY, Kalda A, Linden J, Chen JF. Adenosine A2A receptors in bone marrow-derived cells but not in forebrain neurons are important contributors to 3-nitropropionic acid-induced striatal damage as revealed by cell-type-selective inactivation. J Neurosci. 2006;26(no. 44):11371–11378. doi: 10.1523/JNEUROSCI.1907-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kurokawa M, Aoyama S, Kuwana Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson's disease. J Neurochem. 2002;80:262–70. doi: 10.1046/j.0022-3042.2001.00694.x. [DOI] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, Mateo D, Gimenez-Roldan S. Premorbid smoking, alcohol consumption, and coffee drinking habits in Parkinsons disease: a case control study. Mov Disord. 1992;7(no. 4):339–344. doi: 10.1002/mds.870070407. [DOI] [PubMed] [Google Scholar]
- Joghataie MT, Roghani M, Negahdar F, Hashemi L. Protective effect of caffeine against neurodegeneration in a model of Parkinson's disease in rat: behavioral and histochemical evidence. Parkinsonism Relat Disord. 2004;10:465–468. doi: 10.1016/j.parkreldis.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Johansen KK, Wang L, Aasly JO, White LR, Matson WR, Henchcliffe C, Beal MF, Bogdanov M. Metabolomic profiling in LRRK2-related Parkinson's disease. PLoS One. 2009;4(no. 10):e7551. doi: 10.1371/journal.pone.0007551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B, Georgiev V, Fredholm BB. Distribution and postnatal ontogeny of adenosine A2A receptors in rat brain: comparison with dopamine receptors. Neuroscience. 1997;80(no. 4):1187–207. doi: 10.1016/s0306-4522(97)00143-7. [DOI] [PubMed] [Google Scholar]
- Jones PA, Smith RA, Stone TW. Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res. 1998;800(no. 2):328–335. doi: 10.1016/s0006-8993(98)00540-x. [DOI] [PubMed] [Google Scholar]
- Jones DC, Gunasekar PG, Borowitz JL, Isom GE. Dopamine induced apoptosis is mediated by oxidative stess and is enhanced by cyanide in differentiated PC12 cells. J Neurochem. 2000;74(no. 6):2296–2304. doi: 10.1046/j.1471-4159.2000.0742296.x. [DOI] [PubMed] [Google Scholar]
- Kachroo A, Orlando LR, Grandy DK, Chen JF, Young AB, Schwarzschild MA. Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J Neurosci. 2005;25(no. 45):10414–10419. doi: 10.1523/JNEUROSCI.3660-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo A, Irizarry MC, Schwarzschild MA. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Exp Neurol. 2010 doi: 10.1016/j.expneurol.2010.02.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartzinel R, Shoulson I, Calne DB. Studies with bromocriptine: III. Concomitant administration of caffeine to patients with idiopathic parkinsonism. Neurology. 1976;26:741–743. doi: 10.1212/wnl.26.8.741. [DOI] [PubMed] [Google Scholar]
- Kase H, Aoyama S, Ichimura M, Ikeda K, Ishii A, Kanda T, Koga K, Koike N, Kurokawa M, Kuwana Y, Mori A, Nakamura J, Nonaka H, Ochi M, Saki M, Shimada J, Shindou T, Shiozaki S, Suzuki F, Takeda M, Yanagawa K, Richardson PJ, Jenner P, Bedard P, Borrelli E, Hauser RA, Chase TN. KW-6002 US-001 Study Group. Progress in pursuit of therapeutic A2A antagonists: the adenosine A2A receptor selective antagonist KW6002: research and development toward a novel nondopaminergic therapy for Parkinson's disease. Neurology. 2003;61(no. 11 (Suppl 6)):S97–100. doi: 10.1212/01.wnl.0000095219.22086.31. [DOI] [PubMed] [Google Scholar]
- Kostic VS, Svetel M, Sternic N, Dragasevic N, Przedborski S. Theophylline increases “on” time in advanced parkinsonian patients. Neurology. 1999;52:1916. doi: 10.1212/wnl.52.9.1916. [DOI] [PubMed] [Google Scholar]
- Kulisevsky J, Barbanoj M, Gironell A, Antonijoan R, Casas M, Pascual-Sedano B. A double-blind crossover, placebo-controlled study of the adenosine A2A antagonist theophylline in Parkinson's disease. Clin Neuropharmacol. 2002;25:25–31. doi: 10.1097/00002826-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Kull B, Ferré S, Arslan G, Svenningsson P, Fuxe K, Owman C, Fredholm BB. Reciprocal interactions between adenosine A2A and dopamine D2 receptors in Chinese hamster ovary cells co-transfected with the two receptors. Biochem Pharmacol. 1999;58(no. 6):1035–1045. doi: 10.1016/s0006-2952(99)00184-7. [DOI] [PubMed] [Google Scholar]
- Kyowa Hakko-Kirin Co. [site accessed 2/8/10];2007 Jun 03; URL: http://www.kyowa-kirin.co.jp/english/news/kyowa/er060307.html.
- Kyowa Hakko-Kirin Co. [site accessed 2/15/10];2008 Feb 08; URL: http://www.kyowa-kirin.co.jp/english/news/kyowa/er080228_01.html.
- Kyowa Hakko-Kirin Co. [site accessed 2/8/10];2009 Jan 15; URL: http://www.kyowa-kirin.co.jp/english/news/2009/e2009011501.html.
- Larumbe Ilundain R, Ferrer Valls JV, Vines Rueda JJ, Guerrero D, Fraile P. Case-control study of markers of oxidative stress and metabolism of blood iron in Parkinson's disease. Rev Esp Salud Publica. 2001;75(no. 1):43–53. [PubMed] [Google Scholar]
- Lau YS, Mouradian MM. Protection against acute MPTP-induced dopamine depletion in mice by adenosine A1 agonist. J Neurochem. 1993;60(no. 2):768–771. doi: 10.1111/j.1471-4159.1993.tb03215.x. [DOI] [PubMed] [Google Scholar]
- Lee YC, Chien CL, Sun CN, Huang CL, Huang NK, Chiang MC, Lai HL, Lin YS, Chou SY, Wang CK, Tai MH, Liao WL, Lin TN, Liu FC, Chern Y. Characterization of the rat A2A adenosine receptor gene: a 4.8-kb promoterproximal DNA fragment confers selective expression in the central nervous system. Eur J Neurosci. 2003;18(no. 7):1786–1796. doi: 10.1046/j.1460-9568.2003.02907.x. [DOI] [PubMed] [Google Scholar]
- Lee JK, Won JS, Singh AK, Singh I. Adenosine kinase inhibitor attenuates the expression of inducible nitric oxide synthase in glial cells. Neuropharmacology. 2005;48(no. 1):151–160. doi: 10.1016/j.neuropharm.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci. 2010;10(no. 30(6)):2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeWitt PA, Guttman M, Tetrud JW, Tuite PJ, Mori A, Chaikin P, Sussman NM. 6002-US-005 Study Group. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson's disease: a double-blind, randomized, multicenter clinical trial (6002-US-005) Ann Neurol. 2008;63(no. 3):295–302. doi: 10.1002/ana.21315. [DOI] [PubMed] [Google Scholar]
- Li SC, Schoenberg BS, Wang CC, Cheng XM, Rui DY, Bolis CL, Schoenberg D. A prevalence of survey PD and other movement disorders in the People's Republic of China. Arch of Neurol. 1985;42:655–657. doi: 10.1001/archneur.1985.04060070045013. [DOI] [PubMed] [Google Scholar]
- Lindskog M, Svenningsson P, Pozzi L, Kim Y, Fienberg AA, Bibb JA, Fredholm BB, Nairn AC, Greengard P, Fisone G. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418(no. 6899):774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- Mally J, Stone TW. The effect of theophylline on parkinsonian symptoms. J Pharm Pharmacol. 1994;46:515–517. doi: 10.1111/j.2042-7158.1994.tb03840.x. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Carriba P, Filip M, Borgkvist A, Frankowska M, Bellido I, Tanganelli S, Müller CE, Fisone G, Lluis C, Agnati LF, Franco R, Fuxe K. Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology. 2008;54(no. 5):815–823. doi: 10.1016/j.neuropharm.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Marchi M, Raiteri L, Risso F, Vallarino A, Bonfanti A, Monopoli A. Effects of adenosine A1 and A2A receptor activation on the evoked release of glutamate from rat cerebrocortical synaptosomes. Br J Pharmacol. 2002;136:434–440. doi: 10.1038/sj.bjp.0704712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Diao L, Illes P, Zahniser NR, Larson GA, Johansson B, Fredholm BB, Dunwiddie TV. Modulation of hippocampal glutamatergic transmission by ATP is dependent on adenosine a(1) receptors. J Pharmacol Exp Ther. 2002;303(no. 1):356–363. doi: 10.1124/jpet.102.036731. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord. 2008;23(no. 4):474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- McGill University Health Center. [site accessed 2-7-10]; URL: http://clinicaltrials.gov/ct2/show/NCT00459420.
- Melani A, Pantoni L, Bordoni F, Gianfriddo M, Bianchi L, Vannucchi MG, Bertorelli R, Monopoli A, Pedata F. The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res. 2003;959:243–250. doi: 10.1016/s0006-8993(02)03753-8. [DOI] [PubMed] [Google Scholar]
- Melani A, Gianfriddo M, Vannucchi MG, Cipriani S, Baraldi PG, Giovannini MG, Pedata F. The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006;1073-1074:470–480. doi: 10.1016/j.brainres.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Melani A, Cipriani S, Vannucchi MG, Nosi D, Donati C, Bruni P, Giovannini MG, Pedata F. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain. 2009;132(no. 6):1480–1495. doi: 10.1093/brain/awp076. [DOI] [PubMed] [Google Scholar]
- Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E. Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport. 1998;9(no. 17):3955–3959. doi: 10.1097/00001756-199812010-00034. [DOI] [PubMed] [Google Scholar]
- Morano A, Jimenez-Jimenez FJ, Molina JA, Antolin MA. Risk Factors for Parkinson's disease: case-control study in the province of Caceres, Spain. Acta Neurol Scand. 1994;89(no. 3):164–170. doi: 10.1111/j.1600-0404.1994.tb01655.x. [DOI] [PubMed] [Google Scholar]
- Morelli M, Pinna A, Wardas J, Di Chiara G. Adenosine A2 receptors stimulate cfos expression in striatal neurons of 6-hydroxydopamine-lesioned rats. Neuroscience. 1995;67(no. 1):49–55. doi: 10.1016/0306-4522(94)00602-2. [DOI] [PubMed] [Google Scholar]
- Morelli M, Di Paolo T, Wardas J, Calon F, Xiao D, Schwarzschild MA. Role of adenosine A2A receptors in parkinsonian motor impairment and l-DOPA-induced motor complications. Prog Neurobiol. 2007;83(no. 5):293–309. doi: 10.1016/j.pneurobio.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol. 2009;193:589–615. doi: 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- Moresco RM, Todde S, Belloli S, Simonelli P, Panzacchi A, Rigamonti M, Galli-Kienle M, Fazio F. In vivo imaging of adenosine A2A receptors in rat and primate brain using [11C]SCH442416. Eur J Nucl Med Mol Imaging. 2005;32:405–413. doi: 10.1007/s00259-004-1688-5. [DOI] [PubMed] [Google Scholar]
- Nishi A, Liu F, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P. Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc Natl Acad Sci USA. 2003;100:1322–1327. doi: 10.1073/pnas.0237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaki T. ATP- and adenosine-mediated signaling in the central nervous system: adenosine stimulates glutamate release from astrocytes via A2a adenosine receptors. J Pharmacol Sci. 2004;94(no. 2):100–102. doi: 10.1254/jphs.94.100. [DOI] [PubMed] [Google Scholar]
- Ochi M, Koga K, Kurokawa M, Kase H, Nakamura J, Kuwana Y. Systemic administration of adenosine A(2A) receptor antagonist reverses increased GABA release in the globus pallidus of unilateral 6-hydroxydopamine-lesioned rats: a microdialysis study. Neuroscience. 2000;100(no. 1):53–62. doi: 10.1016/s0306-4522(00)00250-5. [DOI] [PubMed] [Google Scholar]
- Ochiishi T, Chen L, Yukawa A, Saitoh Y, Sekino Y, Arai T, Nakata H, Miyamoto H. Cellular localization of adenosine A1 receptors in rat forebrain: immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J Comp Neurol. 1999;411(no. 2):301–16. [PubMed] [Google Scholar]
- Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol. 2002;19(no. 5):640–653. doi: 10.1093/oxfordjournals.molbev.a004123. [DOI] [PubMed] [Google Scholar]
- Olah ME, Stiles GL. The role of receptor structure in determining adenosine receptor activity. Pharmacol Ther. 2000;85(no. 2):55–75. doi: 10.1016/s0163-7258(99)00051-0. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos S, Borgohain R, Kellett M, Giladi N, Tomic D, Coppell A, Barnard J, Zhu Y, O'Neill G. Efficacy of the Adenosine A2A receptor antagonist BIIB014 in Parkinson's disease (PD) Patients with Motor Fluctuations. Abstract [PD4.004]: 62nd AAN annual meeting; Toronto. 2010. [Google Scholar]
- Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson's disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. [site accessed 2/17/10]; URL: http://clinicaltrials.gov/ct2/show/NCT00833690.
- Pedata F, Gianfriddo M, Turchi D, Melani A. The protective effect of adenosine A2A receptor antagonism in cerebral ischemia. Neurol Res. 2005;27(no. 2):169–174. doi: 10.1179/016164105X21913. [DOI] [PubMed] [Google Scholar]
- Phillis JW. The effects of selective A1 and A2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995;705(no. 1-2):79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- Pierri M, Vaudano E, Sager T, Englund U. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology. 2005;48:517–524. doi: 10.1016/j.neuropharm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Pinna A, Tronci E, Schintu N, Simola N, Volpini R, Pontis S, Cristalli G, Morelli M. A new ethyladenine antagonist of adenosine A(2A) receptors: behavioral and biochemical characterization as an antiparkinsonian drug. Neuropharmacology. 2010;58(no. 3):613–623. doi: 10.1016/j.neuropharm.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Pintor A, Quarta D, Pèzzola A, Reggio R, Popoli P. SCH 58261 (an adenosine A(2A) receptor antagonist) reduces, only at low doses, K(+)-evoked glutamate release in the striatum. Eur J Pharmacol. 2001;421(no. 3):177–180. doi: 10.1016/s0014-2999(01)01058-5. [DOI] [PubMed] [Google Scholar]
- Popoli P, Pintor A, Domenici MR, Frank C, Tebano MT, Pèzzola A, Scarchilli L, Quarta D, Reggio R, Malchiodi-Albedi F, Falchi M, Massotti M. Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J Neurosci. 2002;22:1967–1975. doi: 10.1523/JNEUROSCI.22-05-01967.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post B, Merkus MP, de Haan RJ, Speelman JD CARPA Study Group. Prognostic factors for the progression of Parkinson's disease: a systematic review. Mov Disord. 2007;22:1839–1851. doi: 10.1002/mds.21537. [DOI] [PubMed] [Google Scholar]
- Preux PM, Condet A, Anglade C, Druet-Cabanac M, Debrock C, Macharia W, Couratier P, Boutros-Toni F, Dumas M. Parkinsons's disease and environmental factors. Matched case-control study in the Limousin region, France. Neuroepidemiology. 2000;19(no. 6):333–337. doi: 10.1159/000026273. [DOI] [PubMed] [Google Scholar]
- Proctor P. Similar functions of uric acid and ascorbate in man? Nature. 1970;228(no. 5274):868. doi: 10.1038/228868a0. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Traini C, Cipriani S, Gianfriddo M, Mello T, Giovannini MG, Galli A, Pedata F. The adenosine A2A receptor antagonist ZM241385 enhances neuronal survival after oxygen-glucose deprivation in rat CA1 hippocampal slices. Br J Pharmacol. 2009;157(no. 5):818–830. doi: 10.1111/j.1476-5381.2009.00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragonese P, Salemi G, Morgante L, Aridon P, Epifanio A, Buffa D, Scoppa F, Savettieri G. A case-control study on cigarette, alcohol, and coffee consumption preceeding parkinson's disease. Neuroepidemiology. 2003;22(no. 5):297–304. doi: 10.1159/000071193. [DOI] [PubMed] [Google Scholar]
- Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, Marler JR. Neuroprotective agents for clinical trials in Parkinson's disease: a systematic assessment. Neurology. 2003;60:1234–1240. doi: 10.1212/01.wnl.0000058760.13152.1a. [DOI] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Rodrigues RJ, Alfaro TM, Rebola N, Oliveira CR, Cunha RA. Colocalization and functional interaction between adenosine A(2A) and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J Neurochem. 2005;92(no. 3):433–441. doi: 10.1111/j.1471-4159.2004.02887.x. [DOI] [PubMed] [Google Scholar]
- Romanos E, Planas AM, Amaro S, Chamorro A. Uric acid reduces brain damage and improves the benefits of rt-PA in a rat model of thromboembolic stroke. J Cereb Blood Flow Metab. 2007;27(no. 1):14–20. doi: 10.1038/sj.jcbfm.9600312. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Robeva A, Woodard RL, Guyenet PG, Linden J. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol. 1998;401:163–186. [PubMed] [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H, Morens DM, Grandinetti A, Tung KH, Tanner CM, Masaki KH, Blanchette PL, Curb JD, Popper JS, White L. Association of coffee and caffeine intake with the risk of Parkinson disease. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Saaksjarvi K, Knekt P, Rissanen H, Laaksonen MA, Reunanen A, Mannisto S. Prospective study of coffee consumption and risk of Parkinson's disease. European Journal of Clinical Nutrition. 2008;62:908–915. doi: 10.1038/sj.ejcn.1602788. [DOI] [PubMed] [Google Scholar]
- Salim H, Ferré S, Dalal A, Peterfreund RA, Fuxe K, Vincent JD, Lledo PM. Activation of adenosine A1 and A2A receptors modulates dopamine D2 receptorinduced responses in stably transfected human neuroblastoma cells. J Neurochem. 2000;74(no. 1):432–439. doi: 10.1046/j.1471-4159.2000.0740432.x. [DOI] [PubMed] [Google Scholar]
- Saura J, Angulo E, Ejarque A, Casadó V, Tusell JM, Moratalla R, Chen JF, Schwarzschild MA, Lluis C, Franco R, Serratosa J. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem. 2005;95:919–29. doi: 10.1111/j.1471-4159.2005.03395.x. [DOI] [PubMed] [Google Scholar]
- Schering Plough Co. [site accessed 2/8/10];2008 Nov 24; URL: http://www.merck.com/newsroom/news-releasearchive/schering-plough-news-archive/all/releaseid=1229593.html.
- Schiffmann SN, Jacobs O, Vanderhaeghen JJ. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem. 1991;57:1062–1067. doi: 10.1111/j.1471-4159.1991.tb08257.x. [DOI] [PubMed] [Google Scholar]
- Schubert P, Ogata T, Marchini C, Ferroni S, Rudolphi K. Protective mechanisms of adenosine in neurons and glial cells. Ann N Y Acad Sci. 1997;825:1–10. doi: 10.1111/j.1749-6632.1997.tb48409.x. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Chen JF, Tennis M, Messing S, Kamp C, Ascherio A, Holloway RG, Marek K, Tanner CM, McDermott M, Lang AE. Relating caffeine consumption to Parkinson's disease progression and dyskinesias development. Mov Disord; Seventeenth Annual Symposia on Etiology, pathogenesis, and treatment of PD and other movement disorders; 2003a. pp. 1082–1083. [Google Scholar]
- Schwarzschild MA, Xu K, Oztas E, Petzer JP, Castagnoli K, Castagnoli N, Jr, Chen JF. Neuroprotection by caffeine and more specific A2A receptor antagonists in animal models of Parkinson's disease. Neurology. 2003b;61(no. 11) 6:S55–61. doi: 10.1212/01.wnl.0000095214.53646.72. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson's disease. Trends Neurosci. 2006;29(no. 11):647–54. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Schwid SR, Marek K, Watts A, Lang AE, Oakes D, Shoulson I, Ascherio A the Parkinson Study Group PRECEPT Investigators. Serum urate as a predictor of clinical and radiographic progression in Parkinson's disease. Arch Neurol. 2008;65:716–723. doi: 10.1001/archneur.2008.65.6.nct70003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott GS, Spitsin SV, Kean RB, Mikheeva T, Koprowski H, Hooper DC. Therapeutic intervention in experimental allergic encephalomyelitis by administration of uric acid precursors. Proc Natl Acad Sci. 2002;99(no. 2):16303–16308. doi: 10.1073/pnas.212645999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou T, Richardson PJ, Mori A, Kase H, Ichimura M. Adenosine modulates the striatal GABAergic inputs to the globus pallidus via adenosine A2A receptors in rats. Neurosci Lett. 2003;11(no 352(3)):167–170. doi: 10.1016/j.neulet.2003.08.059. [DOI] [PubMed] [Google Scholar]
- Shoulson I, Chase T. Caffeine and the antiparkinsonian response to levodopa or piribedil. Neurology. 1975;25:722–724. doi: 10.1212/wnl.25.8.722. [DOI] [PubMed] [Google Scholar]
- Simola N, Fenu S, Baraldi PG, Tabrizi MA, Morelli M. Involvement of globus pallidus in the antiparkinsonian effects of adenosine A(2A) receptor antagonists. Exp Neurol. 2006;202(no. 1):255–257. doi: 10.1016/j.expneurol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Simon DK, Swearingen CJ, Hauser RA, Trugman JM, Aminoff MJ, Singer C, Truong D, Tilley BC on behalf of the NET-PD investigators. Caffeine and progression of Parkinson's disease. Clinical Neuropharmacology. 2008;31(no. 4):189–196. doi: 10.1097/WNF.0b013e31815a3f03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy M, Silver D, Mendis T, Sutton J, Mori A, Chaikin P, Sussman NM. A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology. 2008;70(no. 23):2233–2240. doi: 10.1212/01.wnl.0000313834.22171.17. [DOI] [PubMed] [Google Scholar]
- Stromberg U, Waldeck B. Behavioural and biochemical interaction between caffeine and L-dopa. J Pharm Pharmacol. 1973;25:302–308. doi: 10.1111/j.2042-7158.1973.tb10012.x. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Hall H, Sedvall G, Fredholm BB. Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse. 1997;27(no. 4):322–335. doi: 10.1002/(SICI)1098-2396(199712)27:4<322::AID-SYN6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Fourreau L, Bloch B, Fredholm BB, Gonon F, Le Moine C. Opposite tonic modulation of dopamine and adenosine on c-fos gene expression in striatopallidal neurons. Neuroscience. 1999;89:827–837. doi: 10.1016/s0306-4522(98)00403-5. [DOI] [PubMed] [Google Scholar]
- Tan EK, Tan C, Fook-Chong SM, Lum SY, Chai A, Chung H, Shen H, Zhao Y, Teoh ML, Yih Y, Pavanni R, Chandran VR, Wong MC. Dosedependent protective effect of coffee, tea and smoking in Parkinson's disease: a study in ethnic Chinese. J Neurol Sci. 2003;216(no. 1):163–167. doi: 10.1016/j.jns.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Tan LC, Koh WP, Yuan JM, Wang R, Au WL, Tan JH, Tan EK, Yu MC. Differential effects of black versus green tea on risk of PD in the Singapore Chinese health study. Am J Epidemiol. 2007;167:553–560. doi: 10.1093/aje/kwm338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebano MT, Pintor A, Frank C, Domenici MR, Martire A, Pepponi R, Potenza RL, Grieco R, Popoli P. Adenosine A2A receptor blockade differentially influences excitotoxic mechanisms at pre- and postsynaptic sites in the rat striatum. J Neurosci Res. 2004;77(no. 1):100–107. doi: 10.1002/jnr.20138. [DOI] [PubMed] [Google Scholar]
- Tebano MT, Martire A, Chiodi V, Pepponi R, Ferrante A, Domenici MR, Frank C, Chen JF, Ledent C, Popoli P. Adenosine A2A receptors enable the synaptic effects of cannabinoid CB1 receptors in the rodent striatum. J Neurochem. 2009;110(no. 6):1921–1930. doi: 10.1111/j.1471-4159.2009.06282.x. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Takahashi S, Kikuchi T. The urate and xanthine concentrations in the cerebrospinal fluid in patients with vascular dementia of the Binswanger type, Alzheimer type dementia, and Parkinson's disease. J Neural Transm Park Dis Demet Sect. 1993;6(no. 2):119–126. doi: 10.1007/BF02261005. [DOI] [PubMed] [Google Scholar]
- Torvinen M, Marcellino D, Canals M, Agnati LF, Lluis C, Franco R, Fuxe K. Adenosine A2A receptor and dopamine D3 receptor interactions: evidence of functional A2A/D3 heteromeric complexes. Mol Pharmacol. 2005;67(no. 2):400–407. doi: 10.1124/mol.104.003376. [DOI] [PubMed] [Google Scholar]
- Tronci E, Simola N, Carta AR, De Luca MA, Morelli M. Potentiation of amphetamine-mediated responses in caffeine-sensitized rats involves modifications in A2A receptors and zif-268 mRNAs in striatal neurons. J Neurochem. 2006;98(no. 4):1078–1089. doi: 10.1111/j.1471-4159.2006.03943.x. [DOI] [PubMed] [Google Scholar]
- van Calker D, Biber K. The role of glial adenosine receptors in neural resilience and the neurobiology of mood disorders. Neurochem Res. 2005;30(no. 10):1205–1217. doi: 10.1007/s11064-005-8792-1. [DOI] [PubMed] [Google Scholar]
- van der Putten C, Zuiderwijk-Sick EA, van Straalen L, de Geus ED, Boven LA, Kondova I, IJzerman AP, Bajramovic JJ. Differential expression of adenosine A3 receptors controls adenosine A2A receptor-mediated inhibition of TLR responses in microglia. Immunol. 2009;82:12, 7603–7612. doi: 10.4049/jimmunol.0803383. [DOI] [PubMed] [Google Scholar]
- Wallace BA, Ashkan K, Heise CE, Foote KD, Torres N, Mitrofanis J, Benabid AL. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain. 2007;130:2129–2145. doi: 10.1093/brain/awm137. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, O'Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson's disease. Am J Epidemiol. 2007;166:561–567. doi: 10.1093/aje/kwm127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Ketsawatsakul U, Halliwell B. A reassessment of the peroxynitrite scavenging activity of uric acid. Ann N Y Acad Sci. 2002;962:242–259. doi: 10.1111/j.1749-6632.2002.tb04072.x. [DOI] [PubMed] [Google Scholar]
- Willett WC. Nutritional Epidemiology. 2nd. New York: Oxford Univ Press; 1998. [Google Scholar]
- Xu K, Bastia E, Schwarzschild MA. Therapeutic potential of adenosine A2A receptor antagonists in Parkinson's disease. Pharmacol Ther. 2004;105(no. 3):267–310. doi: 10.1016/j.pharmthera.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Xu K, Xu Y, Brown-Jermyn D, Chen JF, Ascherio A, Dluzen DE, Schwarzschild MA. Estrogen prevents neuroprotection by caffeine in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2006;26(no. 2):535–541. doi: 10.1523/JNEUROSCI.3008-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Xu YH, Chen JF, Schwarzschild MA. Neuroprotection by caffeine: Time course and role of its metabolites in the MPTP model of Parkinson Disease. Neuroscience. 2010 doi: 10.1016/j.neuroscience.2010.02.020. [Epub ahead of print] PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeum KJ, Russell RM, Krinsky NI, Aldini G. Biomarkers of antioxidant capacity in the hydrophilic and lipophilic compartments of human plasma. Arch Biochem Biophys. 2004;430:97–103. doi: 10.1016/j.abb.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Bruce-Keller AJ, Goodman Y, Mattson MP. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture and against focal ischemic brain injury in vivo. J Neurosci Res. 1998;53(no. 5):613–625. doi: 10.1002/(SICI)1097-4547(19980901)53:5<613::AID-JNR11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Yu L, Shen HY, Coelho JE, Araujo IM, Huang QY, Day YJ, Rebola N, Canas PM, Rapp EK, Ferrara J, Taylor D, Muller CE, Linden J, Cunha RA, Chen JF. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol. 2008;63(no. 3):338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- Zhang ZX, Roman GC. Worldwide occurrence of PD: an updated review. Neuroepidemiology. 1993;12:195–208. doi: 10.1159/000110318. [DOI] [PubMed] [Google Scholar]